

Abstract
Respiratory modulation seen in the sympathetic nerve activity (SNA) implies that the respiratory and sympathetic networks interact. During hypertension elicited by chronic intermittent hypoxia (CIH), the SNA displays an enhanced respiratory modulation reflecting strengthened interactions between the networks. In this chapter, we review a series of experimental and modeling studies that help elucidate possible mechanisms of sympatho-respiratory coupling. We conclude that this coupling significantly contributes to both the sympathetic baroreflex and the augmented sympathetic activity after exposure to CIH. This conclusion is based on the following findings. (1) Baroreceptor activation results in perturbation of the respiratory pattern via transient activation of postinspiratory neurons in the Bötzinger complex (BötC). The same BötC neurons are involved in the respiratory modulation of SNA, and hence provide an additional pathway for the sympathetic baroreflex. (2) Under hypercapnia, phasic activation of abdominal motor nerves (AbN) is accompanied by synchronous discharges in SNA due to the common source of this rhythmic activity in the retrotrapezoid nucleus (RTN). CIH conditioning increases the CO2 sensitivity of central chemoreceptors in the RTN which results in the emergence of AbN and SNA discharges under normocapnic conditions similar to those observed during hypercapnia in naïve animals. Thus, respiratory–sympathetic interactions play an important role in defining sympathetic output and significantly contribute to the sympathetic activity and hypertension under certain physiological or pathophysiological conditions, and the theoretical framework presented may be instrumental in understanding of malfunctioning control of sympathetic activity in a variety of disease states.
Keywords: respiratory–sympathetic interactions, baroreflex, chronic intermittent hypoxia, hypertension, modeling
1 INTRODUCTION
The respiratory rhythm and sympathetic activity are generated centrally within the brainstem. Neuronal circuits that generate and modulate respiratory and sympathetic activities appear to interact and this interaction depends on various sensory afferents (Gilbey, 2007). Here, we review possible respiratory–sympathetic interactions proposed in our recent experimental and modeling studies. These hypothetical interactions are used to explain the mechanisms of the respiratory modulation seen in sympathetic output (Section 2); the changes in the respiratory patterns due to baroreceptor stimulation (Section 3); the changes in the patterns of respiratory-modulated sympathetic activity (Section 4); and the plasticity seen within brainstem respiratory–sympathetic networks in an animal model of sleep apnea (Section 5). Finally, we discuss the limitations and perspectives of the proposed theoretical framework.
2 RESPIRATORY MODULATION OF SYMPATHETIC ACTIVITY
The respiratory rhythm and coordinated motor pattern is provided by a respiratory central pattern generator (CPG) located in the lower brainstem (Bianchi et al., 1995; Cohen, 1979; Lumsden, 1923). The pre-Bötzinger complex (pre-BötC), located within the medullary ventral respiratory column (VRC) is considered a major source of rhythmic inspiratory activity (Koshiya and Smith, 1999; Paton, 1996; Rekling and Feldman, 1998; Smith et al., 1991). The pre-BötC, interacting with the adjacent Bötzinger complex (BötC) containing mostly expiratory neurons (Ezure, 1990; Ezure et al., 2003; Jiang and Lipski, 1990; Tian et al., 1999) represents a core of the respiratory CPG (Bianchi et al., 1995; Richter, 1996; Richter and Spyer, 2001; Rybak et al., 2004, 2007, 2008; Smith et al., 2007, 2009, 2012; Tian et al., 1999). This core circuitry generates primary respiratory oscillations defined by the intrinsic biophysical properties of respiratory neurons involved, the architecture of network interactions between respiratory neural populations within and between the pre-BötC and BötC, and inputs from other brainstem compartments, including the pons, retrotrapezoid nucleus (RTN), raphé, and nucleus tractus solitarii (NTS) (Smith et al., 2012).
The sympathetic nerve activity (SNA) was shown to display respiratory modulation that persisted after vagotomy and decerebration (Adrian et al., 1932; Barman and Gebber, 1980; Habler et al., 1994; Haselton and Guyenet, 1989; Richter and Spyer, 1990; Simms et al., 2009) supporting the idea of a coupling between brainstem respiratory and sympathetic networks. This coupling may represent an important mechanism for coordination of minute ventilation and vasoconstriction/dilation aimed at increasing the efficiency of oxygen uptake/perfusion at rest, and at boosting vasomotion and assisting with perfusion of tissues for maintaining homeostasis during metabolic challenges (Zoccal et al., 2009b). Recent modeling studies also suggest improved efficiency of cardiac function provided by respiratory–sympathetic interactions (Ben-Tal, 2012; Ben-Tal et al., 2012). Therefore, the respiratory modulation may represent a considerable factor contributing to the dynamic control of SNA.
Under baseline conditions (normoxia/normocapnia) SNA usually exhibits positive modulation during inspiration (Fig. 1, upper traces) (Baekey et al., 2008; Malpas, 1998, 2010; Simms et al., 2010; Zoccal et al., 2008, 2009a,b). It has been suggested that this modulation results from specific interactions between respiratory and sympathetic neurons at the level of ventrolateral medulla, where many of the neurons involved in the generation of respiratory and sympathetic activities are located (Habler et al., 1994; Haselton and Guyenet, 1989; Koshiya and Guyenet, 1996; McAllen, 1987; Richter and Spyer, 1990; Zhong et al., 1997). Specifically in this region, the inspiratory and expiratory neurons of the VRC interact with the presympathetic neurons of the rostral ventrolateral medulla (RVLM) as well as with GABAergic interneurons of caudal ventrolateral medulla (CVLM) inhibiting RVLM neurons (Haselton and Guyenet, 1989; Mandel and Schreihofer, 2006; Richter and Spyer, 1990; Sun et al., 1997). It appears that the pons may play a critical role in these interactions. Ponto-medullary transections in situ were shown to significantly reduce or even eliminate the respiratory modulation of SNA (Fig. 1, “after transection”, see also Baekey et al., 2008). This suggests that pontine projections to medullary respiratory and sympathetic neurons are crucial for the respiratory–sympathetic coupling. Accordingly, pontine neurons may have a direct effect on the activity of presympathetic RVLM neurons or they may act indirectly through respiratory neurons in the VRC (Fig. 2A, blue dashed arrows).
FIGURE 1.

Thoracic sympathetic (thSNA) and phrenic (PNA) nerve activities before and after ponto-medullary transection. Before transection (intact pons), thSNA has a clear respiratory modulation which is attenuated or eliminated after transection.
FIGURE 2.
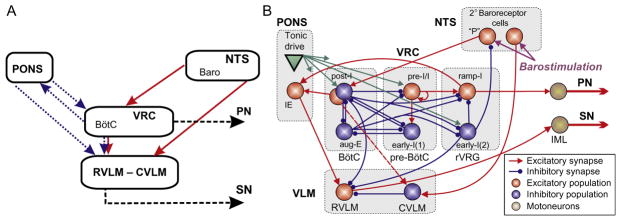
(A) Conceptual model of interaction between respiratory-related activity of the ventral respiratory column (VRC), pontine circuits (PONS), sensory network in the nucleus tractus solitary (NTS), and rostral and caudal ventrolateral medulla (RVLM/CVLM). Dotted arrows represent the effects of VRC and PONS on RVLM providing respiratory modulation of SNA. The sympathetic baroreceptor reflex operates via two pathways (red (gray in the print version) solid arrows): one direct pathway includes baroreceptors, 2nd-order barosensitive cells (Baro) in the NTS and CVLM, which inhibits RVLM and SNA; the other pathway routes via the Bötzinger complex (BötC) in the VRC, whose postinspiratory neurons inhibit RVLM and SNA. (B) Suggested interactions between the respiratory and sympathetic neural populations at the level of brainstem. This model developed by Baekey et al. (2010) was based on the earlier model of Smith et al. (2007) that simulated neural interactions between different populations of respiratory neurons within major brainstem compartments involved in the control of breathing (pons, BötC, pre-BötC, and rVRG). These compartments included the populations of postinspiratory (post-I) and augmenting inspiratory (aug-E) neurons of BötC, preinspiratory/inspiratory (pre-I/I) and early-inspiratory (early-I(1)) neurons of pre-BötC, and ramping inspiratory (ramp-I) and early-I(2) neurons in rVRG. Baekey et al. (2010) extended this model by incorporating NTS containing populations of 2nd-order baroreceptor cells, VLM containing the excitatory RVLM and inhibitory CVLM populations, and the phase-spanning inspiratory–expiratory (IE) population in the pons. Each population (shown as a sphere) consisted of 20–50 single-compartment neurons described in the Hodgkin–Huxley style.
Adapted from Baekey et al. (2010) with permission.
Postinspiratory activity in BötC was shown to be critically dependent on pons (Smith et al., 2007). Baekey et al. (2010) have hypothesized that post-I neurons of BötC are involved in central respiratory–sympathetic interactions by direct inhibition of RVLM during expiration (Fig. 2). They also suggested that the pons participates in these interactions via two pathways. First, it provides a necessary excitatory tonic drive to the post-I neurons, and second, it directly modulates the activity of RVLM neurons by excitatory inputs from the phase-spanning pontine IE population. These two pathways together provide the critical dependence of the respiratory modulation of SNA on the pons, and ponto-medullary transection completely eliminates the respiratory modulation of SNA in their model (Fig. 3B, rightmost panel).
FIGURE 3.
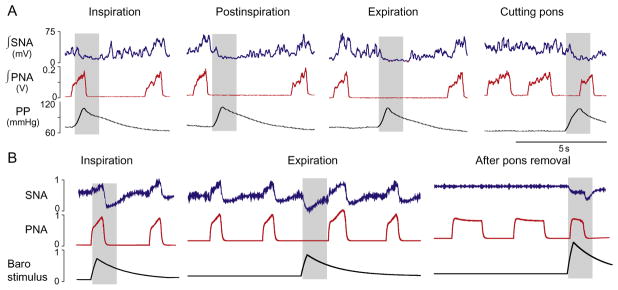
(A) Response of the phrenic (PNA) and sympathetic (SNA) nerve activities to transient increases in perfusion pressure (PP, bottom trace). Stimulation was applied to the intact preparation during inspiration, postinspiration, and late expiration. The stimulus applied during inspiration did not affect the respiratory pattern, whereas the stimuli delivered during the expiratory phase prolonged expiration. After ponto-medullary transection, the applied stimulus shortened the apneustic inspiratory (PNA) bursts. (B) Phase-dependent stimulation of baroreceptors simulated by the model shows a qualitatively similar result. Note that after removal of the pontine compartment in the model the respiratory modulation of SNA is abolished.
Adapted from Baekey et al. (2010) with permission.
3 RESPIRATORY BAROREFLEX
The NTS is the major brainstem region that receives and integrates peripheral cardiovascular and respiratory afferent inputs, including baroreceptor afferents (Loewy and Spyer, 1990). The classical baroreflex control of SNA operates via the 2nd-order baroreceptor neurons that are located in the NTS and project (directly or indirectly) to the CVLM. Through this path, baroreceptor stimulation provides activation of CVLM GABAergic interneurons (Schreihofer and Guyenet, 2002) that in turn inhibit the presympathetic neurons in the RVLM hence lowering both the RVLM activity and SNA (Dampney, 1994; Loewy and Spyer, 1990). This pathway provides a direct negative feedback control of SNA which is an important mechanism adjusting sympathetic outflow in response to arterial pressure excursions.
Respiratory activity is also known to be modulated by the baroreceptor input. Dick and Morris (2004) have shown in vagally intact, decerebrated cats that the activities of approximately 50% of the respiratory-modulated neurons within the VRC responded to transient pressure pulses. Furthermore, it appeared that the expiratory activity was modulated with much greater extent than inspiratory activity (Dick et al., 2005). These observations imply that baroreceptor activation can affect phase-switching mechanisms in the respiratory CPG and, hence, durations of respiratory phase. Baekey et al. (2010) have demonstrated that transient pressure pulses indeed perturb the respiratory pattern. Figure 3 shows the effects of transient increases in the perfusion pressure (PP) in the arterially perfused in situ rat preparation (Baekey et al., 2008). These stimuli were delivered during inspiration, postinspiration, or late expiration and produced phase-dependent effects on the respiratory pattern and, correspondingly, on the respiratory modulation of SNA. With pons intact, the applied barostimulation had almost no effect on the amplitude and duration (i.e., inspiratory period) of the phrenic bursts even when stimuli were delivered during inspiration (Fig. 3A). At the same time, these stimuli suppressed or abolished inspiratory modulation of SNA. In contrast, when the same stimuli were delivered during postinspiration or late expiration, they observed an increase in the expiratory period combined with the sympatho-inhibitory response. The barostimulation-evoked prolongation of expiration was greater if the stimulus was applied later during the expiratory phase (compare second and third columns in Fig. 3A). After pontine transection, the barostimulation shortened the apneustic inspiratory burst (see Fig. 3A, last column).
To create an interface for barostimulation, Baekey et al. (2010) included two populations of 2nd-order baroreceptor neurons in the NTS receiving baroreceptor afferents (Fig. 2B). One of those populations was responsible for the direct baroreflex pathway by sending excitatory projection to CVLM. They hypothesized that barostimulation prolongs expiration by exciting expiratory neurons in BötC. Since this prolongation appears to depend on pons, they suggested that the second barosensitive population (marked “P” in Fig. 2B) excites pontine-dependent post-I populations. Due to the fact that barostimulation had virtually no effect on respiratory patterns when applied during inspiration, they incorporated an inhibitory connection from the inspiratory early-I(2) population in rVRG to the “P” barosensitive population in NTS.
Simulations show the plausibility of this connectivity (Fig. 3B). Transient barostimulation applied to the 2nd-order barosensitive NTS populations produces a temporary reduction of SNA via direct activation of the CVLM population that inhibits the activity of the RVLM population. This represents the direct component of the sympathetic baroreflex. Stimulus application during inspiration does not affect respiratory (PN) activity, because the gain of the input from the 2nd-order barosensitive NTS population to the post-I neurons is suppressed centrally during inspiration by the inhibitory early-I(2) population of rVRG. In contrast, stimulus applied during expiration prolongs expiration via activation of post-I neurons of BötC that inhibit the aug-E population and the RVLM. After pontine removal, the model switches to the apneustic respiratory pattern with elongated bursts of PNA (see Fig. 3B, rightmost panel). As mentioned, in these conditions the rVRG early-I(2) population is no longer active and, hence, releases NTS baroreceptors from inspiratory inhibition. Accordingly, transient barostimulus activates the post-I population during inspiration and prematurely terminates inspiration (compare rightmost panels in Fig. 3).
A closer look at the activity of different populations in the model in response to the transient barostimulation applied during expiration reveals something nontrivial (Fig. 4). Normally, during expiration post-I neurons exhibit a decrementing activity pattern. Aug-E neurons gradually activate, being released from post-I inhibition as it happens in the first breathing cycle in Fig. 4A. As soon as a barostimulus comes, the post-I population (which is already weak by that time) activates again, shuts down the aug-E population, and resets expiration. After post-I activity wanes, the aug-E population reactivates for the second time. Baekey et al. (2010) used this mechanistic explanation for expiratory period prolongation in response to transient pressure pulses applied during expiration as a model prediction. They performed extracellular recording of post-I and aug-E neuronal activity within the BötC using the multielectrode technique and identified neuronal responses elicited by baroreflex stimulations with activation–deactivation patterns in full accordance with the model simulations (Fig. 4B).
FIGURE 4.
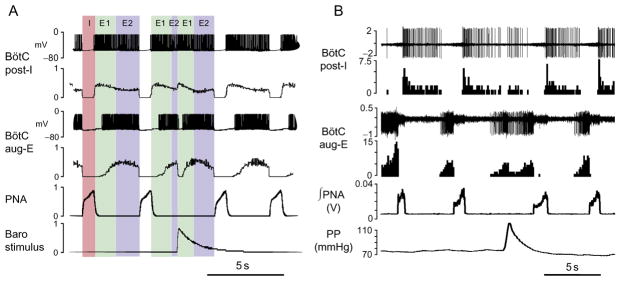
The mechanism of expiration resetting by transient baroreceptor stimulation as predicted by the model (A) and in situ (B). (A) Membrane potentials of post-I and aug-E neurons (first and third traces) and integrated spike histograms of the entire post-I and aug-E populations (second and fourth traces). Shaded intervals indicate I, inspiratory phase; E1, first phase of expiration (postinspiration, post-I); E2, second phase of expiration (late expiration). The applied stimulation (lowest trace) resets expiration by activating post-I population which inhibits aug-E neurons. After the stimulus, the aug-E neuron fires for the second time, which is accompanied by prolongation of expiration. (B) Extracellular recordings and firing rate histograms from post-I (first pair of traces) and aug-E (second pair) neurons in BötC. Below are the integrated phrenic nerve activity (PNA) and perfusion pressure (PP). The response to the applied barostimulation in the in situ preparation is associated with transient increases in post-I and decreases in aug-E activity as predicted by the model.
Adapted from Baekey et al. (2010) with permission.
4 RESPIRATORY–SYMPATHETIC CHEMOREFLEX
At basal conditions (normocapnia/normoxia), the respiratory pattern consists of a phase of active inspiration, with recruitment of diaphragm muscles innervated by phrenic motoneurons; and a phase of passive expiration, in which the reverse air flow is generated by the recoil forces of the thorax/lungs. During hypercapnia, late-expiratory (late-E) discharges emerge in the abdominal nerve (AbN) that occur just prior to phrenic bursts (Abdala et al., 2009a; Molkov et al., 2010), indicating a pattern of active expiration (Fig. 5). With increasing CO2, the frequency of these AbN late-E discharges increases quantally (Fig. 5, 7% CO2) until it reaches a 1:1 ratio with phrenic burst frequency (Fig. 5, 10% CO2) (Abdala et al., 2009a; Molkov et al., 2010). It has been suggested that the abdominal late-E activity originates in the parafacial respiratory group (pFRG) ( Janczewski and Feldman, 2006; Janczewski et al., 2002; Onimaru and Homma, 2003; Onimaru et al., 1988) that appears to anatomically overlap with the RTN. Moreover, neurons were found in the RTN/pFRG region whose rhythmic discharges emerged coincidently with late-E bursts recorded from the AbN during hypercapnic conditions (Abdala et al., 2009a; Molkov et al., 2010). Also, pharmacological inactivation of the RTN/pFRG (Abdala et al., 2009a; Molkov et al., 2010) or inhibition of predominantly Phox2b-expressing neurons in this region (Marina et al., 2010) abolished the hypercapnia-induced late-E bursts in the AbN without affecting the activity of nearby expiratory neurons of the BötC (Abdala et al., 2009a) indicating that the RTN/pFRG is an ultimate site-generating late-E neural activity for active expiration.
FIGURE 5.
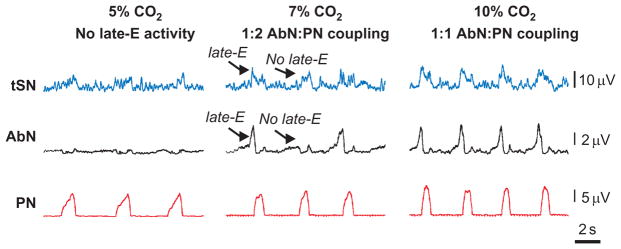
Quantal acceleration of late-E activity in response to progressive hypercapnia. From bottom to top: activity of phrenic (PN), abdominal (AbN) thoracic sympathetic (tSN) nerves at 5% CO2 (normocapnia), and during hypercapnia (7% and 10% CO2). Note the skipping of some late-E bursts in both AbN and tSN at 7% CO2.
Adapted from Molkov et al. (2011) with permission.
FIGURE 7.
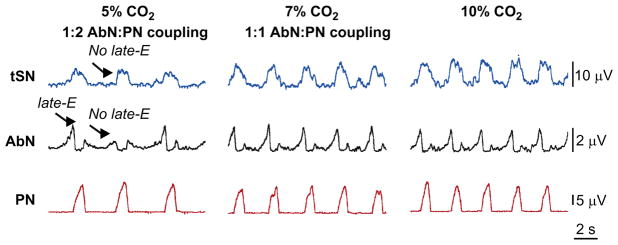
Sympathetic and respiratory responses to hypercapnia in CIH-conditioned rats at normocapnia (5% CO2) and during hypercapnia (7% and 10% CO2). Note the presence of late-E bursts in both AbN and tSN at 5% CO2.
Adapted from Molkov et al. (2011) with permission.
FIGURE 10.

The unified model representing a combined circuitry of schematics from Figs. 2B and 5. RTN/pFRG drive is CO2-dependent. Connections shown by dashed lines and pontine I population represent possible model alternatives (see text for details).
Adapted from Rybak et al. (2012) with permission.
Central chemoreception involves a cluster of Phox2b-expressing neurons located in RTN/pFRG (Guyenet et al., 2008; Stornetta et al., 2006). The RTN chemosensitive neurons are predominantly glutamatergic, as they express the glutamatergic vesicular transporter, and establish connections with respiratory neurons of the VRC, parabrachial, and Kölliker–Fuse nuclei in the pons and dorsal respiratory column (Rosin et al., 2006). These excitatory connections mediate the respiratory response following central chemoreceptor activation. During hypercapnia, the RTN/pFRG provides a source of excitation to bulbospinal expiratory neurons located in cVRG that relay excitatory drive to the lumbar abdominal motoneurons that drive late-E bursting in the AbN (Abdala et al., 2009a; Molkov et al., 2010).
To assimilate these experimental data, Molkov et al. (2010) extended the model of Smith et al. (2007) by incorporating the RTN/pFRG compartment, containing neurons performing central chemoreceptor function whose activity was dependent on CO2 level (Fig. 6). In the model, this compartment includes a late-E population of neurons with intrinsic bursting properties (Abdala et al., 2009a; Molkov et al., 2010). This population outputs to a population of bulbospinal premotor expiratory neurons of cVRG (bs-E) projecting to the abdominal motoneurons that define activity of the AbN (Fig. 5).
FIGURE 6.
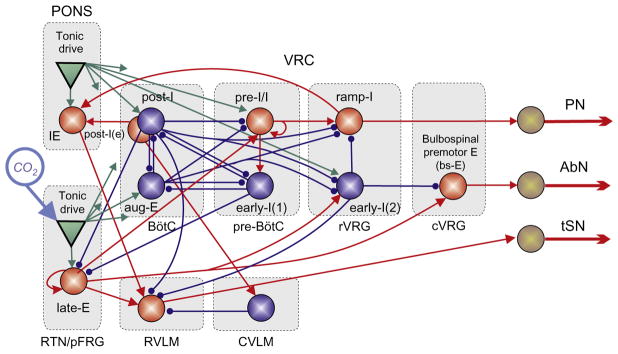
The model by Molkov et al. (2011) combines the circuitry responsible for the respiratory modulation of sympathetic activity from Fig. 5 and the RTN/pFRG oscillator with its interconnections with respiratory CPG. Note excitatory connections from chemosensitive lateE population in RTN/pFRG to the presympathetic neurons in RVLM. See text for more details.
Sympathetic activity was also found to exhibit a late-E discharge during hypercapnia that coincided with AbN late-E activity (Molkov et al., 2011). In Fig. 5 under normocapnic conditions (5% CO2), AbN shows a low-amplitude activity and the integrated tSN activity expresses an augmenting inspiratory modulation. As mentioned, progressive hypercapnia evokes late-E activity with quantally increasing frequency, and at 7% CO2 this frequency reaches a 1:2 ratio, when approximately every second respiratory cycle is skipping in AbN late-E activity. It is important to notice that the tSN late-E discharges coincide with the AbN late-E bursts (Fig. 5). The synchronous activation of abdominal and sympathetic late-E activities is evident at 7% CO2 where sympathetic late-E activity was skipped in the respiratory cycles whenever abdominal late-E activity was also absent.
This strongly supports the idea that the observed late-E activities in both the AbN and tSN have the same source located in the RTN/pFRG. This region contains neurons that are silent during normocapnia (5% CO2) and active during hypercapnia, exhibiting a firing pattern that strongly correlates with AbN late-E activity (Abdala et al., 2009b; Molkov et al., 2010). Molkov et al. (2011) suggested that these late-E neurons of RTN/pFRG are an excitatory source not only to cVRC bulbospinal expiratory (E) neurons but also to presympathetic RVLM neurons (see Fig. 6), culminating in an increase of sympathetic activity correlated with late-E bursts in abdominal motor activity. This hypothesis has a strong experimental support (Guyenet et al., 2008; Moreira et al., 2006; Nattie and Li, 2009). Accordingly, Molkov et al. (2011) combined the models of Baekey et al. (2010) and Molkov et al. (2010) and incorporated this same projection (Fig. 6).
5 CHRONIC INTERMITTENT HYPOXIA
Recent studies indicate that changes in the strength and/or pattern of respiratory–sympathetic coupling may have pathological implications in the control of arterial pressure levels. Such dysfunctions can be observed in the experimental condition of chronic intermittent hypoxia (CIH). CIH is commonly observed in patients suffering from obstructive sleep apnea (OSA) as a consequence of the repetitive episodes of upper airway obstruction during sleep time. It has been suggested that OSA patients are under a risk of the development of arterial hypertension as a result of the exposure to CIH (Caples et al., 2005; Dempsey et al., 2010).
In rats, exposure to CIH produces a sustained increase in arterial pressure (Fletcher et al., 1992a,b) combined with an augmented sympathetic vasomotor tone (Zoccal et al., 2007, 2009a), indicating that the sympathetic nervous system plays a major role in the etiology of CIH-induced hypertension. Moreover, the elevated baseline (under normocapnia/normoxia) SNA of CIH-conditioned rats was shown to exhibit an enhanced respiratory modulation characterized by higher levels of sympathetic activity during late-E phase (Fig. 7, left panel). This effect was independent of the afferent inputs from lungs or peripheral chemoreceptors (Zoccal et al., 2008). Besides, the enhanced sympathetic activity during expiration in CIH rats was associated with the emergence of late-E bursts in AbN and an active expiratory motor pattern. This raises the possibility that central coupling between brainstem respiratory and sympathetic neurons provides a significant contribution to the development of hypertension in CIH-conditioned animals (Zoccal et al., 2009b).
As a result of CIH conditioning, the activities of AbN and tSN are altered in both normocapnia and hypercapnia (Abdala et al., 2009b; Zoccal et al., 2008, 2009a,b). After 10 days exposure to CIH under normocapnic conditions, late-E activity is present in AbN and tSN with a frequency ratio to PN of about 1:2 (Fig. 7, 5% CO2). In contrast to naïve rats (Fig. 5) at 7% CO2, late-E discharges in AbN (and tSN) become 1:1 coupled to PN bursts. By comparing Figs. 5 and 7 one can suggest that 10 days exposure to CIH exaggerates baseline activity of RTN/pFRG chemoreceptors so that they start providing higher output in response to the same CO2 levels. Consequently, it shifts the CO2 threshold for the emergence of late-E oscillations in CIH-conditioned preparations below normocapnic 5% CO2.
Since the presence of sympathetic (and abdominal) late-E activity in CIH-conditioned rats under normal conditions (normocapnia) is similar to that observed in naïve rats during hypercapnia (Abdala et al., 2009a; Molkov et al., 2010), Molkov et al. (2011) hypothesized that the chemosensitive RTN/pFRG neurons involved in the generation of late-E activity get sensitized during CIH conditioning, thereby reducing the CO2 threshold for the emergence of late-E activity in the RTN/pFRG and hence for its appearance in both abdominal and sympathetic motor outflows. In the model (Fig. 5), the RTN/pFRG is considered to be a major central chemoreceptor site whose drive is sensitive to CO2. To implement this property, Molkov et al. (2011) considered RTN/pFRG tonic drive to be variable and dependent on the CO2 level. This putative dependence of RTN/pFRG tonic drive on CO2 is shown in the upper panel of Fig. 8 (solid curve). As hypothesized above, the CO2 sensitivity of RTN/pFRG increases as a result of CIH exposure. This corresponds to the horizontal shift of the CO2-dependent RTN drive by 2% CO2 toward lower CO2 values (to the left, see dashed curve in the upper panel of Fig. 8).
FIGURE 8.

Simulation of the effects of CIH conditioning by shifting the response curve of central chemoreceptors toward lower CO2 levels using the model of Molkov et al. (2011). Top panel shows RTN/pFRG tonic drive as a function of CO2 for the control (black solid curve) and CIH-conditioning (red (gray in the print version) dashed curve) cases. Below are the results of simulations with gradually changing CO2 from 0% (hypocapnia) to 10% (hypercapnia) for control and CIH cases, respectively. Shaded intervals roughly correspond to the experimental conditions shown in Figs. 5, 7, and 9.
Molkov et al. (2011) used the model shown in Fig. 6 with the conditional CO2 dependence illustrated on the top of Fig. 8 to simulate CIH-induced changes in sympathetic and respiratory activities under hyper- and hypocapnic conditions. Lower panels of Fig. 8 show the integrated activity in the PN, AbN, and tSN outputs, when CO2 was gradually increasing from 0% CO2 (hypocapnia) through 5% CO2 (normocapnia) to 10% CO2 (hypercapnia) for both naïve and CIH-conditioned animals. In naïve rats (Fig. 8, “Control” case), progressive hypercapnia (right part of the graph) lead to the emergence and quantal acceleration of late-E bursts in both AbN and tSN, replicating the experimental data shown in Fig. 5. Specifically, the late-E discharges in AbN and tSN appeared at 7% CO2 and reached 1:1 ratio to the PN bursts at 9% CO2. In CIH-conditioned animals, the shifted curve reflecting the CO2 dependence of RTN/pFRG drive was used (dashed line on the upper panel). In contrast to the “control” scenario, the late-E bursts in AbN and tSN emerged at 4% CO2; in the normocapnic state (5% CO2), they showed a stable 1:2 ratio to the PN bursts (Fig. 8, “After CIH”); and at 7% CO2, this ratio reached 1:1, which was also consistent with experimental observations illustrated in Fig. 7.
The simulation results shown in Fig. 8 have an important implication. Note that a reduction of CO2 below 3% (in the control conditions) caused a hypocapnic apnea (a lack of PN activity in Control case in Fig. 8, left part of the graph). After CIH conditioning, the apneic threshold for hypocapnia in the model was reduced by at least 2% CO2, since the PN bursts were still being generated even at 1% CO2 (no apnea label in Fig. 8). To test this modeling prediction, Molkov et al. (2011) exposed the control (naïve) and CIH-conditioned rat preparations to progressive hypocapnia (from normal 5% CO2 to 3% and then to 1%). The naïve rat preparations exhibited a reduction in the integrated PN burst amplitude at 3% CO2 and a hypocapnic apnea at 1% CO2 (Fig. 9, control case). Importantly, these control preparations never expressed late-E activity in AbN or tSN in either normocapnia or hypocapnia. In contrast, in CIH rat preparations (Fig. 9, after CIH), the expressed late-E activity in both AbN and tSN was already present during normocapnia (at 5% CO2), which corresponded to the above simulations (see Fig. 8). This late-E activity, however, disappeared from both nerves at 3% CO2 (no late-E label in Fig. 9 and compare with Fig. 8). At the same time, PN activity with a reduced amplitude (and apparent respiratory modulation of tSN) was still present even at 1% CO2, hence confirming modeling prediction about a reduction of apneic threshold for hypocapnia in CIH-conditioned rats.
FIGURE 9.

Experimental verification of the predicted changes in the sympathetic and respiratory activities in control and CIH-conditioned rats in hypocapnic conditions (3% and 1% CO2). Late-E activity in AbN and tSN in CIH-conditioned preparations is abolished with reduction of CO2 to hypocapnic 3% (marked by no late-E label). Hypocapnia at 1% CO2 eliminates PN activity in control (hypocapnic apnea), but not in CIH-conditioned preparations (no apnea).
Adapted from Molkov et al. (2011) with permission.
One of the yet unmentioned interesting predictions of the model is a CIH-evoked reduction in the synaptic inhibition in the RVLM. Incorporation of this feature allowed Molkov et al. (2011) to reproduce the experimentally observed changes in the tSN burst profile induced by CIH conditioning. This prediction is consistent with raised sympathetic activity and reflex sympathetic responses in CIH rats (Braga et al., 2006) and awaits experimental testing.
6 UNIFIED THEORETICAL FRAMEWORK FOR RESPIRATORY–SYMPATHETIC COUPLING: LIMITATIONS AND PERSPECTIVES
The unified model of sympatho-respiratory circuits shown in Fig. 10 represents a superposition of two previous models describing the emergence of late-E activity in the RTN/pFRG (Molkov et al., 2010, 2011) and the sympathetic–respiratory coupling (Baekey et al., 2010). This model does a good job in providing possible explanations for the origin of respiratory modulation in SNA, respiratory baroreflex, and sympathetic overactivity evoked by hypercapnia and CIH, but it still has uncertainties which have yet to be clarified.
Baekey et al. (2010) focused on two, most straight-forward mechanisms of the respiratory SNA modulation. However, previous studies with the recording from RVLM and CVLM neurons revealed more complexity of respiratory-modulated activity patterns supporting broader interactions between the brainstem respiratory neurons and neurons within RVLM and CVLM. For example, Mandel and Schreihofer (2006) revealed at least four distinct respiratory-modulated discharge patterns in CVLM cells, with inspiratory, expiratory, phase-spanning expiratory–inspiratory, and postinspiratory modulated activity, suggesting that the respiratory modulation of RVLM neurons can be mediated by CVLM. While the model by Baekey et al. (2010) replicates major features of sympatho-respiratory patterning, other plausible solutions (e.g. based on excitatory projections from post-I(e) neurons to CVLM, see Fig. 10) can be considered and comparatively investigated in the future modeling studies.
The unified circuitry in Fig. 10 inherits a special population of 2nd-order barosensitive cells in the NTS projecting to the post-I neurons, whose activity is controlled centrally by early-I(2) population of rVRG (marked by “P” in Fig. 10). Similar control of the NTS pump (P) cells receiving early-inspiratory inhibition was previously described by Miyazaki et al. (1999). Since stimulation of pulmonary afferents also activates post-I neurons via P cells of NTS (Hayashi et al., 1996), these cells may be involved in both Hering–Breuer and barosympathetic reflexes. Yet another possibility for the baroreflex gain control at the level of NTS could be if inhibition during inspiration would come from the pons. Indeed, the electrical stimulation of the parabrachial nucleus is shown to suppress the gain of carotid sinus afferent input to NTS (Felder and Mifflin, 1988), so the control of baroreflex gain can be modeled by incorporating an inhibitory inspiratory population into the pontine compartment projecting to the 2nd-order barosensitive cells in the NTS that excite post-I neurons (Fig. 10). This possibility is also worth considering in future modeling studies.
The unified model defines a neural substrate for the expiratory-facilitatory response to activation of baroreceptors (Brunner et al., 1982; Dove and Katona, 1985; Grunstein et al., 1975; Li et al., 1999a,b; Lindsey et al., 1998; Nishino and Honda, 1982; Richter and Seller, 1975; Speck and Webber, 1983; Stella et al., 2001) and explains previous experimental findings that respiratory neurons, preferentially expiratory neurons, are modulated with the arterial pulse (Dick and Morris, 2004; Dick et al., 2005). This substrate can be used for further extending the model to incorporate interactions with the parasympathetic nervous system, since baroactivated post-I neurons were also found to be crucially involved in the modulation of cardiac vagal motoneurons (Gilbey et al., 1984).
The prolongation of expiration in response to abrupt increases in blood pressure does not seem to have an obvious teleological explanation. On the other hand, volitional slowing breathing or pranayamic breathing is a recognized practice that decreases blood pressure (Jerath et al., 2006). The inclusion of the proposed mechanisms to more integrative models of cardiovascular–respiratory interactions may be instrumental for elucidating the role of expiration prolongation for gas exchange and/or arterial pressure regulation (Ben-Tal, 2012; Ben-Tal et al., 2012).
In our theory of the respiratory baroreflex, we describe how respiratory phase durations are affected by transient increases of blood pressure. However, in cats it has been demonstrated that inspiratory motor output is decreased during longer episodes of elevated blood pressure (Arata et al., 2000; Lindsey et al., 1998; Poliacek et al., 2011). Although it was not relevant in the in situ experiments reviewed here due to very short duration of the stimulus (5 s vs. 60 s and longer in the studies cited), the effect of the reduction in the inspiratory efferent amplitude by sustained hypertension clearly represents another important aspect of the respiratory baroreflex. Combining our findings and the connectivity between 2nd-order baroreceptors and respiratory neurons at the pattern formation level suggested by Lindsey’s group can be instrumental in making another step toward a closed-loop model of cardio-respiratory interactions.
The model implies that the sympathetic baroreceptor reflex, providing negative feedback from baroreceptors to the RVLM and SNA, has two pathways (Fig. 2A). The first, direct path leads from the 2nd-order baroreceptor neurons in NTS to CVLM which inhibits RVLM hence lowering SNA (Dampney, 1994; Guyenet et al., 1990). The second pathway is mediated by respiratory circuits, specifically by the post-I neurons of BötC which inhibit RVLM and prolong expiration. Therefore, our theory suggests that the processing of the baroreceptor afferent information depends on the respiratory–sympathetic interactions and needs an intact respiratory network. The physiological relevance of this concept still requires further experimental investigations.
In reproducing the effect of CIH conditioning on AbN and tSN motor activities observed experimentally, the model relies on a shift of CO2 dependence of RTN drive toward lower CO2 values (Fig. 8) which causes the development of an active expiratory pattern in normoxic/normocapnic conditions due to an exaggerated response of central chemoreceptors after CIH exposure. These observations provide important insights into possible mechanisms involved in the elevation of sympathetic activity and development of arterial hypertension observed after CIH conditioning. However, what causes the underlying sensitization of central chemoreceptors remains unclear. One possible explanation for this may be based on a reduction of inhibitory inputs from the BötC post-I neurons to the RTN/pFRG late-E neurons, which could increase the excitability in the latter (Molkov et al., 2010). A similar effect could be provided by increased excitatory drive from peripheral chemoreceptors (mediated by corresponding neurons in the nucleus of solitary tract) to central chemoreceptors (late-E neurons) (Guyenet et al., 2009; Takakura et al., 2006) and/or by the peripheral chemoreceptor control of the gain of central chemoreceptors (Blain et al., 2010). However, the direct involvement of peripheral chemoreceptor drive would contradict the existing data that carotid body denervation after CIH exposure does not eliminate the late-E sympathetic discharges (Zoccal et al., 2008). Nevertheless, peripheral–central chemoreceptor interaction may be involved in the development of plastic changes in the excitability of central chemoreceptors after CIH conditioning via activation of neuromodulators that enhance the activity of RTN chemosensitive neurons, such as serotonin (Mulkey et al., 2007), ATP (Mulkey et al., 2006), or locally produced oxidative stress ( Jurado-Gamez et al., 2011). These or other, currently unknown mechanisms may be responsible for the observed CIH-evoked sensitization of RTN/pFRG chemoreceptors which requires additional studies (Ben-Tal, 2012; Ben-Tal et al., 2012).
A main message of this review is that the respiratory and sympathetic networks interact and their coupling is plastic and contributes to the baroreceptor reflex control of sympathetic activity and to the elevated sympathetic activity following CIH. The theoretical basis presented explains the complex and intricate circuitry involved in the interplay between the respiratory network and sympathetic nervous system in health and disease. The new knowledge gleaned contributes significantly to our understanding of the control of sympathetic activity and cardiovascular system and assists in novel therapies in a variety of disease states.
Acknowledgments
This work was supported by National Institutes of Health, grant R01 AT008632 to Y.I.M.; grants R33 HL087377; R01 NS057815; and R01 NS069220 to I. A. R. In Brazil, the studies were supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq). In the UK, the studies were supported by the British Heart Foundation, International Retts Syndrome Foundation, and National Institutes of Health.
References
- Abdala AP, Rybak IA, Smith JC, Paton JF. Abdominal expiratory activity in the rat brainstem-spinal cord in situ: patterns, origins and implications for respiratory rhythm generation. J Physiol. 2009a;587:3539–3559. doi: 10.1113/jphysiol.2008.167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdala AP, Rybak IA, Smith JC, Zoccal DB, Machado BH, St-John WM, Paton JF. Multiple pontomedullary mechanisms of respiratory rhythmogenesis. Respir Physiol Neurobiol. 2009b;168:19–25. doi: 10.1016/j.resp.2009.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adrian ED, Bronk DW, Phillips G. Discharges in mammalian sympathetic nerves. J Physiol. 1932;74:115–133. doi: 10.1113/jphysiol.1932.sp002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arata A, Hernandez YM, Lindsey BG, Morris KF, Shannon R. Transient configurations of baroresponsive respiratory-related brainstem neuronal assemblies in the cat. J Physiol. 2000;525 (Pt. 2):509–530. doi: 10.1111/j.1469-7793.2000.t01-1-00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baekey DM, Dick TE, Paton JF. Pontomedullary transection attenuates central respiratory modulation of sympathetic discharge, heart rate and the baroreceptor reflex in the in situ rat preparation. Exp Physiol. 2008;93:803–816. doi: 10.1113/expphysiol.2007.041400. [DOI] [PubMed] [Google Scholar]
- Baekey DM, Molkov YI, Paton JF, Rybak IA, Dick TE. Effect of baroreceptor stimulation on the respiratory pattern: insights into respiratory-sympathetic interactions. Respir Physiol Neurobiol. 2010;174:135–145. doi: 10.1016/j.resp.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barman SM, Gebber GL. Sympathetic nerve rhythm of brain stem origin. Am J Physiol. 1980;239:R42–R47. doi: 10.1152/ajpregu.1980.239.1.R42. [DOI] [PubMed] [Google Scholar]
- Ben-Tal A. Computational models for the study of heart-lung interactions in mammals. Wiley Interdiscip Rev Syst Biol Med. 2012;4:163–170. doi: 10.1002/wsbm.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Tal A, Shamailov SS, Paton JF. Evaluating the physiological significance of respiratory sinus arrhythmia: looking beyond ventilation-perfusion efficiency. J Physiol. 2012;590:1989–2008. doi: 10.1113/jphysiol.2011.222422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi AL, Denavitsaubie M, Champagnat J. Central control of breathing in mammals—neuronal circuitry, membrane-properties, and neurotransmitters. Physiol Rev. 1995;75:1–45. doi: 10.1152/physrev.1995.75.1.1. [DOI] [PubMed] [Google Scholar]
- Blain GM, Smith CA, Henderson KS, Dempsey JA. Peripheral chemoreceptors determine the respiratory sensitivity of central chemoreceptors to CO(2) J Physiol. 2010;588:2455–2471. doi: 10.1113/jphysiol.2010.187211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga VA, Soriano RN, Machado BH. Sympathoexcitatory response to peripheral chemoreflex activation is enhanced in juvenile rats exposed to chronic intermittent hypoxia. Exp Physiol. 2006;91:1025–1031. doi: 10.1113/expphysiol.2006.034868. [DOI] [PubMed] [Google Scholar]
- Brunner MJ, Sussman MS, Greene AS, Kallman CH, Shoukas AA. Carotid sinus baroreceptor reflex control of respiration. Circ Res. 1982;51:624–636. doi: 10.1161/01.res.51.5.624. [DOI] [PubMed] [Google Scholar]
- Caples SM, Gami AS, Somers VK. Obstructive sleep apnea. Ann Intern Med. 2005;142:187–197. doi: 10.7326/0003-4819-142-3-200502010-00010. [DOI] [PubMed] [Google Scholar]
- Cohen MI. Neurogenesis of respiratory rhythm in the mammal. Physiol Rev. 1979;59:1105–1173. doi: 10.1152/physrev.1979.59.4.1105. [DOI] [PubMed] [Google Scholar]
- Dampney RA. Functional organization of central pathways regulating the cardiovascular system. Physiol Rev. 1994;74:323–364. doi: 10.1152/physrev.1994.74.2.323. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Veasey SC, Morgan BJ, O’Donnell CP. Pathophysiology of sleep apnea. Physiol Rev. 2010;90:47–112. doi: 10.1152/physrev.00043.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Morris KF. Quantitative analysis of cardiovascular modulation in respiratory neural activity. J Physiol. 2004;556:959–970. doi: 10.1113/jphysiol.2003.060418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick TE, Shannon R, Lindsey BG, Nuding SC, Segers LS, Baekey DM, Morris KF. Arterial pulse modulated activity is expressed in respiratory neural output. J Appl Physiol. 2005;99:691–698. doi: 10.1152/japplphysiol.01124.2004. [DOI] [PubMed] [Google Scholar]
- Dove EL, Katona PG. Respiratory effects of brief baroreceptor stimuli in the anesthetized dog. J Appl Physiol. 1985;59:1258–1265. doi: 10.1152/jappl.1985.59.4.1258. [DOI] [PubMed] [Google Scholar]
- Ezure K. Synaptic connections between medullary respiratory neurons and considerations on the genesis of respiratory rhythm. Prog Neurobiol. 1990;35:429–450. doi: 10.1016/0301-0082(90)90030-k. [DOI] [PubMed] [Google Scholar]
- Ezure K, Tanaka I, Saito Y. Brainstem and spinal projections of augmenting expiratory neurons in the rat. Neurosci Res. 2003;45:41–51. doi: 10.1016/s0168-0102(02)00197-9. [DOI] [PubMed] [Google Scholar]
- Felder RB, Mifflin SW. Modulation of carotid sinus afferent input to nucleus tractus solitarius by parabrachial nucleus stimulation. Circ Res. 1988;63:35–49. doi: 10.1161/01.res.63.1.35. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Behm R, Miller CC, 3rd, Stauss H, Unger T. Carotid chemoreceptors, systemic blood pressure, and chronic episodic hypoxia mimicking sleep apnea. J Appl Physiol. 1992a;72:1978–1984. doi: 10.1152/jappl.1992.72.5.1978. [DOI] [PubMed] [Google Scholar]
- Fletcher EC, Lesske J, Culman J, Miller CC, Unger T. Sympathetic denervation blocks blood pressure elevation in episodic hypoxia. Hypertension. 1992b;20:612–619. doi: 10.1161/01.hyp.20.5.612. [DOI] [PubMed] [Google Scholar]
- Gilbey MP. Sympathetic rhythms and nervous integration. Clin Exp Pharmacol Physiol. 2007;34:356–361. doi: 10.1111/j.1440-1681.2007.04587.x. [DOI] [PubMed] [Google Scholar]
- Gilbey MP, Jordan D, Richter DW, Spyer KM. Synaptic mechanisms involved in the inspiratory modulation of vagal cardio-inhibitory neurones in the cat. J Physiol. 1984;356:65–78. doi: 10.1113/jphysiol.1984.sp015453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunstein MM, Derenne JP, Milic-Emili J. Control of depth and frequency of breathing during baroreceptor stimulation in cats. J Appl Physiol. 1975;39:395–404. doi: 10.1152/jappl.1975.39.3.395. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Darnall RA, Riley TA. Rostral ventrolateral medulla and sympathorespiratory integration in rats. Am J Physiol. 1990;259:R1063–R1074. doi: 10.1152/ajpregu.1990.259.5.R1063. [DOI] [PubMed] [Google Scholar]
- Guyenet PG, Stornetta RL, Bayliss DA. Retrotrapezoid nucleus and central chemoreception. J Physiol. 2008;586:2043–2048. doi: 10.1113/jphysiol.2008.150870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet PG, Bayliss DA, Stornetta RL, Fortuna MG, Abbott SB, DePuy SD. Retrotrapezoid nucleus, respiratory chemosensitivity and breathing automaticity. Respir Physiol Neurobiol. 2009;168:59–68. doi: 10.1016/j.resp.2009.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habler HJ, Janig W, Michaelis M. Respiratory modulation in the activity of sympathetic neurones. Prog Neurobiol. 1994;43:567–606. doi: 10.1016/0301-0082(94)90053-1. [DOI] [PubMed] [Google Scholar]
- Haselton JR, Guyenet PG. Central respiratory modulation of medullary sympathoexcitatory neurons in rat. Am J Physiol. 1989;256:R739–R750. doi: 10.1152/ajpregu.1989.256.3.R739. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Coles SK, McCrimmon DR. Respiratory neurons mediating the Breuer-Hering reflex prolongation of expiration in rat. J Neurosci. 1996;16:6526–6536. doi: 10.1523/JNEUROSCI.16-20-06526.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski WA, Feldman JL. Distinct rhythm generators for inspiration and expiration in the juvenile rat. J Physiol Lond. 2006;570:407–420. doi: 10.1113/jphysiol.2005.098848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczewski WA, Onimaru H, Homma I, Feldman JL. Opioid-resistant respiratory pathway from the preinspiratory neurones to abdominal muscles: in vivo and in vitro study in the newborn rat. J Physiol. 2002;545:1017–1026. doi: 10.1113/jphysiol.2002.023408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerath R, Edry JW, Barnes VA, Jerath V. Physiology of long pranayamic breathing: neural respiratory elements may provide a mechanism that explains how slow deep breathing shifts the autonomic nervous system. Med Hypotheses. 2006;67:566–571. doi: 10.1016/j.mehy.2006.02.042. [DOI] [PubMed] [Google Scholar]
- Jiang C, Lipski J. Extensive monosynaptic inhibition of ventral respiratory group neurons by augmenting neurons in the Botzinger complex in the cat. Exp Brain Res. 1990;81:639–648. doi: 10.1007/BF02423514. [DOI] [PubMed] [Google Scholar]
- Jurado-Gamez B, Fernandez-Marin MC, Gomez-Chaparro JL, Munoz-Cabrera L, Lopez-Barea J, Perez-Jimenez F, Lopez-Miranda J. Relationship of oxidative stress and endothelial dysfunction in sleep apnoea. Eur Respir J. 2011;37:873–879. doi: 10.1183/09031936.00027910. [DOI] [PubMed] [Google Scholar]
- Koshiya N, Guyenet PG. Tonic sympathetic chemoreflex after blockade of respiratory rhythmogenesis in the rat. J Physiol. 1996;491 (Pt. 3):859–869. doi: 10.1113/jphysiol.1996.sp021263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiya N, Smith JC. Neuronal pacemaker for breathing visualized in vitro. Nature. 1999;400:360–363. doi: 10.1038/22540. [DOI] [PubMed] [Google Scholar]
- Li Z, Morris KF, Baekey DM, Shannon R, Lindsey BG. Responses of simultaneously recorded respiratory-related medullary neurons to stimulation of multiple sensory modalities. J Neurophysiol. 1999a;82:176–187. doi: 10.1152/jn.1999.82.1.176. [DOI] [PubMed] [Google Scholar]
- Li Z, Morris KF, Baekey DM, Shannon R, Lindsey BG. Multimodal medullary neurons and correlational linkages of the respiratory network. J Neurophysiol. 1999b;82:188–201. doi: 10.1152/jn.1999.82.1.188. [DOI] [PubMed] [Google Scholar]
- Lindsey BG, Arata A, Morris KF, Hernandez YM, Shannon R. Medullary raphe neurones and baroreceptor modulation of the respiratory motor pattern in the cat. J Physiol. 1998;512 (Pt. 3):863–882. doi: 10.1111/j.1469-7793.1998.863bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewy AD, Spyer KM. Central Regulation of Autonomic Functions. Oxford University Press; New York: 1990. [Google Scholar]
- Lumsden T. Observations on the respiratory centres in the cat. J Physiol. 1923;57:153–160. doi: 10.1113/jphysiol.1923.sp002052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malpas SC. The rhythmicity of sympathetic nerve activity. Prog Neurobiol. 1998;56:65–96. doi: 10.1016/s0301-0082(98)00030-6. [DOI] [PubMed] [Google Scholar]
- Malpas SC. Sympathetic nervous system overactivity and its role in the development of cardiovascular disease. Physiol Rev. 2010;90:513–557. doi: 10.1152/physrev.00007.2009. [DOI] [PubMed] [Google Scholar]
- Mandel DA, Schreihofer AM. Central respiratory modulation of barosensitive neurones in rat caudal ventrolateral medulla. J Physiol. 2006;572:881–896. doi: 10.1113/jphysiol.2005.103622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina N, Abdala AP, Trapp S, Li A, Nattie EE, Hewinson J, Smith JC, Paton JF, Gourine AV. Essential role of Phox2b-expressing ventrolateral brainstem neurons in the chemosensory control of inspiration and expiration. J Neurosci. 2010;30:12466–12473. doi: 10.1523/JNEUROSCI.3141-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllen RM. Central respiratory modulation of subretrofacial bulbospinal neurones in the cat. J Physiol. 1987;388:533–545. doi: 10.1113/jphysiol.1987.sp016630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Tanaka I, Ezure K. Excitatory and inhibitory synaptic inputs shape the discharge pattern of pump neurons of the nucleus tractus solitarii in the rat. Exp Brain Res. 1999;129:191–200. doi: 10.1007/s002210050889. [DOI] [PubMed] [Google Scholar]
- Molkov YI, Abdala AP, Bacak BJ, Smith JC, Paton JF, Rybak IA. Late-expiratory activity: emergence and interactions with the respiratory CpG. J Neurophysiol. 2010;104:2713–2729. doi: 10.1152/jn.00334.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkov YI, Zoccal DB, Moraes DJ, Paton JF, Machado BH, Rybak IA. Intermittent hypoxia-induced sensitization of central chemoreceptors contributes to sympathetic nerve activity during late expiration in rats. J Neurophysiol. 2011;105:3080–3091. doi: 10.1152/jn.00070.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira TS, Takakura AC, Colombari E, Guyenet PG. Central chemoreceptors and sympathetic vasomotor outflow. J Physiol. 2006;577:369–386. doi: 10.1113/jphysiol.2006.115600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Mistry AM, Guyenet PG, Bayliss DA. Purinergic P2 receptors modulate excitability but do not mediate pH sensitivity of RTN respiratory chemoreceptors. J Neurosci. 2006;26:7230–7233. doi: 10.1523/JNEUROSCI.1696-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulkey DK, Rosin DL, West G, Takakura AC, Moreira TS, Bayliss DA, Guyenet PG. Serotonergic neurons activate chemosensitive retrotrapezoid nucleus neurons by a pH-independent mechanism. J Neurosci. 2007;27:14128–14138. doi: 10.1523/JNEUROSCI.4167-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nattie E, Li A. Central chemoreception is a complex system function that involves multiple brain stem sites. J Appl Physiol. 2009;106:1464–1466. doi: 10.1152/japplphysiol.00112.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino T, Honda Y. Changes in pattern of breathing following baroreceptor stimulation in cats. Jpn J Physiol. 1982;32:183–195. doi: 10.2170/jjphysiol.32.183. [DOI] [PubMed] [Google Scholar]
- Onimaru H, Homma I. A novel functional neuron group for respiratory rhythm generation in the ventral medulla. J Neurosci. 2003;23:1478–1486. doi: 10.1523/JNEUROSCI.23-04-01478.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onimaru H, Arata A, Homma I. Primary respiratory rhythm generator in the medulla of brainstem-spinal cord preparation from newborn rat. Brain Res. 1988;445:314–324. doi: 10.1016/0006-8993(88)91194-8. [DOI] [PubMed] [Google Scholar]
- Paton JF. The ventral medullary respiratory network of the mature mouse studied in a working heart-brainstem preparation. J Physiol. 1996;493 (Pt. 3):819–831. doi: 10.1113/jphysiol.1996.sp021425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poliacek I, Morris KF, Lindsey BG, Segers LS, Rose MJ, Corrie LW, Wang C, Pitts TE, Davenport PW, Bolser DC. Blood pressure changes alter tracheo-bronchial cough: computational model of the respiratory-cough network and in vivo experiments in anesthetized cats. J Appl Physiol. 2011;111:861–873. doi: 10.1152/japplphysiol.00458.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekling JC, Feldman JL. PreBotzinger complex and pacemaker neurons: hypothesized site and kernel for respiratory rhythm generation. Annu Rev Physiol. 1998;60:385–405. doi: 10.1146/annurev.physiol.60.1.385. [DOI] [PubMed] [Google Scholar]
- Richter DW. Neural regulation of respiration: rhythmogenesis and afferent control. In: Greger R, Windhorst U, editors. Comprehensive Human Physiology. Springer-Verlag; Berlin: 1996. pp. 2079–2095. [Google Scholar]
- Richter DW, Seller H. Baroreceptor effects on medullary respiratory neurones of the cat. Brain Res. 1975;86:168–171. doi: 10.1016/0006-8993(75)90651-4. [DOI] [PubMed] [Google Scholar]
- Richter DW, Spyer KM. Cardiorespiratory control. In: Loewy AD, Spyer KM, editors. Central Regulation of Autonomic Functions. Oxford University Press; USA: 1990. [Google Scholar]
- Richter DW, Spyer KM. Studying rhythmogenesis of breathing: comparison of in vivo and in vitro models. Trends Neurosci. 2001;24:464–472. doi: 10.1016/s0166-2236(00)01867-1. [DOI] [PubMed] [Google Scholar]
- Rosin DL, Chang DA, Guyenet PG. Afferent and efferent connections of the rat retrotrapezoid nucleus. J Comp Neurol. 2006;499:64–89. doi: 10.1002/cne.21105. [DOI] [PubMed] [Google Scholar]
- Rybak IA, Shevtsova NA, Paton JF, Dick TE, St-John WM, Morschel M, Dutschmann M. Modeling the ponto-medullary respiratory network. Respir Physiol Neurobiol. 2004;143:307–319. doi: 10.1016/j.resp.2004.03.020. [DOI] [PubMed] [Google Scholar]
- Rybak IA, Abdala AP, Markin SN, Paton JF, Smith JC. Spatial organization and state-dependent mechanisms for respiratory rhythm and pattern generation. Prog Brain Res. 2007;165:201–220. doi: 10.1016/S0079-6123(06)65013-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, O’Connor R, Ross A, Shevtsova NA, Nuding SC, Segers LS, Shannon R, Dick TE, Dunin-Barkowski WL, Orem JM, Solomon IC, Morris KF, Lindsey BG. Reconfiguration of the pontomedullary respiratory network: a computational modeling study with coordinated in vivo experiments. J Neurophysiol. 2008;100:1770–1799. doi: 10.1152/jn.90416.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybak IA, Molkov YI, Paton JFR, Abdala APL, Zoccal DB. Chapter 143—modeling the autonomic nervous system. In: David R, Italo B, Geoffrey B, Phillip AL, Julian FRP, editors. Primer on the Autonomic Nervous System. 3. Academic Press; San Diego: 2012. pp. 681–687. [Google Scholar]
- Schreihofer AM, Guyenet PG. The baroreflex and beyond: control of sympathetic vasomotor tone by GABAergic neurons in the ventrolateral medulla. Clin Exp Pharmacol Physiol. 2002;29:514–521. doi: 10.1046/j.1440-1681.2002.03665.x. [DOI] [PubMed] [Google Scholar]
- Simms AE, Paton JF, Pickering AE, Allen AM. Amplified respiratory-sympathetic coupling in the spontaneously hypertensive rat: does it contribute to hypertension? J Physiol. 2009;587:597–610. doi: 10.1113/jphysiol.2008.165902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms AE, Paton JF, Allen AM, Pickering AE. Is augmented central respiratory-sympathetic coupling involved in the generation of hypertension? Respir Physiol Neurobiol. 2010;174:89–97. doi: 10.1016/j.resp.2010.07.010. [DOI] [PubMed] [Google Scholar]
- Smith JC, Ellenberger HH, Ballanyi K, Richter DW, Feldman JL. Pre-Botzinger complex: a brainstem region that may generate respiratory rhythm in mammals. Science. 1991;254:726–729. doi: 10.1126/science.1683005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Koizumi H, Rybak IA, Paton JF. Spatial and functional architecture of the mammalian brain stem respiratory network: a hierarchy of three oscillatory mechanisms. J Neurophysiol. 2007;98:3370–3387. doi: 10.1152/jn.00985.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Rybak IA, Paton JF. Structural and functional architecture of respiratory networks in the mammalian brainstem. Philos Trans R Soc Lond B Biol Sci. 2009;364:2577–2587. doi: 10.1098/rstb.2009.0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JC, Abdala AP, Borgmann A, Rybak IA, Paton JF. Brainstem respiratory networks: building blocks and microcircuits. Trends Neurosci. 2012;36 (3):152–162. doi: 10.1016/j.tins.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck DF, Webber CL., Jr Baroreceptor modulation of inspiratory termination by intercostal nerve stimulation. Respir Physiol. 1983;52:387–395. doi: 10.1016/0034-5687(83)90093-2. [DOI] [PubMed] [Google Scholar]
- Stella MH, Knuth SL, Bartlett D. Respiratory response to baroreceptor stimulation and spontaneous contractions of the urinary bladder. Respir Physiol. 2001;124:169–178. doi: 10.1016/s0034-5687(00)00215-2. [DOI] [PubMed] [Google Scholar]
- Stornetta RL, Moreira TS, Takakura AC, Kang BJ, Chang DA, West GH, Brunet JF, Mulkey DK, Bayliss DA, Guyenet PG. Expression of Phox2b by brainstem neurons involved in chemosensory integration in the adult rat. J Neurosci. 2006;26:10305–10314. doi: 10.1523/JNEUROSCI.2917-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QJ, Minson J, Llewellyn-Smith IJ, Arnolda L, Chalmers J, Pilowsky P. Botzinger neurons project towards bulbospinal neurons in the rostral ventrolateral medulla of the rat. J Comp Neurol. 1997;388:23–31. doi: 10.1002/(sici)1096-9861(19971110)388:1<23::aid-cne2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Takakura AC, Moreira TS, Colombari E, West GH, Stornetta RL, Guyenet PG. Peripheral chemoreceptor inputs to retrotrapezoid nucleus (RTN) CO2-sensitive neurons in rats. J Physiol. 2006;572:503–523. doi: 10.1113/jphysiol.2005.103788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian GF, Peever JH, Duffin J. Botzinger-complex, bulbospinal expiratory neurones monosynaptically inhibit ventral-group respiratory neurones in the decerebrate rat. Exp Brain Res. 1999;124:173–180. doi: 10.1007/s002210050612. [DOI] [PubMed] [Google Scholar]
- Zhong S, Zhou SY, Gebber GL, Barman SM. Coupled oscillators account for the slow rhythms in sympathetic nerve discharge and phrenic nerve activity. Am J Physiol. 1997;272:R1314–R1324. doi: 10.1152/ajpregu.1997.272.4.R1314. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Oliveira FR, Antunes-Rodrigues J, Machado BH. Increased sympathetic activity in rats submitted to chronic intermittent hypoxia. Exp Physiol. 2007;92:79–85. doi: 10.1113/expphysiol.2006.035501. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Simms AE, Bonagamba LG, Braga VA, Pickering AE, Paton JF, Machado BH. Increased sympathetic outflow in juvenile rats submitted to chronic intermittent hypoxia correlates with enhanced expiratory activity. J Physiol. 2008;586:3253–3265. doi: 10.1113/jphysiol.2008.154187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoccal DB, Bonagamba LG, Paton JF, Machado BH. Sympathetic-mediated hypertension of awake juvenile rats submitted to chronic intermittent hypoxia is not linked to baroreflex dysfunction. Exp Physiol. 2009a;94:972–983. doi: 10.1113/expphysiol.2009.048306. [DOI] [PubMed] [Google Scholar]
- Zoccal DB, Paton JF, Machado BH. Do changes in the coupling between respiratory and sympathetic activities contribute to neurogenic hypertension? Clin Exp Pharmacol Physiol. 2009b;36:1188–1196. doi: 10.1111/j.1440-1681.2009.05202.x. [DOI] [PubMed] [Google Scholar]