

Abstract
Introduction
Small-amplitude, short-duration motor unit action potentials are non-specific findings seen in myopathies and neuromuscular junction (NMJ) disorders. NMJ studies (repetitive nerve stimulation and single-fiber electromyography) can determine if such findings are related to NMJ abnormalities but are not considered routinely in atypical cases.
Methods
Medical records of 338 patients with confirmed NMJ disorders were reviewed to identify cases with a clinical or electrodiagnostic impression of myopathy during initial evaluation. A history of muscle biopsy with findings that did not support a myopathic process was required for inclusion.
Results
Four patients met the inclusion criteria. NMJ studies were abnormal in all cases. One patient had elevated acetylcholine receptor antibodies. Three patients were antibody negative: 2 demonstrated immunotherapy responsiveness, and 1 had a Rapsyn mutation.
Conclusions
NMJ disorders may mimic myopathies, and NMJ studies should be performed to clarify so-called “myopathic” electromyographic findings to avoid unnecessary testing and delayed diagnosis.
Keywords: action potentials, electromyography, myasthenia gravis, neuromuscular junction disorders, repetitive nerve stimulation
Although electrodiagnostic (EDx) studies are indispensable for evaluating patients with neuromuscular disorders, they occasionally yield nonspecific or misleading findings that can lead to a delay in diagnosis.1–3 This is particularly true with myopathic disorders, where needle electromyography (EMG) findings overlap with those affecting the neuromuscular junction (NMJ).4 Short-duration, small-amplitude motor unit action potentials (MUAPs) can be seen in NMJ disorders from intermittent blocking of some muscle fiber action potentials due to failed neuromuscular transmission; clinicians commonly associate this appearance with myopathic disorders in which degenerating muscle fibers prevent the transmission of action potentials along their length (Fig. 1A). The presence of MUAP instability can suggest abnormal neuromuscular transmission, but these findings are often subtle and overlooked.
Figure 1.
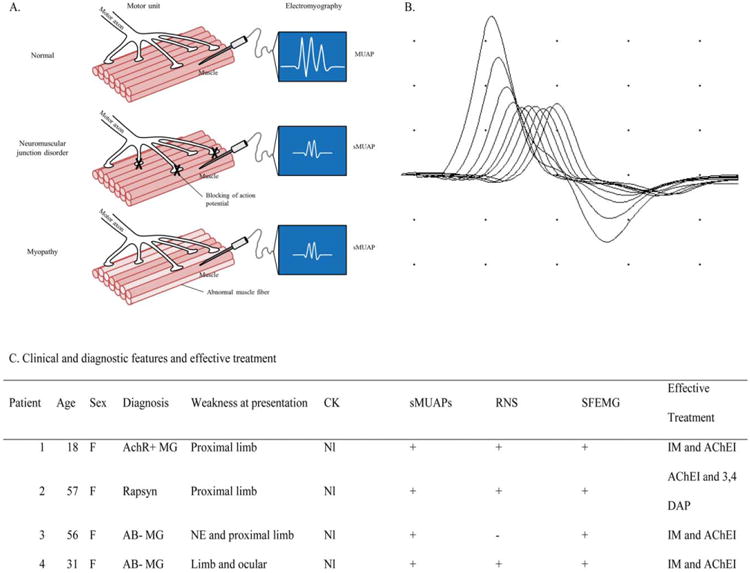
(A) Diagram of motor units and EMG findings in neuromuscular junction disorder patients compared with normal. NMJ dysfunction produces blocking of some action potentials, resulting in a motor unit action potential (MUAP) of shorter duration and smaller amplitude. In myopathy, it is the disruption of muscle fibers that results in small-amplitude, short-duration MUAPs. (B) RNS at 3 Hz of the right ulnar nerve in patient 1 with >50% compound muscle action potential amplitude decrement. (C) Table shows clinical, diagnostic, and treatment characteristics of identified patients with NMJ disorders mimicking the clinical and EDx pattern of myopathy. MUAP, motor unit action potential; sMUAP, small-amplitude, short-duration motor unit action potential; AchR+, acetylcholine receptor antibody positive; MG, myasthenia gravis; IM, immunomodulatory treatment; AChEI, acetylcholinesterase inhibitors; CK, creatine kinase; Rapsyn, congenital myasthenia related to Rapsyn mutation; RNS, >10% decrement of CMAP amplitude (fourth compared with initial response) with repetitive nerve stimulation at 3 Hz; SFEMG, increased jitter and blocking on single-fiber electromyography; 3,4 DAP, 3,4-diaminopyridine; AB-, seronegative for acetylcholine receptor and muscle-specific tyrosine kinase antibodies; NE, neck extensor.
Misidentification of an NMJ disorder as a primary myopathy may result in unnecessary diagnostic procedures and delay appropriate treatment. In this case report and retrospective chart review, we highlight the pitfalls of reliance on EMG alone in distinguishing between myopathic and NMJ disorders. We propose inclusion of repetitive nerve stimulation (RNS) or single-fiber electromyography (SFEMG) in the evaluation of patients who present with weakness.
Methods
Patients were identified from billing records over 5 years at a large tertiary medical center using the diagnostic codes of myasthenia gravis (358.0), myasthenic syndrome (358.1), and myopathy (359). We performed a retrospective analysis of EDx and clinical records on all patients. Cases were included if they had undergone a negative/normal muscle biopsy for clinically suspected myopathy and were subsequently shown to have an NMJ disorder on the basis of serologic or repetitive nerve stimulation/ single-fiber nerve electromyography (RNS/SFEMG) findings. Patients were excluded if the biopsy was abnormal or diagnostic of a specific muscle disorder, as we were not interested in studying coexisting NMJ dysfunction and myopathy.
Case
An 18-year-old woman presented with a limb-girdle pattern of weakness lacking ocular or bulbar weakness or clinical evidence of fatigability. She had undergone muscle biopsy prior to referral to our center based on myopathic MUAPs on needle EMG of proximal muscles. Muscle biopsy was unrevealing for a degenerative or inflammatory muscle disorder, which led to her referral. RNS demonstrated a prominent compound muscle action potential amplitude decrement typical for an NMJ disorder (Fig. 1B). Serum acetylcholine antibody assay performed subsequent to RNS was positive, and a diagnosis of myasthenia gravis (MG) was made. The patient responded to immunomodulatory therapy with significant improvement in strength.
Results
Four patients with the eventual diagnosis of an NMJ disorder were found to meet all inclusion criteria. The clinical features are shown in Figure 1C. Three patients were diagnosed with MG: 1 was seropositive for acetylcholine receptor antibodies (AChR), and 2 patients were seronegative for both AChR and muscle-specific tyrosine kinase antibodies. The fourth patient was diagnosed with congenital myasthenic syndrome related to a Rapsyn mutation. Prominent weakness (at least 1 muscle graded ≤4 on manual muscle testing) was evident in a proximal limb distribution suggestive of a myopathy in all 4 patients. Oculobulbar weakness was present in only 1 patient. SFEMG studies were positive in all 4 patients, whereas RNS studies were positive for decrement (52–70%) in 3 of the 4 patients (Fig. 1C).
Two additional patients with coexistent antibody-positive MG and biopsy-proven inflammatory myopathy (1 polymyositis, 1 dermatomyositis) were excluded due to abnormal muscle biopsies diagnostic of inflammatory myopathy. Both patients had elevated creatine kinase (CK) [467 U/L and 713 U/L (reference: 26–140 U/L)].
Discussion
Screening for NMJ disorders is not always included in the EDx evaluation of suspected myopathy, particularly if features that suggest this possibility (i.e., fatigability, fluctuating weakness, or oculobulbar symptoms) are lacking. In 3 of the 4 patients described, the presenting symptom of the NMJ defect was proximal weakness. In a prior study, 12% of patients with MG presented with a limb-girdle pattern of weakness, and 2% of patients had weakness that spared oculobulbar muscles; these patients are at increased risk of mis-diagnosis.7 The treatment ramifications of misdiagnosis of an NMJ defect as a myopathic disorder are significant, given the availability of specific effective therapies for MG. Without a specific diagnosis, decisions regarding effective treatments cannot be made, and symptomatic treatments, such as use of acetylcholinesterase inhibitors, will not be considered if a neuromuscular transmission defect goes unidentified. Although rare, NMJ disorders and myopathies can coexist, as evidenced by the 2 excluded patients, and would also not be discovered without additional testing.
In our cohort, 4 patients (1.2% of reviewed cases) presented with features that mimicked myopathy sufficient to prompt muscle biopsy but had normal biopsy results. Although reference texts for neuromuscular and EDx medicine often recommend considering RNS or SFEMG in the assessment of myopathy, the utility of NMJ testing in patients with suspected myopathy has not been examined. This small case series suggests that evaluation of a weak patient suspected of having a myopathy should include RNS or SFEMG or both to avoid delays in accurate diagnosis and to prevent unnecessary diagnostic work-up, such as muscle biopsy. With an estimated cost of $2500, not including surgical fees, such an approach would lead to significant cost savings.
When short-duration or small-amplitude MUAPs occur because of abnormal neuromuscular transmission, motor unit instability (jiggle) should be present and is best appreciated on triggered MUAP analysis. Blocking on SFEMG, the correlate of MUAP instability on needle EMG, was evident in all 4 patients, but MUAP instability was only noted in 2. Therefore, where there is a high index of suspicion, SFEMG should be performed prior to biopsy, even if RNS is normal and motor unit instability is not noted. In our series, fibrillation potentials did not preclude the presence of an NMJ defect, but normal CK may be helpful. Our findings support the notion that these EDx components should be included when assessing patients with proximal limb weakness and small, short-duration MUAPs on needle EMG, especially in patients with normal CK. This has the potential to diminish unnecessary or invasive testing and to assist in developing effective treatment strategies.
Abbreviations
- AChR
acetylcholine receptor
- CK
creatine kinase
- EDx
electrodiagnostic
- EMG
electromyography
- MG
myasthenia gravis
- MUAP
motor unit action potential
- NMJ
neuromuscular junction
- RNS
repetitive nerve stimulation
- SFEMG
single-fiber electromyography
- sMUAP
short-duration, small-amplitude motor unit action potential
References
- 1.Dabby R, Lange DJ, Trojaborg W, Hays AP, Lovelace RE, Brannagan TH, et al. Inclusion body myositis mimicking motor neuron disease. Arch Neurol. 2001;58:1253–1256. doi: 10.1001/archneur.58.8.1253. [DOI] [PubMed] [Google Scholar]
- 2.Kinali M, Beeson D, Pitt MC, Jungbluth H, Simonds AK, Aloysius A, et al. Congenital myasthenic syndromes in childhood: diagnostic and management challenges. J Neuroimmunol. 2008;202:6–12. doi: 10.1016/j.jneuroim.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 3.Warmolts JR, Mendell JR. Open-biopsy electromyography. Direct correlation of a pattern of excessively recruited, pathologically small motor unit potentials with histologic evidence of neuropathy. Arch Neurol. 1979;36:406–409. doi: 10.1001/archneur.1979.00500430036004. [DOI] [PubMed] [Google Scholar]
- 4.Stalberg EV, Sonoo M. Assessment of variability in the shape of the motor unit action potential, the “jiggle,” at consecutive discharges. Muscle Nerve. 1994;17:1135–1144. doi: 10.1002/mus.880171003. [DOI] [PubMed] [Google Scholar]
- 5.Engel WK. Brief, small, abundant motor-unit action potentials. A further critique of electromyographic interpretation. Neurology. 1975;25:173–176. doi: 10.1212/wnl.25.2.173. [DOI] [PubMed] [Google Scholar]
- 6.Daube JR. The description of motor unit potentials in electromyography. Neurology. 1978;28:623–625. doi: 10.1212/wnl.28.7.623. [DOI] [PubMed] [Google Scholar]
- 7.Wirtz PW, Sotodeh M, Nijnuis M, van Doorn PA, van Engelen BG, Hintzen RQ, et al. Difference in distribution of muscle weakness between myasthenia gravis and the Lambert-Eaton myasthenic syndrome. J Neurol Neurosurg Psychiatry. 2002;73:766–768. doi: 10.1136/jnnp.73.6.766. [DOI] [PMC free article] [PubMed] [Google Scholar]