

Abstract
Identification of new reactions expands our knowledge of chemical reactivity and enables new synthetic applications. Accelerating the pace of this discovery process remains challenging. We describe a highly effective and simple platform for screening a large number of potential chemical reactions in order to discover and optimize previously unknown catalytic transformations thereby revealing new chemical reactivity. Our strategy is based on labeling one of the reactants with a polyaromatic chemical tag, which selectively undergoes photoionization-desorption process upon laser irradiation without the assistance of an external matrix and enables rapid mass spectrometric detection of any products originating from such labeled reactants in complex reaction mixtures without any chromatographic separation. This method was successfully employed for high-throughput discovery and subsequent optimization of two previously unknown benzannulation reactions.
High-throughput reaction-screening approaches that enable rapid and accurate detection of new products with unanticipated structures can substantially expand our knowledge of chemical reactivity. While several innovative strategies to address this general problem have been reported (1-11), development of a highly efficient, broadly useful and preparatively simple reaction-discovery platform remains challenging. We have recently employed matrix-assisted laser desorption/ionization and time-of-flight mass spectrometry (MALDI-TOF-MS) to analyze chemical transformations on the surface of self-assembled monolayers of alkanethiolates on gold (12). Despite the high throughput of the primary reaction screen and its ability to detect products with unanticipated structures, subsequent translation of the initially identified interfacial reactions to preparative, solution-phase processes often required substantial effort. We now describe the development of a new reaction-discovery strategy that features not only the excellent screening throughput but also a highly efficient translation of the initial “hits” into catalytic, synthetically useful transformations. The reactions are rapidly analyzed in solution using label-assisted laser desorption/ionization and time-of-flight mass spectrometry (LA-LDI-TOF-MS). This simple and highly effective approach is based on the incorporation of a readily available polyaromatic tag into the structure of a reactant, thereby greatly facilitating the desorption/ionization process and enabling rapid and selective MS analysis of hundreds of chemical reactions in solution under matrix-free conditions with excellent efficiency. After validation of the concept through monitoring the course of several known transformations, the technology was utilized to evaluate the outcome of 696 different reactant combinations, and led to the discovery of two previously unknown benzannulations.
Results and Discussion
Rapid screening of chemical reactions by MALDI-TOF-MS is attractive for two main reasons. First, the efficiency and throughput of this approach compares favorably to commonly used LCMS and GCMS methods since reaction mixtures are analyzed directly without any chromatographic fractionation. Second, the high sensitivity of this technique enables MS analysis of reactions performed on exceedingly small scale, enabling highly efficient miniaturization of experimental design. Indeed, accurate analytical data can be readily obtained using only pmols of analyte. Despite such desirable features, development of the solution-based MALDI-TOF-MS reaction-discovery platform presents a substantial challenge since ionization of the matrix commonly used for the desorption/ionization process, substantially complicates accurate detection of analytes with low molecular weights. A notable exception was reported by Senkan, who used a resonance-enhanced multiphoton ionization to selectively detect benzene in the presence of a cyclohexane (13). While this method was used to screen a relatively small library of heterogeneous catalysts for their ability to promote dehydrogenation, the approach is based on the detection of a specific reaction product and is not easily applicable to monitoring efficiency of many other reactions. In contrast, our main objective was to develop a broadly useful, practical reaction-discovery platform that can be readily employed to identify and optimize a range of new chemical transformations. We envisioned that introduction of an appropriate MS label into the structure of one of the reactants could promote a selective desorption/ionization process and enable accurate detection of products originating from such labeled analytes, completely eliminating the need for a matrix and greatly simplifying spectral analysis (Fig. 1A). While the use of MS labeling approach to facilitate the ionization process has been recognized (14-18) and used to optimize at least two established reactions (16,18), this powerful concept has not been employed for high-throughput discovery of new chemical transformations.
Figure 1. Use of LA-LDI-TOF-MS to monitor progress of a representative known reaction.

a, General strategy for monitoring the progress of chemical reactions using LA-LDI-TOF-MS, which entails labeling one of the reactants with a tag that permits matrix-free laser-induced desorption/ionization and rapid detection of any products originating from such labeled analyte. b, Reaction scheme of a representative chemical transformation of 1 to 2, which was studied using LA-LDI-TOF-MS. c, MS spectra for conversion of alcohol 1 to ester 2. d, Plot of relative ion intensity ratio (I2/I1) vs mole ratio (M2/M1) (y = 0.1324x + 0.0216, R2 = 0.99365). Error bars represent standard deviations.
Since commercial MALDI-TOF-MS instruments are typically equipped with lasers that irradiate in the UV region of electromagnetic spectrum, the effective MS label must readily undergo photoionization-desorption process upon laser irradiation without the assistance of an external matrix. In addition, such MS label should be chemically inert under a range of commonly used reaction conditions in organic and organometallic chemistry. It has been established that many polyaromatic compounds efficiently undergo photoionization-desorption process upon laser-induced irradiation in the UV region presumably due to their high molar absorptivity and ability to form radical cations that can be detected by MS (19,20). We initially examined a range of polyaromatic compounds and identified pyrene as an effective label for selective ionization. The progress of each reaction can be readily analyzed by monitoring conversion of a MS-labeled reactant A into the expected MS-labeled product AB in the presence of regent(s) C (Figure 1A), enabling selective detection of only two species in crude reaction mixtures under matrix free conditions. To validate the utility and generality of this method, we analyzed the progress of several known transformations. Treatment of pyrene-containing alcohol 1 with Fmoc-protected valine under standard esterification conditions produced the expected ester 2 (Figure 1B). The course of this reaction was readily analyzed by the disappearance of the peak of reactant with m/z 274.5 (Figure 1C) and formation of the product peak with m/z 596.6 (M+1). In addition to the qualitative assessment of the reaction progress, LA-LDI-TOF-MS could be readily used to quantify the conversion of 1 to 2 by measuring intensities of the MS peaks corresponding to the two compounds (Figure 1D). Having examined several other known reactions (Supplementary Fig. 1), we clearly established that LA-LDI-TOF-MS enabled efficient and accurate detection of pyrene-labeled products, validating the generality of this analytical method and setting the stage for its further implementation to search for new chemical reactivity.
Electron rich alkynes, especially siloxy alkynes, represent a fertile ground for developing new carbon-carbon and carbon-heteroatom bond-forming reactions (12, 21-29). Such reactions can be catalyzed by a range of transition metals as well as Brønsted acids or bases and typically proceed under mild conditions. To further explore reactivity of this important functional group, we employed LA-LDI-TOF-MS reaction screening platform using a pyrene-containing siloxy alkyne 3 (Fig. 2A). This labeled substrate A was treated with 23 different reactants B as well as a negative control. Each of the reactions was evaluated in the absence and presence of 29 individual reagents C including a negative solvent control, corresponding to 696 discrete experiments, which were set up in 1,2-dichloroethane at ambient temperature in a 96-well format using a conventional robotic liquid handler. The progress of each experiment was analyzed after 1 h, 1 day and 4 days by spotting a 0.8 μL aliquot of each reaction mixture onto a standard stainless steel plate used by MALDI-TOF instruments. Following solvent evaporation, each spot was analyzed directly using LDI-TOF-MS in a positive-ion reflector mode. The high throughput of this screening platform is highly noteworthy as 696 spectra were typically collected within two hours by a conventional MALDI-TOF spectrometer working in an automated data acquisition mode.
Figure 2. Reaction screen using LA-LDI-TOF-MS and identification of two catalytic benzannulations.
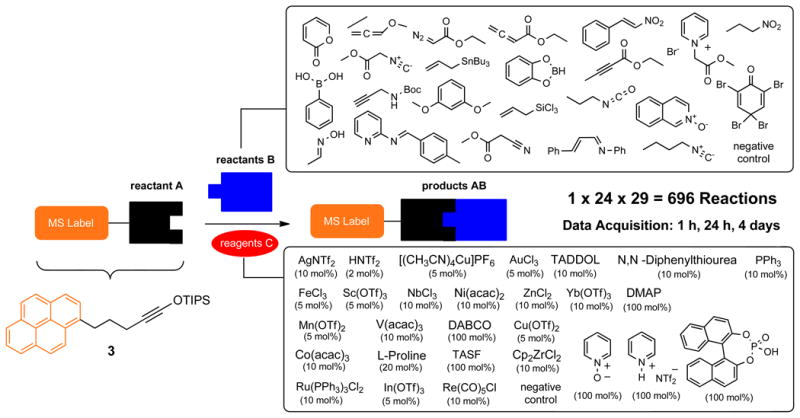
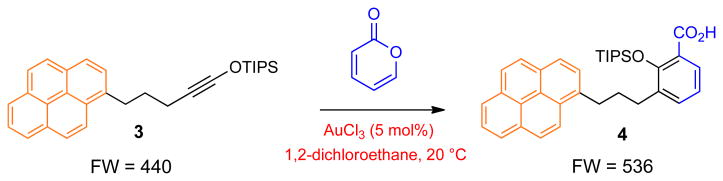
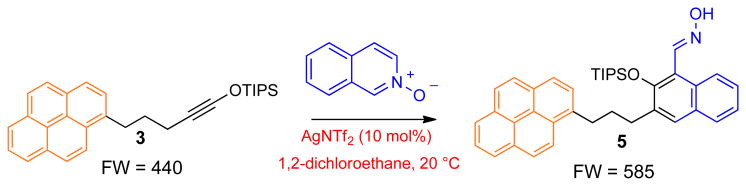
a, High-throughput screening of 696 potential reactions. Siloxy alkyne 3 contains a pyrene tag, which enables matrix-free detection of any products originating from this compound using LA-LDI-TOF-MS platform. TADDOL: (α,α,α,α-tetraaryl-1,3-dioxolane-4,5-dimethanol); DMAP: 4-Dimethylaminopyridine; DABCO: 1,4-diazabicyclo[2.2.2]octane; TASF: tris(dimethylamino)sulfonium difluorotrimethylsilicate. b, Initial identification of the benzannulation of siloxy alkyne 3 with 2-pyrone in the presence of AuCl3 to give carboxylic acid 4. c, Initial identification of the benzannulation of siloxy alkyne 3 with N-isoquinoline oxide in the presence of AgNTf2 to give oxime 5.
We analyzed all of the MS spectra and repeated several reactions that produced unanticipated products on larger scale in order to enable their complete structural characterization by other commonly used analytical methods. This effort identified two benzannulation reactions, which have not been previously described. Treatment of alkyne 3 with 2-pyrone in the presence of 5 mol% of AuCl3 afforded carboxylic acid 4 with m/z of 536, which was isolated in 75% yield (Fig. 2B and Supplementary Fig. 2). In addition, reaction of alkyne 3 with isoquinoline N-oxide in the presence of 10 mol% of AgNTf2 gave oxime 5 with m/z 585 in 52% yield (Fig. 2C and Supplementary Fig. 3). While initial structural assignments of 4 and 5 relied on NMR spectroscopy, the structures of both products were ultimately secured by X-ray crystallography. It is also noteworthy that neither of the two transformations occurred in the absence of a catalyst even at elevated temperatures.
Our next efforts centered on optimizing the efficiency of the transformation shown in Figure 2B. To this end, we examined several known gold and silver complexes and monitored each experiment using either LDI-TOF-MS or more conventional NMR spectrometry (Supplementary Table 1). This study demonstrated that LA-LDI-TOF-MS could be used for rapid reaction optimization and revealed that gold(I) complex 8 containing highly sterically congested Johnphos ligand proved excellent in catalyzing the reaction between siloxy alkynes 6 and 2-pyrones 7 to give siloxy acid 9 (Fig. 3). Subsequent one-flask desilylation with HF-pyridine afforded the corresponding salicylic acids 10. The initial step proceeded presumably via a formal [4+2] cycloaddition to give bicyclic intermediate A, which underwent subsequent fragmentation of the C-O bond and aromatization. While 2-pyrones are known to undergo [4+2] cycloadditions, such reactions generally require high temperatures and proceed typically with complete loss of CO2 from the initially produced cycloadducts (21). However, the tandem cycloaddition/fragmentation pathway described in Figure 3 has not been reported. This process successfully tolerated various substitution patterns of siloxy alkynes (R1) and 2-pyrones (R2 and R3). Reactions of unsubstituted 2-pyrone (R2=R3=H) with two alkyl-substituted siloxy alkynes resulted in efficient formation of the corresponding salicylic acids 10a and 10b. Introduction of electron-withdrawing groups (R3=CO2Me) into the 2-pyrone structure gave the expected benzannulation products 10c and 10d. Presence of the aromatic substituent (R3=Ph) was also well-tolerated and afforded biaryl product 10e. Finally, the use of 5-chloro-2-pyrone (R3=Cl) allowed us to test a wide range of substitution patterns on siloxy alkynes including various alkyl and aryl substituents. All reactions proceeded efficiently to give the corresponding benzannulation products 10f-l. The structures of 10a and 10e were verified by X-ray crystallography.
Figure 3. Mechanism and scope of Au-catalyzed benzannulation of siloxy alkyne with 2-pyrones.
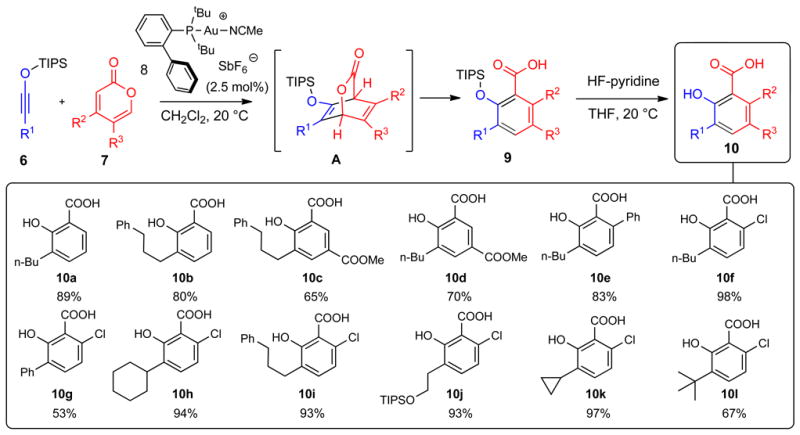
Siloxy alkyne 6 undergoes a [4+2] cycloaddition with 2-pyrone 7 to give a putative intermediate A, which undergoes subsequent fragmentation to deliver carboxylic acid 9. Compound numbers are shown in bold. Isolated yields are shown below each compound number. R is a generic alkyl or aryl substituent.
While the initial discovery or the reaction between siloxy alkyne 3 and isoquinoline N-oxide was made using Ag-based catalyst, we found during subsequent optimization studies (Supplementary Table 2) that the same gold(I) complex 8 proved to be optimal for catalyzing this benzannulation process (Fig. 4). In this case, the optimization study was performed using conventional NMR methods due to the high propensity for ion fragmentation of the major reaction product under LDI-TOF-MS conditions. This process represents another example of a formal [4+2] cycloaddition/fragmentation pathway, which begins presumably via the formation of tricyclic intermediate B, followed by C-N bond fragmentation and aromatization to give oxime 12. Subsequent one-flask desilylation can be efficiently achieved using tetrabutyl ammonium fluoride (TBAF) to give 2-naphthols 13. This reaction tolerated a wide range of substitutions of the siloxy alkyne (R1= alkyl) as exemplified by the efficient formation of benzannulation products 13a-d. Furthermore, the use of 1-methyl isoquinoline N-oxide (R2=Me) afforded the corresponding naphthol derivative 13e. Finally, a variety of halogenated isoquinoline N-oxides successfully afforded the expected products 13f-l, representing a range of highly functionalized, synthetically useful naphthalene derivatives. The structures of 13a and 13f were established by X-ray crystallography.
Figure 4. Mechanism and scope of Au-catalyzed benzannulation siloxy alkynes with isoquinoline N-oxides.
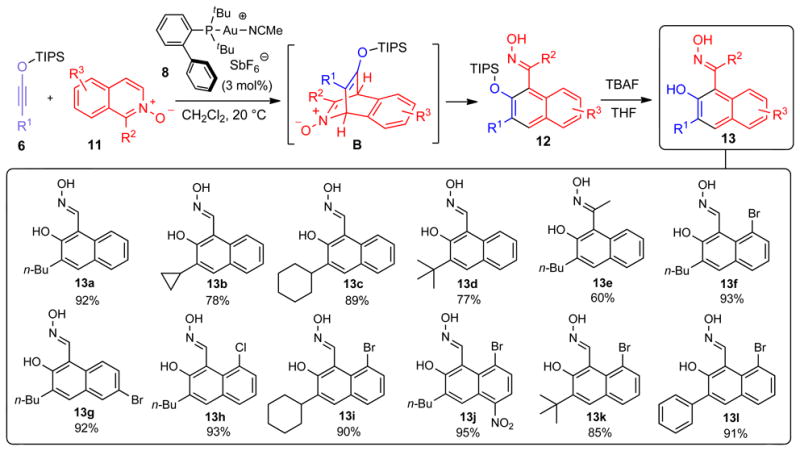
Siloxy alkyne 6 undergoes a [4+2] cycloaddition with isoquinoline N-oxide 11 to give a putative intermediate B, which undergoes subsequent fragmentation to deliver oxime 12. Compound numbers are shown in bold. Isolated yields are shown below each compound number. R is a generic alkyl or aryl substituent. Due to instability, the yield of compound 13h was determined by NMR using an internal standard. The product of this reaction was subsequently isolated as the corresponding imine in 82% yield following treatment of 13h with MoCl5 and Zn in acetonitrile as described in Supplementary Information.
In summary, we have described a broadly useful platform for rapid reaction discovery. Our general approach is based on introduction of a polyaromatic label into the structure of one of the reactants. As a result, any conversion of such compound into any other products can be easily monitored using matrix-free LDI-TOF-MS even in complex reaction mixtures without any chromatographic fractionation. We demonstrated a direct application of this screening strategy to the discovery of two benzannulation reactions, which proceed via initial [4+2] cycloaddition, followed by ring-opening and aromatization. Such reactions represent previously unknown modes of benzannulation reactivity of alkynes (28,29) and provide a simple and efficient synthetic entry into substituted salicylic acids and highly functionalized naphthols. We envision that this broadly useful reaction-discovery platform will find a range of future applications for identification and optimization of chemical transformations.
Methods
LA-LDI-TOFF-MS reaction screen
Solutions of alkyne 3 (reactant A, 0.3 M in 30 mL of 1,2-dichloromethane), each of the 23 reactants B (0.3 M in 8 mL of 1,2-dichloromethane) and each of the 28 reagents C (0.006 M-0.3 M in 3 mL of 1,2-dichloroethane) were prepared manually. Using PerkinElmer Multiprobe liquid handler, 30 μL of the above solution of alkyne 3 was dispensed into 696 wells in eight 96-deep well plates (Axygen Scientific P-DW-500-C). Using the same liquid handler, each well was treated with solutions of 23 reactants B (30 μL per well) and 28 reagents C (30 μL per well), as well as the negative controls for both B and C containing only the solvent, so that each well received a unique pairwise combination of B and C corresponding to 696 reaction mixtures. The plates were sealed with aluminum foil and left for 1 hour at room temperature. The seals were removed. Using Perkin Elmer Janus automated workstation equipped with a 96-channel pipetting head, 0.8 μL aliquot of each reaction mixture was transferred onto standard stainless steel plates used by MALDI-TOF instruments. The plates were allowed to air dry, and were analyzed in automatic mode on a Bruker Ultraflextreme MALDI-TOF/TOF mass spectrometer equipped with a 355 nm Bruker smartbeam-II laser, using positive ion reflector mode. The reaction plates were resealed and MS analysis was repeated after 24 h and 4 days as described above.
Benzannulation of siloxy alkynes with 2-pyrones
A mixture of 2-pyrone 7 (0.15 mmol) and (Johnphos)AuNCMe-SbF6 (8, 2.5 mol%) in dichloromethane (0.2 mL) at 0 °C was treated dropwise with siloxyalkyne 6 (1.5 eq.) dissolved in dichloromethane (0.3 mL) over two hours. The reaction mixture was warmed to room temperature and stirred until the reaction was complete (typically 2-12 hours). The solvent was removed under reduced pressure. The residue was dissolved in dichloromethane (2.5 mL), placed in an ice-bath and treated dropwise with a solution of HF-pyridine (0.05 mL, 70% aqueous HF and 30% pyridine). Following warming to room temperature, the reaction mixture was treated with water (1 mL) and extracted with ethyl acetate. The combined organic layers were dried over anhydrous MgSO4, concentrated under reduced pressure and subjected to column chromatography on silica gel to deliver benzannulation product 10.
Benzannulation of siloxy alkynes with isoquinoline N-oxides
A mixture of isoquinoline N-oxide 11 (0.15 mmol) and (Johnphos)AuNCMe-SbF6 (8, 3.0 mol%) in dichloromethane (0.3 mL) was treated with siloxyalkyne 6 (1.5 eq.) dissolved in dichloromethane (0.3 mL). The reaction mixture was stirred for 12 h and concentrated under reduced pressure. The residue was dissolved in dichloromethane (2.5 mL), placed in an ice-bath and treated drop-wise with TBAF (1.1 eq. 1M in THF). Following warming to room temperature, the reaction mixture was diluted with water (1 mL) and extracted with ethyl acetate. The combined organic layers were dried over anhydrous MgSO4, concentrated under reduced pressure and subjected to column chromatography on silica gel to deliver benzannulation product 13.
Supplementary Material
Acknowledgments
This work was funded by the National Institutes of Health (P50 GM086145) and the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust. We thank Ian Steele for X-ray crystallographic analysis.
Footnotes
Author Contribution: J.R.C.-P. developed the reaction-screening platform, performed and analyzed all reactions using MS. D.I.C and S.L carried out the reaction optimization and scope studies. M.M. provided assistance with instrumentation and data analysis. S.A.K. provided overall management of the project. The manuscript was written by S.A.K, M. M., J.R.C.-P and D.I.C.
Additional Information: The authors declare no competing financial interests.
References
- 1.Kanan MW, Rozenman MM, Sakurai K, Snyder TM, Liu DR. Reaction discovery enabled by DNA-templated synthesis and in vitro selection. Nature. 2004;431:545–549. doi: 10.1038/nature02920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beeler AB, Su S, Singleton CA, Porco JA., Jr Discovery of chemical reactions through multidimensional screening. J Am Chem Soc. 2007;129:1413–1419. doi: 10.1021/ja0674744. [DOI] [PubMed] [Google Scholar]
- 3.Rozenman MM, Kanan MW, Liu DR. Development and initial application of a hybridization-independent, DNA-encoded reaction discovery system compatible with organic solvents. J Am Chem Soc. 2007;129:14933–14938. doi: 10.1021/ja074155j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodell JR, McMullen JP, Zaborenko N, Maloney JR, Ho CX, Jensen KF, Porco JA, Jr, Beeler AB. Development of an automated microfluidic reaction platform for multidimensional screening: reaction discovery employing bicyclo[3.2.1]octanoid scaffolds. J Org Chem. 2009;74:6169–6180. doi: 10.1021/jo901073v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mueller CA, Markert C, Teichert AM, Pfaltz A. Mass spectrometric screening of chiral catalysts and catalyst mixtures. Chem Commun. 2009;13:1607–1618. doi: 10.1039/b822382c. [DOI] [PubMed] [Google Scholar]
- 6.Wassenaar J, Jansen E, van Zeist WJ, Bickelhaupt FM, Siegler MA, Spek AL, Reek JNH. Catalyst selection based on intermediate stability measured by mass spectrometry. Nature Chem. 2010;2:417–420. doi: 10.1038/nchem.614. [DOI] [PubMed] [Google Scholar]
- 7.Ebner C, Muller CA, Markert C, Pfaltz A. Determining the enantioselectivity of chiral catalysts by mass spectrometric screening of their racemic forms. J Am Chem Soc. 2011;133:4710–4713. doi: 10.1021/ja111700e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Kamlet AS, Steinman JB, Liu DR. A biomolecule-compatible visible-light-induced azide reduction from a DNA-encoded reaction-discovery system. Nature Chem. 2011;3:146–153. doi: 10.1038/nchem.932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robbins DW, Hartwig JF. A Simple, Multidimensional approach to high-throughput discovery of catalytic reactions. Science. 2011;333:1423–1427. doi: 10.1126/science.1207922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McNally A, Prier CK, MacMillan DWC. Discovery of an α-amino C–H arylation reaction using the strategy of accelerated serendipity. Science. 2011;334:1114–1117. doi: 10.1126/science.1213920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friest JA, Broussy S, Chung WJ, Berkowitz DB. Combinatorial catalysis employing a visible enzymatic reacon in real time: Synthetically versatile (pseudo)halometalation/carbocyclizations. Angew Chem Int Ed Engl. 2011;50:8895–8899. doi: 10.1002/anie.201103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montavon TJ, Li J, Cabrera-Pardo JR, Mrksich M, Kozmin SA. Three-component reaction discovery enabled by mass spectrometry of self-assembled monolayers. Nature Chem. 2012;4:45–51. doi: 10.1038/nchem.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Senkan SM. High-throughput screening of solid-state catalyst libraries. Nature. 1998;394:350–353. [Google Scholar]
- 14.Yoshino K, Takao T, Murata H, Shimonishi Y. Use of the derivatizing agent, 4-aminobenzoic acid 2-(diethylamino)ethyl ester, for high-sensitivity detection of oligosaccharides by electrospray ionization mass spectrometry. Anal Chem. 1995;67:4028–4031. doi: 10.1021/ac00117a034. [DOI] [PubMed] [Google Scholar]
- 15.Ahn YH, Yoo JS. Malononitrile as a new derivatizing reagent for high-sensitivity analysis of oligosaccharides by electrospray ionization mass spectrometry. Rapid Commun Mass Spectrom. 1998;12:2011–2015. doi: 10.1002/(SICI)1097-0231(19981230)12:24<2011::AID-RCM429>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 16.Szewczyk JW, Zuckerman RL, Bergman RG, Ellman JA. A mass spectrometric labeling strategy for high-throughput reaction evaluation and optimization: Exploring C–H Activation. Angew Chem Int Ed Engl. 2001;40:216–219. doi: 10.1002/1521-3773(20010105)40:1<216::AID-ANIE216>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 17.Pablo L, Bernad J, Khan S, Korshun VA, Southern EM, Shchepinov MS. S(O)-Pixyl protecting group as efficient mass-tag. Chem Commun. 2005:3466–3468. doi: 10.1039/b504913j. [DOI] [PubMed] [Google Scholar]
- 18.Thuong MBT, Catala C, Colas C, Schaeffer C, Dorsselaer AV, Mann A, Wagner A. Trimethoxyarene as highly ionizable tag for reaction analysis by atmospheric pressure photoionization mass spectrometry (APPI/MS) : Exploration of heterocylic synthesis. Eur J Org Chem. 2012:85–92. [Google Scholar]
- 19.McCarley TD, McCarley RL, Limbach PA. Electron-transfer ionization in matrix-assisted laser desorption/ionization mass spectrometry. Anal Chem. 1998;70:4376–4379. [Google Scholar]
- 20.Macha SF, McCarley TD, Limbach PA. Influence of ionization energy on charge-transfer ionization in matrix-assisted laser desorption/ionization mass spectrometry. Analytica Chim Acta. 1999;397:235–245. [Google Scholar]
- 21.Zhang L, Kozmin SA. Brønsted acid-promoted cyclizations of siloxyalkynes with arenes and alkenes. J Am Chem Soc. 2004;126:10204–10205. doi: 10.1021/ja046586x. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Kozmin SA. Gold-catalyzed cycloisomerizations of siloxy enynes to cyclohexadienes. J Am Chem Soc. 2004;126:11806–11807. doi: 10.1021/ja046112y. [DOI] [PubMed] [Google Scholar]
- 23.Sweis R, Schramm MP, Kozmin SA. Silver-catalyzed [2+2] cycloadditions of siloxyalkynes. J Am Chem Soc. 2004;126:7442–7443. doi: 10.1021/ja048251l. [DOI] [PubMed] [Google Scholar]
- 24.Sun J, Kozmin SA. Brønsted Acid-Promoted Cyclizations of 1-Siloxy-1,5-diynes. J Am Chem Soc. 2005;127:13512–13513. doi: 10.1021/ja055054t. [DOI] [PubMed] [Google Scholar]
- 25.Sun J, Kozmin SA. Silver-catalyzed hydroamination of siloxy alkynes. Angew Chem Int Edn Engl. 2006;45:4991–4993. doi: 10.1002/anie.200601276. [DOI] [PubMed] [Google Scholar]
- 26.Turkmen Y, Montavon TJ, Kozmin SA, Rawal VH. Silver-catalyzed inverse electron-deman Diels-Alder reaction of 1,2-diazines with siloxy alkynes. J Am Chem Soc. 2012;134:9062–9065. doi: 10.1021/ja302537j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Afarinkia K, Vinader V, Nelson TD, Posner GH. Diels-Alder cycloadditions of 2-pyrones and 2-pyridones. Tetrahedron. 1992;48:9111–9171. [Google Scholar]
- 28.Danheiser RL, Gee SK. A regiocontrolled annulation approach to highly substituted aromatic compounds. J Org Chem. 1984;49:1672–1674. [Google Scholar]
- 29.Danheiser RL, Nishida AN, Savariar S, Trova MP. Trialkylsilyloxyalkynes: Synthesis and aromatic annulation reactions. Tetrahedron Lett. 1988;29:4917–4920. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.