

Abstract
Purpose of review
Primary (immotile) cilia are specialized organelles present on most cell types. Almost all of proteins associated with a broad spectrum of human cystic kidney diseases have been localized to the region in or around the cilia. Abnormal cilia structure and/or function have been reported in animal models and human cystic kidneys. The goal of this review is to discuss current understanding of the mechanisms by which abnormal genes/proteins and cilia interact to potentially influence renal cystogenesis.
Recent findings
Novel direct recording of cilia calcium levels/channel activity suggest that cilia form a calcium-mediated signaling microenvironment separate from the cytoplasm, which could provide a mechanism for cilia-specific downstream signaling. Genetic-based studies confirm that cilia are not required for cystogenesis but modulate cystic kidney disease severity through a novel, undefined mechanism. Mechanisms by which both cilia-associated and non-cilia associated proteins can alter cilia structure/function have also been identified.
Summary
Considerable progress has been made in defining the mechanisms by which abnormal genes and proteins affect cilia structure and function. However, the exact mechanisms by which these interactions cause renal cyst formation and progression of cystic kidney disease are still unknown.
Keywords: cilia, polycystic kidney disease, calcium, ciliopathies
Introduction
In the almost 15 years since a connection was first made between the primary cilium and polycystic kidney diseases (PKD) [1], there has been a substantial and growing interest in this organelle once thought to be “vestigial.” Studies in normal and cystic cells, animals and human kidneys have provided important insights into the role of cilia in normal kidney physiology and cystogenesis. This review will provide an overview of normal cilia structure and function, evidence for the role of cilia in the cystic kidney diseases collectively known as “ciliopathies” and ongoing areas of investigation and controversy as highlighted in recent published literature.
Structure and localization of primary cilia
Cilia are specialized cell organelles that protrude out from the cell membrane and are tethered to the membrane by basal bodies (Figure 1). They are comprised of microtubules aligned along the longitudinal axis and are categorized based on the structure of their microtubules and their function [2]. Motile cilia have a “9+2” structure or a “9+0 structure” and beat in rhythmic fashion in order to facilitate cell or fluid movement. Immotile, or primary cilia, have a “9+0” structure and do not move constitutively, but can bend in response to flow or mechanical stressors. Primary cilia are present on almost all cell types, including epithelial cells, endothelial cells, and fibroblasts [3]. Only a single primary cilium is typically present on each cell, although multiple cilia may be present in pathogenic states. In the kidney, primary cilia are present on the apical (urinary /luminal) surface of epithelial cells from all tubular segments.
Figure 1. Localization of Human Cystic Renal Disease Proteins.
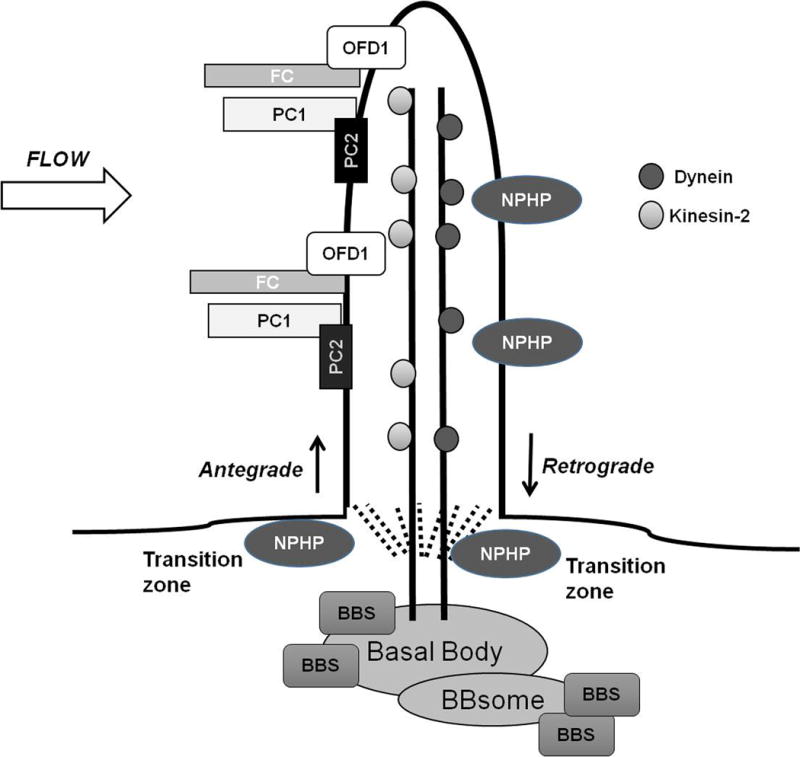
Proteins associated with ARPKD, ADPKD, Nephronophthisis, Bardet-Biedl Syndrome and Oro-facial-digital syndrome type 1 localize to primary cilia on renal epithelial cells. Proteins are transported in and out of the cilia by Kinesin-2 or IFT-dynein motors, respectively. Several NPHP proteins localize to the transition zone, the region between the cilia and the basal body. BBS -related proteins localize to the basal bodies where some aggregate to form the BBsome. Abnormal ciliary proteins are thought to interact with cilia to affect downstream signaling pathways including those related to cyclic AMP, mTOR, Canonical Wnt, G-protein coupled receptors, epidermal growth factor receptor family, MAP/ERK, planar cell polarity, calcium and cell cycle signaling. The exact mechanisms by which these interactions occur remain poorly defined.
(FC= fibrocystin; PC1 = polycystin 1; PC2 = polycystin 2; NPHP= nephrocystin; BBS = Bardet-Biedl proteins; OFD1= orofacial digital syndrome type 1)
The basal bodies (mother centrioles) serve as docking sites for proteins targeted to the cilia. Multiple proteins (cargo) traffic into and out of the cilia through a tightly regulated process called intraflagellar transport (IFT). Antegrade movement of into the cilia is facilitated by the carrier proteins, Kinesin-2 motors. Retrograde movement out of the cilia is facilitated by the carrier proteins, IFT-dynein motors [4, 5].
The renal “ciliopathies”
A potential role of cilia in the pathogenesis of PKD was first suggested in 1999, when the C. Elegans (worm) genes, LOV1 and PKD-2, homologues of the human ADPKD genes, PKD1 and PKD2, respectively, were found to encode proteins that localized to flagella [1]. Subsequently, fibrocystin (FC), the protein product of the ARPKD gene, PKHD1, was also found to localize to cilia [6]. Additional evidence for common pathogenic mechanisms in ARPKD and ADPKD was provided by the finding that PC1 and PC2 colocalized in cilia with the proteins from two ARPKD mouse models, the orpk and cpk mice [7, 8]. Proteins associated with other cystic kidney diseases have also been localized to the cilia or basal bodies, further expanding the family of “ciliopathies” (Table 1). These include proteins associated with Nephronophthisis, Bardet-Biedl Syndrome, Meckel-Gruber Syndrome and Joubert Syndrome [9].
Table 1.
Cilia-associated Human Cystic Kidney Disease Genes and Clinical Features
| Disease | Gene(s) | Clinical features |
|---|---|---|
| Autosomal Dominant Polycystic Kidney Disease (ADPKD) |
PKD1 PKD2 |
|
| Autosomal Recessive Polycystic Kidney Disease (ARPKD) | PKHD1 |
|
| Juvenile Nephronophthisis (JN) |
NPHP 1-11 GLIS2 NEK8 CEP164 TMEM67 TTC21B WDR19 ZNF423 ANKS6 CEP83 |
|
| Joubert syndrome (with renal disease, JS-Ren) |
RPGRIP1L CC2D2A CEP290 NPHP1 AHI1 TMEM216 TMEM138 TMEM237 OFD1 |
|
| Bardet-Biedl Syndrome (BBS) |
BBS 1-12 MKS1 MKS3 CEP290 SDCCAG8 SEPT7 |
|
| Oro-facial digital syndrome type I (OFD1) | OFD1 |
|
| Jeune syndrome/Short rib thoracic dysplasia |
IFT80 IFT 140 DYNC2H1 TTC21B WDR19 WDR60 NEK1 |
|
| Meckel-Gruber syndrome (MGS) |
MKS1-11 CEP290 NPHP3 RPGRIP1L CC2D2A TCTN2 B9D1 B9D2 TMEM67 TMEM216 TMEM231 |
|
Generated from information provided by Gene Reviews® (http://www.ncbi.nlm.nih.gov/books/NBK1116) and OMIM (http://www.ncbi.nlm.nih.gov/omim). Accessed November 9, 2014.
Cilia in PKD Pathogenesis
The localization of multiple PKD-associated proteins to the cilia and basal bodies was an important step towards defining the pathogenesis of cystic kidney diseases [10]. However, the mechanisms by which cilia participate in normal physiology and PKD pathogenesis were largely undefined [3, 11]. Numerous subsequent studies have attempted to identify links that connect cystic kidney disease genes, cilia and cystogenic processes. Multiple downstream/effector process implicated in cystogenesis have been implicated in cilia-associated signaling. These include pathways related to cyclic AMP, mTOR, Canonical Wnt, G-protein coupled receptors, epidermal growth factor receptor family, MAP/ERK, planar cell polarity, calcium and cell cycle signaling [9]. Despite these advances, there remain many unanswered questions about how mutant kidney proteins, cilia and downstream signaling pathways interconnect to produce cysts.
Several animal models have implicated abnormal cilia structure in PKD pathogenesis. The orpk mouse is an ARPKD model with a mutation in the gene that encodes polaris (alternatively called IFT88), an IFT protein. Renal tubular cells from orpk mice have short cilia and targeted disruption of IFT88 gene expression results in absent cilia in another animal model (chlamydomonas) [12]. Mice that lack kidney-specific expression of KIF3A, one of the essential IFT-associated kinesin motors [13], are born with normal kidneys but develop progressive renal cystic disease in the postnatal period. Renal tubular epithelial cells from this model lack cilia and have characteristics typical of cystic epithelia, including increased cell proliferation, apoptosis and abnormal epidermal growth factor receptor (EGFR) localization [14–16]. These findings suggested that abnormal or absent cilia can produce polycystic kidneys, but cilia are not required for normal kidney development. In contrast, PKD1 null mice form normal-appearing cilia, suggesting that polycystin is not required for normal cilia assembly [17].
Abnormal cilia function has also been implicated in cystic kidney disease pathogenesis. The postulated mechanism is that cilia function as mechanosensors, transmitting flow signals into chemical signaling pathways in the cell. Abnormal cilia structure or function would be expected to cause abnormal response to flow, thereby altering downstream signaling processes and promoting activation of procystogenic pathways. Because of the putative role of PKD2 as a calcium channel [18], many key observations have centered around the role of cilia and polycystins in flow-induced changes in intracellular calcium, an important component of second messenger signaling pathways. Normal kidney cells with intact cilia show increases in intracellular calcium in response to flow-induced cilia bending [19] and chemical removal of cilia blocks that response [20]. In contrast, cells from PKD1 knockout mice have normal-appearing cilia but exhibit impaired flow-mediated calcium influx responses [17]. Flow-induced calcium signaling has also been shown to be dependent on PC2-mediated calcium influx and removal of cilia abolishes the response [17]. Interestingly, studies in the orpk mouse (which have stunted cilia) showed that renal tubule epithelia from mutant mice have increased, dysregulated calcium entry and abnormal localization of PC2 to the entire apical membrane instead of only the cilia [21]. Taken together, these studies show that normal flow-induced intracellular calcium signaling requires not only intact cilia and polycystins/related PKD proteins but also regulation of PC2 localization to maintain tight control of calcium entry.
Studies in other ciliopathies have also supported a role of cilia in the pathogenesis of not only cystic kidney disease but also extrarenal manifestations, particularly those involving the skeleton, liver and eyes [2]. Nephronophthisis and Bardet-Biedl syndrome (BBS) are both heterogeneous autosomal recessive cystic kidney disorders with multiple causative genes and phenotypes distinct from ARPKD and ADPID (Table 1). Nephronophthisis is associated with significant ocular and other extra-renal manifestations [22] and multiple NPHP proteins have been localized to the cilia, specifically to the transition zone, the region at the base of the cilia that connects to the basal body [11] (Figure 1). Notably, patients with NPHP2, which is caused by mutations in the inversin (inv) gene, have polycystic kidneys and situs invertus, highlighting the role of cilia in mediating not only cystic kidney disease but also left-right axis determination during development [23]. BBS also has prominent extrarenal manifestations, including retinal abnormalities, obesity, diabetes mellitus, cognitive delays and polydactyly [24]. Multiple BBS proteins localize to the basal body, including 7 that form a complex called the BBsome, which has been shown to be essential for ciliagenesis [25]. Basal body dysfunction in multiple cell types (e.g., retinal, renal, pulmonary) is thought to underlie the pleiotropic aspects of BBS [26].
Recent insights and controversies
Although a central role for cilia in the pathogenesis of PKD remains undisputed, recent studies have called into question several purported mechanisms by which cilia participate in cystogenesis. These studies highlight the complexities of these processes, and may help explain the widely variable clinical features and disease course among the ciliopathies.
Interrelationship of Polycystins and Cilia
To study the interrelationship between loss of polycystins and loss of cilia, Ma and colleagues [27] created mice that lacked postnatal expression of PKD1 or PKD2, KIF3A or IFT20 or a combination of Pkd1/Kif3a or Pkd2/Ift2. As predicted, mice that lacked Pkd1 or Pkd2 but had intact cilia had severe cystic kidney disease by postnatal day 24; mice that had intact polycystins but lacked Kif3a or Ift20 had mild cystic kidney disease. Unexpectedly, they found that mice that lacked Pkd1 or Pkd2 and lacked intact cilia (“double knockouts”) had cystic kidney disease that was milder than the animals that lacked polycystin but had intact cilia. This suggested that the loss of cilia conveyed a protective effect by slowing cystogenesis. In further experiments, they found that altering polycystin expression levels did not alter the mild cystic kidney disease seen in the cilia null mice. Thus, the severity of cystic kidney disease after polycystin loss was dependent on the presence of intact cilia, but the severity of disease after loss of cilia was independent of polycystin levels. Similar findings were confirmed in later (adult)-onset ADPKD models. This “cilia-dependent cyst-activating mechanism” was found to be independent of several of the known pro-cystogenic pathways, including MAPK/ERK, mTOR and cAMP-related signaling. The identification of a new, mechanism for altering cystic kidney disease severity and progression raises the possibility of new pathways and targets for pharmacologic inhibition.
Cilia Assembly/Disassembly and Cystogenesis
Mice lacking max-interaction protein 1 (Mx1), a transcription factor, develop slowly progressive polycystic kidney disease. However, Mx1 is not expressed in the cilia or basal bodies, so the relationship of Mx1 to cystogenesis was not evident. To explore potential mechanisms, Ko et al [28] examined kidneys from Mx1-deficient mice and confirmed they had a typical polycystic phenotype, including truncated cilia and increased activation of the pro-proliferative bRAF/MEK/ERK signaling pathway. Normal cilia length could be restored by ectopic re-expression of Mx1. They also found that Mx1-deficient kidneys have decreased expression of several cilia structural genes, including Ift20. Further studies showed that Mx1 is a positive regulator Ift20 expression via effects on the transcription factor, Ets. This study identified a novel mechanism by which non-ciliary genes could impact cilia assembly and structure.
Cilia assembly and disassembly during cell division are tightly regulated by acetylases and deacetylases, which also regulate the assembly of centrosomes during cell division. PKD epithelia have increased polyploidy consistent with abnormal centrosome regulation. Zhou et al [29] found that overexpression of SIRT2, a NAD+-dependent protein deacetylase that regulates centrosomes and cilia disassembly decreased the number of renal epithelial cells with cilia, whereas SIRT2 inhibition increased the number. SIRT2 overexpression also increased polyploidy. These findings suggested dysregulated SIRT2 expression in cilia and centrosome could induce polyploidy and cilia defects characteristic of PKD and suggest a new potential therapeutic target.
Cilia-Associated Calcium Signaling
Flow-mediated intracellular calcium release regulated by polycystins is one of the earliest pathways implicated in cystogenesis [17]. However, direct measurements of cilia calcium concentrations and channel activity have not been possible. Decaen and colleagues overcame this barrier and provided the first report of direct recording of cilia calcium channel activity [30]. They then applied this novel methodology to further understand calcium concentration and movement in the cell cytoplasm and cilia [31]. They found that basal cilia calcium concentrations were several times higher than those of the cytoplasm and that changes in ciliary calcium concentration could occur without causing changes in cytoplasmic calcium. They further identified a unique heteromeric TRP channel, PKD1L1-PKD2L1 that was responsible for regulating the calcium concentration in cilia. The findings of this study were in direct contrast to previously published studies that suggested that PKD1/PKD2 mediated calcium entry into the cilia was transmitted to the cytoplasm to influence intracellular calcium concentrations. Live cell imaging studies by Jin et al [32], provided additional evidence that calcium signaling could be modulated in the “ciliaplasm” separately from the cell body cytoplasm.
Kuo et al examined the role of cilia and calcium signaling in cystogenesis by studying the impact of intracellular calcium channel disruption on signaling [33]. Stable cell lines lacking PC2 (which resides on endoplasmic reticulum and the cilia membrane) or one of two inositol 1,4,5-trisphosphate receptors (InsP3R) subsets all developed cysts with intact cilia by 2 weeks. By 8 weeks, two cell lines (PC2 and InsP3R3) had intact cilia and cysts but one cell line (InsP3R1) lost cilia and had larger cysts than the other two. These studies suggest that disruption of intracellular calcium at the ER alone is sufficient to cause cysts and that intact cilia are not required for cyst initiation or growth, but may modulate the severity of cystic kidney disease in this model system.
Interactions of Cilia-Associated PKD Proteins
PC1, PC2, fibrocystin and several other proteins form a complex at the cilia cell membrane [34]. The interrelationships of the protein members of this purported signaling complex have not been well established, although PC1 and PC2 interact at their respective carboxy termini via a coiled-coiled domain [35]. In order to model interactions in human cells, Freedman et al [36] established inducible pluripotent stem (iPS) cell lines from fibroblasts of ADPKD patients and healthy controls. The 3 ADPKD samples all had confirmed Pkd1 mutations. iPS cells from ADPKD and healthy subjects elaborated cilia and had similar rates of apoptosis and proliferation. However, ADPKD iPS cells had decreased amount of PC2 on the cilia. Reintroduction of normal PC1 “rescued” the PC2 defect, suggesting that PC1 modulates cilia expression of PC2.
Oro facial digital syndrome type 1, caused by mutations in OFD1 (Table 1), is a ciliopathy, but its relationship to the other cilia associated PKD proteins was not known. Jerman et al [37] found that OFD1 is part of the large signaling complex that contains PC1, PC2, EGFR, and flotillins. In human ADPKD cells, they further showed that loss of PC1 cilia localization was associated with loss of cilia localization of the other protein complex members. These studies highlight the interrelatedness of the various ciliopathy proteins and suggest possible mechanisms by which altered expression or localization of a mutant protein can impact expression or localization of the other (normal) cilia-associated proteins.
Conclusion
A large body of research over the last 15 years has provided important insights into the role of cilia in the pathogenesis of cystic kidney diseases, including localization of multiple cystic kidney disease proteins to the cilia or basal bodies and recognition that the “ciliopathies” share many pathophysiologic features. Recent studies, however, have highlighted the complexity of the interplay of mutant proteins, cilia and cystogenesis. Although many ciliopathies demonstrate abnormal or absent cilia, cilia are not required for cyst formation. The presence of intact cilia may, in fact, promote worsening cystic kidney disease in the presence of abnormal PKD proteins [27]. New mechanisms whereby non-cilia associated proteins impact cilia structure and function have been identified [29, 33]. The interactions of diverse cilia-associated cystic kidney disease proteins continue to be investigated, with an expanding list of proteins that comprise the polycystin cilia signaling complex [36, 37]. A new technique to directly measure cilia calcium concentrations and channel activity demonstrated that cilia have a separate calcium signaling microenvironment modulated by a unique calcium channel [30, 31]. These and other studies provided important insights into the role of cilia in the pathogenesis of cystic kidney disease but the exact mechanisms by which mutant cystic kidney disease genes and cilia actually cause cysts remain poorly defined. Elucidation of these mechanisms may provide new targets for therapeutic interventions in these diseases.
Key Points.
Cilia are complex organelles present on most cell types in the body, including renal epithelial cells and participate in processes that may contribute to cyst formation such as flow-mediated intracellular calcium release.
Proteins from nearly all of the human cystic kidney diseases have been localized to the cilium or basal body, and abnormalities in cilia structure and/or function are common features of the “ciliopathies.”
Newer studies suggest that cilia form a distinct calcium-mediated signaling microenvironment separate from the cytoplasm, are capable of modulating the severity of PKD, and their structure or function may be modulated by both cilia-associated and non-cilia associated proteins.
There remain many unanswered questions about how the individual gene defects interact with cilia and downstream signaling processes to initiate cyst development and progressive cystic kidney disease.
Acknowledgments
None
Financial support and sponsorship: Dr. Dell receives funding support from NIH/NIDDK (R01DK085099 and DCC Pilot and Feasibility Program)
Footnotes
Conflicts of Interest: None
References
- 1.Barr MM, Sternberg PW. A polycystic kidney-disease gene homologue required for male mating behaviour in C. elegans. Nature. 1999;401(6751):386–9. doi: 10.1038/43913. [DOI] [PubMed] [Google Scholar]
- 2.Ferkol TW, Leigh MW. Ciliopathies: the central role of cilia in a spectrum of pediatric disorders. J Pediatr. 2012;160(3):366–71. doi: 10.1016/j.jpeds.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3*.Patel A. The Primary cilium calcium channels and their role in flow sensing. Pflugers Archiv: European journal of physiology. 2014 doi: 10.1007/s00424-014-1516-0. This review article presents an overview of cilia and their relationship to PKD with a particular emphasis on recent studies that have examined the relationship between cilia, flow and calcium channels. [DOI] [PubMed] [Google Scholar]
- 4.Scholey JM, Anderson KV. Intraflagellar transport and cilium-based signaling. Cell. 2006;125(3):439–42. doi: 10.1016/j.cell.2006.04.013. [DOI] [PubMed] [Google Scholar]
- 5.Mencarelli C, Mitchell A, Leoncini R, Rosenbaum J, Lupetti P. Isolation of intraflagellar transport trains. Cytoskeleton. 2013;70(8):439–52. doi: 10.1002/cm.21121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang MZ, Mai W, Li C, Cho SY, Hao C, Moeckel G, et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc Natl Acad Sci U S A. 2004;101(8):2311–6. doi: 10.1073/pnas.0400073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13(10):2508–16. doi: 10.1097/01.asn.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 8.Pazour GJ, San Agustin JT, Follit JA, Rosenbaum JL, Witman GB. Polycystin-2 localizes to kidney cilia and the ciliary level is elevated in orpk mice with polycystic kidney disease. Current biology: CB. 2002;12(11):R378–80. doi: 10.1016/s0960-9822(02)00877-1. [DOI] [PubMed] [Google Scholar]
- 9.Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533–43. doi: 10.1056/NEJMra1010172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pazour GJ. Intraflagellar transport and cilia-dependent renal disease: the ciliary hypothesis of polycystic kidney disease. J Am Soc Nephrol. 2004;15(10):2528–36. doi: 10.1097/01.ASN.0000141055.57643.E0. [DOI] [PubMed] [Google Scholar]
- 11.Yoder BK. Role of primary cilia in the pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2007;18(5):1381–8. doi: 10.1681/ASN.2006111215. [DOI] [PubMed] [Google Scholar]
- 12.Pazour GJ, Dickert BL, Vucica Y, Seeley ES, Rosenbaum JL, Witman GB, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151(3):709–18. doi: 10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A. 2003;100(9):5286–91. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Orellana SA, Sweeney WE, Neff CD, Avner ED. Epidermal growth factor receptor expression is abnormal in murine polycystic kidney. Kidney Int. 1995;47(2):490–9. doi: 10.1038/ki.1995.62. [DOI] [PubMed] [Google Scholar]
- 15.Dell KM, Nemo R, Sweeney WE, Jr, Avner ED. EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 2004;65(6):2018–29. doi: 10.1111/j.1523-1755.2004.00623.x. [DOI] [PubMed] [Google Scholar]
- 16.Wilson PD. Mouse models of polycystic kidney disease. Current topics in developmental biology. 2008;84:311–50. doi: 10.1016/S0070-2153(08)00606-6. [DOI] [PubMed] [Google Scholar]
- 17.Nauli SM, Alenghat FJ, Luo Y, Williams E, Vassilev P, Li X, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33(2):129–37. doi: 10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 18.Koulen P, Cai Y, Geng L, Maeda Y, Nishimura S, Witzgall R, et al. Polycystin-2 is an intracellular calcium release channel. Nature cell biology. 2002;4(3):191–7. doi: 10.1038/ncb754. [DOI] [PubMed] [Google Scholar]
- 19.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. The Journal of membrane biology. 2001;184(1):71–9. doi: 10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 20.Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. The Journal of membrane biology. 2003;191(1):69–76. doi: 10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 21.Siroky BJ, Ferguson WB, Fuson AL, Xie Y, Fintha A, Komlosi P, et al. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol. 2006;290(6):F1320–8. doi: 10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- 22.Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18(6):1855–71. doi: 10.1681/ASN.2006121344. [DOI] [PubMed] [Google Scholar]
- 23.Otto EA, Schermer B, Obara T, O’Toole JF, Hiller KS, Mueller AM, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34(4):413–20. doi: 10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24*.Forsythe E, Beales PL. Bardet-Biedl syndrome. European journal of human genetics: EJHG. 2013;21(1):8–13. doi: 10.1038/ejhg.2012.115. This review article presents an overview of Bardet Biedl Syndrome and includes a discusssion of the clinical features, pathogenesis, diagnostic approach, genetic counselling and disease management of this rare disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, et al. A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell. 2007;129(6):1201–13. doi: 10.1016/j.cell.2007.03.053. [DOI] [PubMed] [Google Scholar]
- 26.Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, et al. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425(6958):628–33. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- 27**.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S. Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet. 2013;45(9):1004–12. doi: 10.1038/ng.2715. The study examines the role of cilia and polycystins in the mechanism of cyst formation. Using a genetic-based approach the authors created mice that have post natal inactivation of polcystins (PC1 and PC2), cilia structural proteins or both. They found that the presence of cilia actually promoted cystogenesis in animals that lacked polcystin 1 or 2 and that loss of cilia in those mice was actually protective. This cilia-depedent cyst growth was not explained by activaton of common cystogenic pathways. These observations have important implications in PKD pathogenesis and also identify a potential novel cyst-modulating pathway. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28*.Ko JY, Yoo KH, Song SA, Kim do Y, Kong HK, Ahn C, et al. Inactivation of max-interacting protein 1 induces renal cilia disassembly through reduction in levels of intraflagellar transport 20 in polycystic kidney. J Biol Chem. 2013;288(9):6488–97. doi: 10.1074/jbc.M112.413302. This study identifies a novel mechanism whereby a non-cilia associated protein, Mx1, is able to modify cilia structure and demonstrate that Mx1 deficient mice develop ciliary defects due to los of Mx1 mediated expression of one of the intraflagellar transport proteins, IFT20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Zhou X, Fan LX, Li K, Ramchandran R, Calvet JP, Li X. SIRT2 regulates ciliogenesis and contributes to abnormal centrosome amplification caused by loss of polycystin-1. Hum Mol Genet. 2014;23(6):1644–55. doi: 10.1093/hmg/ddt556. This study examines the relationship of cilia and centrosome regulation and cystogenesis and presents for the first time evidence that SIRT2, a nicotinamide adenine dinucleotide (NAD)-dependent deacetylase, regulates both ciliogenesis and centrosome amplification. The study demonstrates that upregulation of SIRT2 in renal epithelia cells causes both ciliary defects and polyploidy, two features of cystic epithelia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30**.DeCaen PG, Delling M, Vien TN, Clapham DE. Direct recording and molecular identification of the calcium channel of primary cilia. Nature. 2013;504(7479):315–8. doi: 10.1038/nature12832. This study is the first to demonstrate direct recording of calcium channel activity in primary cilia. This methodology has important implications for studying the role of polycystins and other cilia-associated proteins in flow-induced calcium signaling and activation of downstream cystogenic mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31**.Delling M, DeCaen PG, Doerner JF, Febvay S, Clapham DE. Primary cilia are specialized calcium signalling organelles. Nature. 2013;504(7479):311–4. doi: 10.1038/nature12833. This study utilizes a direct cilia calcium channel recording technique recently developed by this same group to demonstrate that cilia are a unique calcium compartment separate from the cell body. They also show that calcium is regulated by a unique heterotrimeric TRP channel, PKD1L1-PKD2L1. This study contrasts with previous published studies of cilia-associated calcium signalling by demonstrating that changes in ciliary calcium concentration occur without changes in global cytoplasmic clacium. The results of this study have important implications for the understanding of mechanisms underlying cilia associated calcium signalling and its role in cystogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32*.Jin X, Mohieldin AM, Muntean BS, Green JA, Shah JV, Mykytyn K, et al. Cilioplasm is a cellular compartment for calcium signaling in response to mechanical and chemical stimuli. Cellular and molecular life sciences: CMLS. 2014;71(11):2165–78. doi: 10.1007/s00018-013-1483-1. This study utilizes live cell imaging to demonstrate differences in calcium concentrations in the cilium versus cell cytoplasm, and suggests that “cilioplasm” may function as a separate cellular compartment fo calcium signalling in response to mechanical or chemical stimuli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33**.Kuo IY, DesRochers TM, Kimmerling EP, Nguyen L, Ehrlich BE, Kaplan DL. Cyst formation following disruption of intracellular calcium signaling. Proc Natl Acad Sci U S A. 2014;111(39):14283–8. doi: 10.1073/pnas.1412323111. This study utilizes 2D and 3D cell/tissue culture systems to demonstrate that knockdown of endoplasmic reticulum calcium channels (including PC2) is sufficient to induce cystogenesis without impacting cilia formation. These techniques will be valuable in helping to define how PC2 mutations alter calcium signalling and promote cystogenesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang S, Zhang J, Nauli SM, Li X, Starremans PG, Luo Y, et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Molecular and cellular biology. 2007;27(8):3241–52. doi: 10.1128/MCB.00072-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qian F, Germino FJ, Cai Y, Zhang X, Somlo S, Germino GG. PKD1 interacts with PKD2 through a probable coiled-coil domain. Nat Genet. 1997;16(2):179–83. doi: 10.1038/ng0697-179. [DOI] [PubMed] [Google Scholar]
- 36*.Freedman BS, Lam AQ, Sundsbak JL, Iatrino R, Su X, Koon SJ, et al. Reduced ciliary polycystin-2 in induced pluripotent stem cells from polycystic kidney disease patients with PKD1 mutations. J Am Soc Nephrol. 2013;24(10):1571–86. doi: 10.1681/ASN.2012111089. This study reports a methodology for establishing induced pluripotent stem cells from fibroblasts of ADPKD patients and utilizes this technique to study the impact of PKD1 mutations on PC2 expression levels. This system may prove very valuable for investigating the functional effect of PKD mutations without the need for isolation of primary renal or other epithelial cell lines from affected patients. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37*.Jerman S, Ward HH, Lee R, Lopes CA, Fry AM, MacDougall M, et al. OFD1 and flotillins are integral components of a ciliary signaling protein complex organized by polycystins in renal epithelia and odontoblasts. PloS one. 2014;9(9):e106330. doi: 10.1371/journal.pone.0106330. This study reports for the first time that OFD1 proteins are part of a ciliary complex that includes polycystins, flotillin proteins and EGFR and provides the molecular basis for the shared renal cystic kidney disease phenotypes caused by mutations in OFD1 and PKD proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]