

Abstract
Mammals express four arrestin subtypes, three of which have been shown to self-associate. Cone photoreceptor-specific arrestin-4 is the only one that is a constitutive monomer. Visual arrestin-1 forms tetramers both in crystal and in solution, but the shape of its physiologically relevant solution tetramer is very different from that in the crystal. The biological role of the self-association of arrestin-1, expressed at very high levels in rod and cone photoreceptors, appears to be protective, reducing the concentration of cytotoxic monomers. The two nonvisual arrestin subtypes are highly homologous, and self-association of both is facilitated by IP6, yet they form dramatically different oligomers. Arrestin-2 apparently self-associates into “infinite” chains, very similar to those observed in IP6-soaked crystals, where IP6 connects the concave sides of the N- and C-domains of adjacent protomers. In contrast, arrestin-3 only forms dimers, in which IP6 likely connects the C-domains of two arrestin-3 molecules. Thus, each of the three self-associating arrestins does it in its own way, forming three different types of oligomers. The physiological role of the oligomerization of arrestin-1 and both nonvisual arrestins might be quite different, and in each case it remains to be definitively elucidated.
Keywords: Arrestin, Self-association, Structure, Crystal, EPR, Cytotoxicity
1 Visual Arrestin-1: The Discovery of Oligomerization
The beginning of arrestin history is rather convoluted: the first member of what we now call the arrestin family was originally discovered as an antigen against which patients with uveitis have antibodies (Wacker et al. 1977). Therefore, this protein was named S-antigen, and its gene is still called Sag in the HUGO database. The ability of this protein to oligomerize was described when it was identified, isolated, and characterized (Wacker et al. 1977). A soluble protein with an apparent molecular weight of ~48 kDa was later found to bind light-activated phosphorylated rhodopsin (P-Rh*) (Kuhn et al. 1984) and suppress its signaling (Wilden et al. 1986a). Later it was established that the 48-kDa protein and S-antigen are one and the same protein; it was named arrestin for its ability to “arrest” rhodopsin signaling. Despite active functional work with this protein, its oligomerization was largely ignored until two groups independently found that arrestin crystallizes as a tetramer under different conditions (Granzin et al. 1998; Hirsch et al. 1999) (Fig. 1). Its self-association was further analyzed by analytical centrifugation, which suggested that arrestin-11 forms dimers and tetramers in solution (Schubert et al. 1999). This was taken as an indication that the solution tetramer is likely similar to that in the crystal, and the data were interpreted accordingly (Schubert et al. 1999). Since it was clearly demonstrated earlier that at low nanomolar concentrations, where no self-association would be possible, arrestin-1 binds P-Rh* (Gurevich and Benovic 1992, 1993, 1995, 1997; Gurevich et al. 1995), oligomers were hypothesized to be an inactive storage form (Schubert et al. 1999). Two subsequent studies of arrestin-1 oligomerization by small-angle X-ray scattering yielded surprisingly different self-association constants (Imamoto et al. 2003; Shilton et al. 2002). Since the wavelength of X-rays is comparable to the size of arrestin, the small-angle X-ray scattering data could provide information about the shape of the solution tetramer, which was concluded to be the same as that in the crystal. One of these studies (Imamoto et al. 2003) proposed that visual arrestin-1 forms tetramers according to: 2M ⇄ D (K1), 2D ⇄ T (K2), where M, D, and T are monomer, dimer, and tetramer, respectively (MDT model). The oligomerization was found to be cooperative in the sense that the association constant K2 > K1. When the dimerization constant is much greater than the tetramerization constant, the concentration of dimers in the equilibrium mixture is small: it is dominated by tetramers.
Fig. 1.

The crystallographic tetramer of arrestin-1. In the structure [PDB ID: 1CF1 (Hirsch et al. 1999)] each protomer is shown in a different color. The crystallographic tetramer is a dimer of dimers, where individual dimers are held together via C-to-N-domain interfaces (CN), and the two dimers form a tetramer via C-to-C-domain interfaces (CC). The interfaces are enlarged on the right, with residues in positions probed by site-directed spin labeling EPR (Hanson et al. 2007c, 2008a) shown as stick models. Color coding: the residues in positions where the behavior of the spin label was consistent with predictions based on the crystal structure are shown in orange; those in positions where the behavior of the spin label was inconsistent with crystal structure are shown in yellow. Note that at least half of the positions fall into the latter category
2 Crystal and Solution Tetramers of Arrestin-1 Have Nothing in Common
Next, self-association of arrestin-1 in solution was analyzed by multi-angle laser light scattering (MALLS) (Hanson et al. 2007c). The advantages of this method include high resolution to within a few hundred Daltons, wide molecular mass range, relatively small sample size, and high sample throughput. Importantly, because the wavelength of light is large compared to the dimensions of arrestin-1 monomer or any oligomer, no assumptions regarding the shape of solution tetramer are necessary for data interpretation (Mogridge 2004). The results confirmed the earlier proposed MDT model (Imamoto et al. 2003) of monomer–dimer–tetramer equilibrium and the cooperativity of self-association, although it yielded different constants for the same bovine arrestin-1: K1 = 2.7 ± 0.1 × 104, K2 = 1.3 ± 0.1 × 105, which translates into KD,dim = 1/K1 = 37 μM and KD,tet = 1/K2 = 7.5 μM (Hanson et al. 2007c). Interestingly, mutations that were predicted to disrupt self-association based on the crystal tetramer did not affect oligomerization, whereas many others that would not be expected to affect protomer interactions in the crystal had profound effects (Hanson et al. 2007c).
Continuous wave (CW) electron paramagnetic resonance (EPR) spectroscopy can be used to monitor the mobility of a spin label on the surface of a protein (Hanson et al. 2006b). If a particular element happens to be on the protomer–-protomer interaction interface, its mobility would decrease upon oligomer formation. A spin-label side chain (R1) introduced at many positions where significant immobilization was expected based on the crystal structure showed little to no change in mobility, whereas spin labels in several positions that are not on the crystal interfaces were immobilized upon tetramer formation (Hanson et al. 2007c). Collectively, the light scattering and EPR data showed that residues 79, 85, 173, 197, 244, and 348 are involved in inter-subunit interactions in the solution tetramer. While this result would be expected for 79, 85, 197, and 348 based on the crystal tetramer, the strong immobilization of 173R1 and the strong perturbation of self-association due to 244R1 were not predicted by the crystal tetramer (Fig. 1). Neither the native Leu173 and Val244 nor the R1 side chain modeled at these positions in the crystal tetramer makes contacts with neighboring subunits (Hirsch et al. 1999).
Relatively small perturbations and lack of immobilization of R1 at sites 60, 272, and 344, which are deeply buried at the CN, CC, and CN interfaces, respectively, were also inconsistent with the crystal tetramer, where residue 344 is buried to the extent that the R1 side chain cannot be modeled without major rearrangement of the structure (Fig. 1). Importantly, the 344R1 does not perturb the formation of oligomers (Hanson et al. 2007c). The weak perturbation of self-association by 89R1 and lack of spectral change of 89R1, located directly at the NN interface in the crystal (Fig. 1), do not support its existence. These results clearly indicate that the tetramer in solution is quite different from that observed in the crystal.
Double electron–electron resonance (DEER), a pulse EPR technique (Jeschke 2002), is a powerful method for measuring distances between paramagnetic centers in the range of ~19–60 Å (Pannier et al. 2000), complementing CW EPR methods that determine distances between 10 and 20 Å (Altenbach et al. 2001; Hanson et al. 2006b). DEER was used to measure distances between unique spin labels on each protomer within the solution tetramer, which were placed at eight non-perturbing or mildly perturbing sites in the tetramer (74, 108, 139, 173, 240, 272, 273, and 344). Only in one case (273R1) were the experimentally determined inter-spin distances close to the predictions based on the crystal structure, whereas the data for the other sites were clearly incompatible with the crystal tetramer (Hanson et al. 2007c). Thus, several lines of evidence independently suggested that the shape of the solution tetramer must be different.
These unexpected findings made it necessary to elucidate the structure of the physiologically relevant solution tetramer, which holds clues to the functional role of arrestin-1 self-association. Since crystallography was misleading in this regard, the shape of the solution tetramer was deduced using inter-spin distances in the oligomer and the positions where the spin label was immobilized upon self-association (Hanson et al. 2007c, 2008a). These data were used as inputs for Rosetta modeling (Gray et al. 2003a, b; Schueler-Furman et al. 2005; Wang et al. 2005). Several iterations yielded a model for a tetramer consistent with all experimental data (Hanson et al. 2008a), which turned out to be symmetrical diamond shaped, with two nearly identical CC and NN interfaces, where all the interaction interfaces on each protomer are engaged by sister subunits (Fig. 2). Since modeling per se does not yield unambiguous information, this model was subjected to rigorous post hoc testing.
Fig. 2.
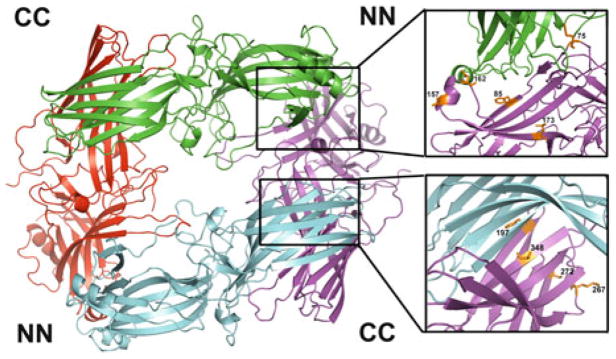
Solution tetramer of arrestin-1. Studies using site-directed spin labeling EPR, long-range inter-subunit distance measurements by DEER spectroscopy, site-directed mutagenesis, Rosetta modeling, and inter-subunit disulfide bridge formation (Hanson et al. 2007c, 2008a) lead to the conclusion that the solution tetramer of arrestin-1 is a symmetrical closed diamond, where adjacent protomers interact via two types of interfaces: C-to-C domain (CC) and N-to-N domain (NN). Enlarged interfaces are shown on the right, with residues in positions experimentally tested by various methods shown as stick models (see text for details)
First, the R1 side chain was introduced either directly at the putative interface (position 75) or outside it (positions 376 and 381). CW EPR showed immobilization of the label at 75, with no evidence of immobilization at 376 or 381, consistent with the model (Hanson et al. 2008a). The residues Phe197 and Ala348 in the CC interface and Thr157 and Asp162 in the NN interface in the model are very close to their counterparts in the adjacent monomer (Fig. 2). In the crystal tetramer, all of these residues are far from their counterparts in other protomers (>20 Å). In contrast, residue Leu173 in the NN interface and Ser272 in the CC interface are far from their counterparts in the model. To test these predictions, single cysteine mutants were created and their ability to form inter-subunit disulfide bonds in solution was determined. In the presence of DTT, each arrestin ran as a single band on SDS-PAGE at a molecular weight (MW) corresponding to the arrestin monomer. However, in the absence of DTT, the Thr157Cys, Asp162Cys, Phe197Cys, and Ala348Cys mutants showed a second band corresponding to the expected mobility of the arrestin dimer (Hanson et al. 2008a). This suggests that residues 157, 162, 197, and 348 are close enough to their counterparts in the arrestin oligomer to self-cross-link in solution. As predicted, the absence of DTT did not induce cross-linking of Leu173Cys and Ser272Cys. These data strongly support the orientation of the NN and CC interfaces in the model, since disulfide cross-linking only occurs at very short (~5 Å) Cβ–Cβ distances between the two residues.
Finally, the model was tested via targeted disruption of arrestin-1 self-association by mutations directly affecting predicted inter-subunit interfaces. Since the introduction of a spin label per se constitutes a mutation, first the effects of cysteine substitution followed by spin labeling at positions 85 in the predicted NN interface, as well as at positions 197 and 267 in the predicted CC interface, were evaluated. The labeling at all three positions reduced arrestin-1 self-association, confirming that these residues are in the inter-protomer interfaces. Importantly the effects of spin labeling at 197 and 267 were not additive, so that simultaneous labeling of both produced the same effect as the more detrimental to self-association 197R1 (Hanson et al. 2008a). This is consistent with both side chains being at the same interface. In contrast, the combination of 85R1 and 197R1 was much more disruptive than the labeling of either of these two sites alone, consistent with their localization in two different interfaces (Hanson et al. 2008a). Interestingly, the resulting 85R1/197R1 protein virtually lost the ability to self-associate. In the native structure both positions are occupied by phenylalanines. The replacement of Phe85 or Phe197 with alanine reduces self-association, whereas simultaneous substitution of both yields arrestin-1 that is essentially unable to oligomerize (Hanson et al. 2008a). Thus, three independent lines of evidence strongly support the model of solution tetramer (Fig. 2). Most importantly, these studies lead to the generation of a constitutively monomeric form of arrestin-1, which is necessary to elucidate the biological role of arrestin-1 self-association.
The proposed structure of the solution tetramer explains several observations that were inconsistent with the crystal tetramer. First, it explains the observed cooperativity (Hanson et al. 2007c; Imamoto et al. 2003): the interaction between two dimers engages two interfaces, whereas dimer formation involves only one. In contrast, in the crystal tetramer interfaces of comparable size mediate both dimerization and the interaction between two dimers in the tetramer (Granzin et al. 1998; Hirsch et al. 1999). Second, the circular “closed” configuration engages all self-association interfaces, explaining why arrestin-1 self-association stops at tetramer, so that larger oligomers are never formed. In contrast, in the crystal tetramer two protomers are left “dangling” with unused potential interaction interfaces (Granzin et al. 1998; Hirsch et al. 1999) that could mediate the binding of additional monomers. Finally, in the solution tetramer all arrestin-1 elements implicated in receptor binding, which were identified by numerous groups using a variety of methods (Dinculescu et al. 2002; Gimenez et al. 2012a; Gurevich and Benovic 1993; Gurevich et al. 1993; Hanson et al. 2006b; Hanson and Gurevich 2006; Kim et al. 2012; Ohguro et al. 1994; Pulvermuller et al. 2000; Vishnivetskiy et al. 2004, 2011; Zhuang et al. 2010, 2013), are either directly engaged or shielded by sister protomers, which explains why only monomeric arrestin-1 can bind rhodopsin (Hanson et al. 2007c). Moreover, the proposed structure of the solution tetramer (Fig. 2) adequately explains the recent finding that manipulation of the receptor-binding surface of arrestin-1 to enhance its ability to interact with unphosphorylated rhodopsin significantly changes self-association parameters (Vishnivetskiy et al. 2013a).
3 The Mechanism of Arrestin-1 Self-Association Is Conserved in Mammalian Evolution
All these mechanistic studies were performed with bovine arrestin-1, which was purified first both from its native source (Wilden et al. 1986b) and upon overexpression in Escherichia coli (Gray-Keller et al. 1997). However, most of the physiological insights into rod function have been obtained in genetically modified mice (Arshavsky and Burns 2012; Makino et al. 2003), with the ultimate goal of translating the findings to human therapy (Song et al. 2009) (Chapter 7). The key biologically relevant facts about arrestin-1 were established in mice: (1) that it is the second (after rhodopsin) most abundant protein in rods (Hanson et al. 2007b; Song et al. 2011; Strissel et al. 2006) (see Chaps. 4 and 5); (2) that it undergoes light-dependent redistribution in rod photoreceptors (Hanson et al. 2007b; Nair et al. 2005) (Chaps. 4–6); and (3) that it is unexpectedly abundant in cones, where it represents ~98 % of total arrestin complement, whereas cone-specific arrestin-4 accounts for only ~2 % (Nikonov et al. 2008) (see Chap. 6). Thus, it was critically important to test whether mouse and human arrestin-1 self-associate and to determine the parameters of its oligomerization in these species.
Purified mouse arrestin-1 was found to form dimers and tetramers, similar to its bovine homolog (Kim et al. 2011). Interestingly, both dimerization (KD,dim = 57.5 ± 0.6 μM) and tetramerization (KD,tet = 63.1 ± 2.6 μM) dissociation constants of mouse protein were significantly higher than the corresponding values for bovine arrestin-1 [37.2 ± 0.2 μM and 7.4 ± 0.1 μM, respectively (Hanson et al. 2007c, 2008a)]. Moreover, whereas self-association of bovine arrestin-1 is cooperative (KD,tet < KD,dim) (Hanson et al. 2007c; Imamoto et al. 2003), both constants are roughly equal for mouse arrestin-1, eliminating cooperativity. The dramatic differences in self-association constants of arrestin-1 from these two mammalian species made it imperative to determine the properties of human arrestin-1. Purified human arrestin-1 was also found to self-associate and form dimers and tetramers. However, it demonstrated strikingly different constants compared to bovine and mouse proteins: remarkably low KD,dim = 2.95 ± 0.02 μM and relatively high KD,tet = 224 ± 5 μM (Kim et al. 2011). Importantly, if the overall concentration of arrestin-1 in the cell body of mammalian dark-adapted rod photoreceptors is similar to that measured in mouse (~2 mM (Song et al. 2011)), it greatly exceeds all measured dissociation constants. Therefore, despite these differences in self-association parameters the concentration of monomeric arrestin-1 in human, bovine, and mouse rods would be in a fairly narrow range, 30–90 μM. As the majority of arrestin-1 would exist in the form of tetramer in all three species, the tetramer concentration in the rod would vary by no more than 30 %, and the most striking difference would be in the expected dimer concentrations, varying from ~60 μM in bovine to ~280 μM in human rod (Kim et al. 2011).
Nonetheless, measured KD,dim between human and mouse arrestin-1 differs ~20-fold, and KD,tet of bovine and human proteins is ~30-fold different. The magnitude of these differences raises the possibility that arrestin-1 in these three species could use distinct interaction interfaces. In this scenario phenomenological similarity of self-association could represent convergent evolution, rather than direct conservation of the molecular mechanism. The nature of the interaction interfaces in the solution tetramer of bovine arrestin-1 was strongly supported by the observation that the combination of two mutations predicted to disrupt NN (Phe85Ala) and CC (Phe197Ala) self-association interfaces makes the protein essentially a constitutive monomer, with KD,dim = 525 μM and no detectable tetramerization (Hanson et al. 2008a). To test whether interaction interfaces are conserved in mouse protein, self-association of the double mutant carrying homologous substitutions Phe86Ala + Phe198Ala was tested. This mutation yielded the same phenotype as in bovine protein: mouse arrestin-1-Phe86Ala + Phe198Ala demonstrated dramatically impaired self-association, with KD,dim = 537 μM and no tetramer formation (Kim et al. 2011). The finding that homologous mutations in bovine and mouse arrestin-1 similarly disrupt their self-association strongly suggests that both proteins use the same interfaces for oligomerization. Thus, strikingly different self-association constants reflect the difference in the energy of interactions between the subunits, whereas the organization of the solution tetramer is likely the same in all mammals. Importantly, the elimination of these two phenylalanines does not appreciably affect arrestin-1 binding to its two best-characterized partners, P-Rh* and microtubules (Kim et al. 2011). This finding suggests that these residues are strictly conserved in all mammalian arrestin-1 proteins (Gurevich and Gurevich 2006) because they facilitate self-association, indicating that robust arrestin-1 oligomerization is a biologically important aspect of its function in photoreceptor cells (Gurevich et al. 2011).
4 Possible Biological Role of Arrestin-1 Self-Association
Unambiguous demonstration that only monomeric arrestin-1 is capable of binding rhodopsin (Hanson et al. 2007c) confirmed the earlier hypothesis that dimers and tetramers are storage forms (Schubert et al. 1999). Although rod photoreceptors express many signaling proteins at levels several orders of magnitude higher than “normal” cells (Pugh and Lamb 2000), and arrestin-1 is the second most abundant protein in the rod (Hanson et al. 2007b; Song et al. 2011; Strissel et al. 2006), no other signaling protein in photoreceptors has an inactive storage form. Thus, arrestin-1 propensity to form inactive oligomers calls for an explanation.
The first glimpse into a possible role of this phenomenon emerged from unexpected quarters. In an attempt to compensate for defects in rhodopsin phosphorylation, two transgenic lines were created expressing the enhanced phosphorylation-independent arrestin-1-3A mutant (Gurevich 1998) at ~50 and ~240 % of normal WT level (Song et al. 2009). It turned out that the lower expressor line actually showed the expected compensation, whereas rod photoreceptors in the other degenerated even faster than in arrestin-1 knockout mice (Song et al. 2009, 2013). To achieve light sensitivity at the physical limit of single photons (Baylor et al. 1979), rods express very high levels of all signaling proteins (Pugh and Lamb 2000), maintaining a fairly precarious balance. As a result, overexpression of a perfectly normal WT protein can often lead to photoreceptor death, as has been shown for rhodopsin (Tan et al. 2001). However, it was found that the expression of WT arrestin-1 at essentially the same level, ~220 % of WT, is harmless (Song et al. 2011), indicating that it is the mutant nature of arrestin-1-3A that makes it toxic for rods. The analysis of the 3A mutant by MALLS showed that while this enhanced mouse arrestin-1 binds Rh* much better than WT, its self-association is partially compromised: KD,dim increased from 57.5 ± 0.6 μM of WT protein (Kim et al. 2011) to 135 ± 2 μM, with a simultaneous increase of KD,tet from 63.1 ± 2.6 μM to 380 ± 79 μM (Song et al. 2013). Calculations based on these constants, relative volumes of rod compartments (Peet et al. 2004), and arrestin-1 distribution in dark-adapted rod (Hanson et al. 2007b; Nair et al. 2005; Song et al. 2011; Strissel et al. 2006) indicate that the concentration of arrestin-1 monomer in the cell body of WT mouse rod is ~95 μM (out of ~2,000 μM of total arrestin-1). Due to robust self-association, a 2.2-fold increase of WT arrestin-1 to ~4,400 μM results in only a modest increase in free monomer, to ~104 μM. In contrast, the expression of arrestin-1-3A at 240 % of WT level would yield ~270 μM of monomer, almost three times more than in WT rods (Song et al. 2013). Importantly, the expression of the same mutant at ~50 % of WT level yields only ~115 μM monomer, which is not dramatically different from WT overexpressors, consistent with the relatively good health of photoreceptors in these animals (Song et al. 2009, 2013), at least until they reach the age of 32 weeks (Song et al. 2013). WT arrestin-1 was shown to effectively recruit mutants with partially compromised oligomerization into tetramers (Hanson et al. 2007c). Thus, if too high monomer concentration adversely affects rods, co-expression of WT arrestin-1 with the mutants would be expected to protect them. Indeed, it was shown that WT arrestin-1 expressed in rods with high levels of arrestin-1-3A affords partial protection against the mutant, slowing down photoreceptor death (Song et al. 2013). Interestingly, arrestin-1 was shown to interact with N-ethylmaleimide-sensitive factor (NSF) (Huang et al. 2010), a protein involved in exocytosis of neurotransmitter in the synapses. Indeed, synaptic terminals of rods expressing high levels of 3A mutant showed early damage, and their protection by WT arrestin-1 was very robust (Song et al. 2013). Collectively, the existing evidence is consistent with the idea that a relatively low level of monomeric arrestin-1 is optimal for photoreceptor health, whereas an excess of monomer induces cell death (see Chap. 16).
This hypothesis explains why arrestin-1 developed the ability to self-associate: rods need sufficient amounts of arrestin-1 to quench virtually all rhodopsin (Chap. 4 and 5), yet can tolerate only fairly low levels of monomer (Song et al. 2013). Thus, to solve this problem rods store the bulk of arrestin-1 in the form of “safe” oligomers. Cytotoxicity of the monomer can also explain the relatively low expression of arrestin-4 (Chan et al. 2007), which is outnumbered by arrestin-1 in cones by ~50:1 (Nikonov et al. 2008). Arrestin-4, a cone-specific subtype, appeared early in vertebrate evolution (Gurevich and Gurevich 2006). In contrast to other subtypes that form tight relatively long-lived complexes with their cognate GPCRs (Bayburt et al. 2011; Gurevich et al. 1995, 1997), arrestin-4 forms only low-affinity fairly transient complexes with cone opsins (Sutton et al. 2005). Functionally, this is perfectly suited for cones operating at high levels of illumination, which makes recycling and immediate reuse of cone opsins a necessity. Like rods, cones need enough arrestin to stop the signaling by all expressed photopigment. However, arrestin-4 is the only subtype that is self-association deficient, a natural constitutive monomer (Hanson et al. 2008b). If the monomer is toxic, cones simply cannot afford to express sufficient amounts of arrestin-4 and therefore keep the majority of their arrestin complement in the form of safely self-associating arrestin-1.
Thus, it appears that self-association of arrestin-1 is a cytoprotective mechanism, reducing the concentration of toxic monomer in photoreceptor cells. While it remains to be elucidated whether monomer toxicity arises from excessive binding to NSF (Huang et al. 2010), inappropriate engagement of clathrin adaptor AP2 (Moaven et al. 2013), or some other partner, it appears that arrestin-1 oligomerization prevents harmful interactions (Song et al. 2013).
5 Oligomerization of Nonvisual Arrestins: Mechanism and Consequences
Whereas arrestin-1 is expressed at very high levels in rods (Hanson et al. 2007b; Strissel et al. 2006) and cones (Nikonov et al. 2008), with concentrations reaching ~2 mM in the body of dark-adapted photoreceptors (Song et al. 2011), intracellular concentrations of nonvisual arrestins are much lower. Even in mature neurons, which express both at higher levels than most cells, the concentrations of arrestin-3 and -2 reach only ~30 and 200 nM, respectively (Gurevich et al. 2002, 2004). However, arrestins are fairly evenly distributed only in the non-stimulated cell (Song et al. 2006). Both arrestin-2 and -3 are recruited to active phosphorylated GPCRs in all cell types and were shown to become concentrated in the vicinity of the plasma membrane and endosomes upon GPCR activation (Barak et al. 1997). By virtue of their binding to polymerized tubulin (Hanson et al. 2006a), nonvisual arrestins also appear to be concentrated in the vicinity of microtubules (Hanson et al. 2007a). Thus, local concentration in particular cell compartments under certain circumstances can greatly exceed estimated averages. Indeed, arrestin-2 and -3 expressed at near-physiological levels were reported to form homo- and hetero-oligomers in cells (Milano et al. 2006; Storez et al. 2005). Hetero-oligomerization of arrestin-3, which has a functional nuclear export signal in it C-terminus (Scott et al. 2002; Song et al. 2006; Wang et al. 2003), with arrestin-2 that does not, appears to help the removal of arrestin-2 from the nucleus (Storez et al. 2005).
Inositol-hexakisphosphate (IP6, a.k.a. phytic acid), an abundant metabolite present in many cells in concentrations of 15–100 μM (Shears 2001), was shown to greatly enhance self-association of both nonvisual arrestins (Milano et al. 2006). Even though full-length arrestin-2 crystallizes as a monomer (Milano et al. 2002), solving the structure of crystals soaked with IP6 revealed that IP6 bridges neighboring molecules in a head-to-tail configuration via interactions with two sites, one in the N-domain and the other in the C-domain of arrestin-2 (Milano et al. 2006). Direct binding studies combined with extensive mutagenesis showed that the C-domain site has a much higher affinity (KD ~ 40 nM) than the N-domain site (KD ~ 1 μM) for IP6, but both are well within the range of physiological IP6 concentrations in the cell (Milano et al. 2006).
Elimination of positively charged residues critical for IP6 binding increased the arrestin-2 presence in the nucleus, suggesting that oligomers are largely cytoplasmic (Milano et al. 2006). Both IP6 binding sites appear to be localized on the receptor-binding surface of arrestins identified by many groups using various methods (Dinculescu et al. 2002; Gimenez et al. 2012b; Gurevich and Benovic 1993, 1995; Gurevich et al. 1995; Hanson et al. 2006b; Hanson and Gurevich 2006; Kim et al. 2012; Ohguro et al. 1994; Pulvermuller et al. 2000; Vishnivetskiy et al. 2011; Zhuang et al. 2013), indicating that simultaneous binding of receptor and IP6 is impossible. This finding suggested that, as in the case of arrestin-1, oligomers represent an inactive storage form, whereas monomeric arrestins are recruited to GPCRs, as well as translocated to the nucleus (Milano et al. 2006). Interestingly, while IP6 greatly increases self-association of nonvisual subtypes (Hanson et al. 2008b; Milano et al. 2006), it significantly inhibits the oligomerization of arrestin-1 (Hanson et al. 2008b), indicating that the interfaces involved and overall shape of the oligomers formed by visual and nonvisual arrestins are different.
Experiments with purified proteins and cells expressing IP6 binding-deficient mutants of both arrestin-2 and -3 also suggested that they form oligomers larger than dimer (Milano et al. 2006). However, while arrestin-1 was shown to stop at tetramer (Hanson et al. 2007c), in which all interaction interfaces are engaged by sister subunits (Hanson et al. 2008a), it remained unclear whether arrestin-2 and -3 also stop at a particular size of oligomer, or can form “infinite” chains, as suggested by IP6-soaked arrestin-2 crystal structure (Milano et al. 2006). This issue was addressed using pure arrestins in the presence of IP6 by MALLS (Chen et al. 2013). In the absence of IP6, arrestin-2 and -3 have a low tendency to self-associate with a KD around 100 μM (Fig. 3). IP6 promotes their self-association, and the KDs decrease to 5.5 and 7.8 μM for arrestin-2 and -3, respectively (Chen et al. 2013).
Fig. 3.
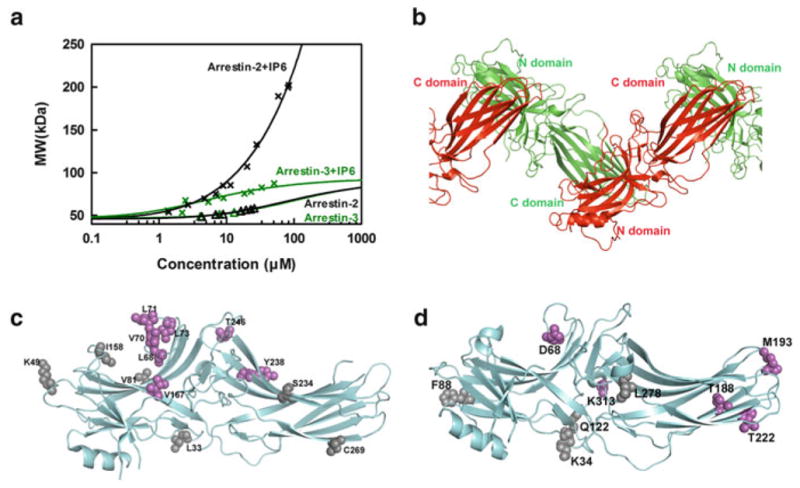
The two nonvisual arrestins form distinct oligomers. (a) The average molecular weight of arrestin-2 and arrestin-3 in the presence (crosses) and absence (triangles) of 100 μM IP6 as a function of total arrestin concentration was measured by MALLS. Arrestin-2 data were fit by a linear polymerization model (black line), while arrestin-3 data were fit by a monomer–dimer model (green line). Neither nonvisual arrestin showed a propensity to self-associate at physiologically relevant concentrations in the absence of IP6. (b) The crystal structure of arrestin-2 in complex with IP6 [PDB ID: 1ZSH (Milano et al. 2006)] shows that arrestin-2 forms “infinite” chains through C-to-N-domain interactions mediated by IP6. (c) The positions of spin-labeled sites are shown as spheres on the crystal structure of arrestin-2 [PDB ID: 1ZSH (Milano et al. 2006)]. The sites with inter-subunit distances shorter than 50 Å (Leu68, Val70, Leu71, Leu73, Val167, Tyr238, and Thr246), as measured by DEER spectroscopy in the presence of IP6, are colored magenta, and the ones with inter-subunit distance longer than 50 Å (Leu33, Lys49, Val81, Ile158, Ser234, and Cys269) are colored gray [data from Chen et al. (2013)]. (d) The positions of spin-labeled sites are shown as spheres on the crystal structure of arrestin-3 [PDBID: 3P2D (Zhan et al. 2011)]. The sites with inter-subunit distance shorter than 50 Å (Asp68, Thr188, Met193, Thr222, and Lys313), as measured by DEER spectroscopy in the presence of IP6, are colored magenta, while the ones with inter-subunit distance longer than 50 Å (Lys34, Phe88, Gln122, and Leu278) are colored gray [data from Chen et al. (2013)]
Despite high homology arrestin-2 and -3 form distinct oligomers in the presence of IP6: arrestin-3 forms dimers; in contrast, arrestin-2 forms long chains that go beyond tetramer. The average molecular weight of arrestin-2 keeps growing without obvious saturation. At the highest concentration tested (84 μM), the average molecular weight of arrestin-2 oligomers reached ~202 kDa, which exceeded the expected molecular weight for the arrestin-2 tetramer (184 kDa) (Fig. 3). Due to the formation of higher order oligomers MALLS data do not fit into the MDT model. Instead, a liner polymerization model (M[n] + M ↔ M[n + 1]) fit arrestin-2 oligomerization data very well (Chen et al. 2013). This suggested that arrestin-2 might form an infinite chain mediated by IP6, as in the arrestin-2 crystals soaked with IP6 (Milano et al. 2006). However, the crystal structure does not necessarily reflect that which exists in solution, since in that study the orientation of arrestin-2 molecules relative to each other was fixed by crystallization in the absence of IP6 (Milano et al. 2006). Therefore, DEER was used to probe the structure of the solution oligomer of arrestin-2 in the presence of IP6 (Chen et al. 2013). Thirteen sites on arrestin-2 were selected on both the N-domain (Leu33, Lys49, Leu68, Val70, Leu71, Leu73, Val81, Ile158, and Val167) and the C-domain (Ser234, Tyr238, Thr246, and Cys269). These sites were located on the receptor-binding concave side (Lys49, Ile158, Val81, Leu68, Val70, Leu71, Leu73, Val167, Thr246, Tyr238, and Ser234) and the convex side (Leu33 and Cys269) to obtain a comprehensive characterization of the solution oligomer of arrestin-2 in the presence of IP6. A nitroxide spin label (R1) at selected sites in arrestin-2 was introduced by chemical modification of these unique cysteines and the inter-subunit distances were measured using the DEER spectroscopy. The measured DEER distances (Chen et al. 2013) matched remarkably well with the expected nitroxide-to-nitroxide distances between adjoining protomers in the crystal structure (Milano et al. 2006). All but two sets of data matched to within 3 Å of the expected crystallographic distances. Importantly, the sites with closer distances (<50 Å) clustered in the central parts of the concave receptor-binding side, which suggested that IP6 mediates the interaction between the N- and C-domains of arrestin-2, so that only the central parts of the concave side come close together (Fig. 3). Collectively, these data clearly suggest that the arrangement of protomers in the arrestin-2 crystals soaked with IP6 closely resembles the structure of the solution oligomer of arrestin-2, further supporting the hypothesis that arrestin-2 forms “infinite” chains in the presence of IP6 in solution, similar to those observed in the crystal (Chen et al. 2013; Milano et al. 2006). In contrast, MALLS data showed that the average molecular weight of arrestin-3 oligomers in the presence of IP6 did not exceed that of a dimer (Chen et al. 2013) (Fig. 3). Since the saturation was not reached due to concentration limitations, a higher order oligomer could not be excluded. However, the fact that the data could not be fit to either an MDT model or a polymerization model, but fit well to monomer–dimer equilibrium model suggested that the formation of higher order oligomers of arrestin-3 in the presence of IP6 was not favored. DEER was used to probe the structure of arrestin-3 oligomers in solution in the presence of IP6. The distances measured with several arrestin-3 mutants in the presence of IP6 aligned moderately well with the expected distances based on the arrestin-2 IP6 crystallographic oligomer, but they were not as clearly matched as the arrestin-2 data (Chen et al. 2013). Interestingly, the sites in arrestin-3 with distances shorter than 50 Å are clustered not only in the central crest (Asp68, Lys313), but also in the C-domain, including the distal part of the C-domain (Thr188, Met193, Thr222), while the sites in the N-domain (Lys34, Phe88, and Gln122) had much longer distances, beyond the range of reliable measurement by DEER spectroscopy (Chen et al. 2013). These data suggested that IP6 might mediate the interaction between the C-domains of two arrestin-3 molecules, so that the sites on the C-domain are in close contact, whereas the sites on the N-domain remain far apart. This model would explain why arrestin-3 stops at a dimer, since the interfaces mediating IP6-assisted interaction are no longer exposed upon the formation of the C-to-C-domain dimer. This is in contrast to arrestin-2, in which only the sites in the central crest on the concave side come close to each other in the presence of IP6. Though more data are needed to generate a high-resolution model for the arrestin-3 dimer (or larger oligomer, if it exists), it is very clear that in the presence of IP6 arrestin-2 and arrestin-3 form structurally distinct oligomers (Chen et al. 2013). Since nonvisual arrestins were reported to form mixed oligomers (Milano et al. 2006; Storez et al. 2005), it remains to be elucidated whether these resemble arrestin-2 chains or arrestin-3 C-to-C dimers or have a unique shape and size distinct from both.
6 Do Arrestin Oligomers Have Specific Functions?
It was shown that mutations disrupting self-association of arrestin-1 do not significantly affect its ability to bind its preferred form of rhodopsin, P-Rh*, or micro-tubules (Kim et al. 2011). However, the same mutations somewhat reduced the binding of an enhanced phosphorylation-independent mutant to Rh* (Vishnivetskiy et al. 2013a), in agreement with the finding that distinct arrestin-1 elements are involved in its interactions with different functional forms of rhodopsin (Zhuang et al. 2013). This difference might also reflect distinct stoichiometry of the arrestin-1–rhodopsin interactions in these cases. While arrestin-1 was shown to bind the P-Rh* monomer in nanodiscs (Bayburt et al. 2011; Kim et al. 2012; Singhal et al. 2013; Vishnivetskiy et al. 2013b) and bicelles (Zhuang et al. 2013), a possibility of an alternative mode of interaction was reported in native disc membranes with a high fraction of light-activated rhodopsin, where arrestin-1 appears to engage two rhodopsin molecules simultaneously (Sommer et al. 2011, 2012) (Chap. 5). Even though in these situations arrestin-1 binds only one rhodopsin molecule with high enough affinity to stabilize its active conformation (Sommer et al. 2011, 2012), the engagement of one or two rhodopsin molecules, one of which might be unphosphorylated, could be one of the mechanistic differences in arrestin-1 binding to P-Rh* and Rh*. In either case, it appears that only monomeric arrestin-1 can bind rhodopsin. Interestingly, while rhodopsin binding induces the dissociation of all arrestin-1 oligomers, indicating that only monomeric arrestin-1 can bind the receptor (Hanson et al. 2007c), the monomer (Hanson et al. 2006a, 2007a) and all oligomers appear to bind tubulin comparably, so that in the presence of a sufficient concentration of microtubules to bind all arrestin-1 the inter-subunit distances reporting the presence of oligomers do not appear to be affected (Hanson et al. 2007c).
In the case of nonvisual arrestins, we know even less about specific functions of the oligomeric forms. Oligomerization-deficient mutants were found to bind clathrin, clathrin adaptor AP2, and ERK1/2 normally (Milano et al. 2006), in agreement with the localization of binding sites for these partners (Coffa et al. 2011; Goodman et al. 1996; Laporte et al. 1999) away from residues that mediate IP6 binding (Milano et al. 2006). However, an arrestin-3 mutant that did not bind IP6 was found to lack tight association with another partner, ubiquitin ligase Mdm2, and in contrast to WT arrestin-3, these presumably monomeric mutants did not suppress Mdm2-dependent degradation of p53 (Boularan et al. 2007). While it was proposed that arrestin-3 oligomers provide more interaction sites for putative dimers of Mdm2 (Boularan et al. 2007), another plausible explanation is that since Mdm2 preferentially binds arrestins in the basal conformation (Ahmed et al. 2011; Song et al. 2006, 2007), oligomerization might simply stabilize this conformational state of nonvisual arrestins, which are inherently more flexible than arrestin-1 (Han et al. 2001; Hirsch et al. 1999; Zhan et al. 2011). One study suggested that monomeric nonvisual arrestins are more likely to enter the nucleus (Milano et al. 2006), whereas another found comparable levels of arrestin oligomers in the nucleus and cytoplasm (Boularan et al. 2007), although in the latter case it remained unclear whether arrestin oligomers can enter the nucleus, or arrestins self-associate after entering it as monomers and/or dimers.
It is entirely possible that nonvisual arrestins self-associate for the same reason as arrestin-1 to prevent the buildup of a cytotoxic monomeric form (Song et al. 2013), but this idea needs to be tested experimentally. It is clear that more experimentation is necessary before we will be able to unambiguously determine specific functions of nonvisual arrestin oligomers and sort out cellular processes affected by their impaired self-association.
Footnotes
Different systems of arrestin names are used in the field and in this book. We use the systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database).
References
- Ahmed MR, Zhan X, Song X, Kook S, Gurevich VV, Gurevich EV. Ubiquitin ligase parkin promotes Mdm2-arrestin interaction but inhibits arrestin ubiquitination. Biochemistry. 2011;50:3749–3763. doi: 10.1021/bi200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Oh K-J, Trabanino RJ, Hideg K, Hubbell WL. Estimation of inter-residue distances in spin labeled proteins at physiological temperatures: experimental strategies and practical limitations. Biochemistry. 2001;40:15471–15482. doi: 10.1021/bi011544w. [DOI] [PubMed] [Google Scholar]
- Arshavsky VY, Burns ME. Photoreceptor signaling: supporting vision across a wide range of light intensities. J Biol Chem. 2012;287:1620–1626. doi: 10.1074/jbc.R111.305243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak LS, Ferguson SS, Zhang J, Caron MG. A beta-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J Biol Chem. 1997;272:27497–27500. doi: 10.1074/jbc.272.44.27497. [DOI] [PubMed] [Google Scholar]
- Bayburt TH, Vishnivetskiy SA, McLean M, Morizumi T, Huang C-C, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV. Rhodopsin monomer is sufficient for normal rhodopsin kinase (GRK1) phosphorylation and arrestin-1 binding. J Biol Chem. 2011;286:1420–1428. doi: 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baylor DA, Lamb TD, Yau KW. Responses of retinal rods to single photons. J Physiol. 1979;288:613–634. [PMC free article] [PubMed] [Google Scholar]
- Boularan C, Scott MG, Bourougaa K, Bellal M, Esteve E, Thuret A, Benmerah A, Tramier M, Coppey-Moisan M, Labbé-Jullié C, et al. beta-arrestin 2 oligomerization controls the Mdm2-dependent inhibition of p53. Proc Natl Acad Sci USA. 2007;104:18061–18066. doi: 10.1073/pnas.0705550104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Rubin WW, Mendez A, Liu X, Song X, Hanson SM, Craft CM, Gurevich VV, Burns ME, Chen J. Functional comparisons of visual arrestins in rod photoreceptors of transgenic mice. Invest Ophthalmol Vis Sci. 2007;48:1968–1975. doi: 10.1167/iovs.06-1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Zhuo Y, Francis DJ, Vishnivetskiy SA, Hanson SM, Zhan X, Brooks EK, Iverson TI, Altenbach C, Hubbell WL, et al. The two non-visual arrestins form distinct oligomers. J Biol Chem. 2013;288 in review. [Google Scholar]
- Coffa S, Breitman M, Spiller BW, Gurevich VV. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry. 2011;50:6951–6958. doi: 10.1021/bi200745k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinculescu A, McDowell JH, Amici SA, Dugger DR, Richards N, Hargrave PA, Smith WC. Insertional mutagenesis and immunochemical analysis of visual arrestin interaction with rhodopsin. J Biol Chem. 2002;277:11703–11708. doi: 10.1074/jbc.M111833200. [DOI] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Enhancing receptor specificity of non-visual arrestins by targeting receptor-discriminator residues. J Biol Chem. 2012a;287:29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimenez LE, Vishnivetskiy SA, Baameur F, Gurevich VV. Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J Biol Chem. 2012b;287:29495–29505. doi: 10.1074/jbc.M112.366674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- Granzin J, Wilden U, Choe HW, Labahn J, Krafft B, Buldt G. X-ray crystal structure of arrestin from bovine rod outer segments. Nature. 1998;391:918–921. doi: 10.1038/36147. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Moughon S, Wang C, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein–protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J Mol Biol. 2003a;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Moughon SE, Kortemme T, Schueler-Furman O, Misura KM, Morozov AV, Baker D. Protein-protein docking predictions for the CAPRI experiment. Proteins. 2003b;52:118–122. doi: 10.1002/prot.10384. [DOI] [PubMed] [Google Scholar]
- Gray-Keller MP, Detwiler PB, Benovic JL, Gurevich VV. Arrestin with a single amino acid sustitution quenches light-activated rhodopsin in a phosphorylation-independent fasion. Biochemistry. 1997;36:7058–7063. doi: 10.1021/bi963110k. [DOI] [PubMed] [Google Scholar]
- Gurevich VV. The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J Biol Chem. 1998;273:15501–15506. doi: 10.1074/jbc.273.25.15501. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J Biol Chem. 1992;267:21919–21923. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin: sequential multisite binding ensures strict selectivity towards light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Visual arrestin binding to rhodopsin: diverse functional roles of positively charged residues within the phosphorylation-recignition region of arrestin. J Biol Chem. 1995;270:6010–6016. doi: 10.1074/jbc.270.11.6010. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Benovic JL. Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol Pharmacol. 1997;51:161–169. doi: 10.1124/mol.51.1.161. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Gurevich VV. Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 2006;7:236. doi: 10.1186/gb-2006-7-9-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich VV, Richardson RM, Kim CM, Hosey MM, Benovic JL. Binding of wild type and chimeric arrestins to the m2 muscarinic cholinergic receptor. J Biol Chem. 1993;268:16879–16882. [PubMed] [Google Scholar]
- Gurevich VV, Dion SB, Onorato JJ, Ptasienski J, Kim CM, Sterne-Marr R, Hosey MM, Benovic JL. Arrestin interaction with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, b2-adrenergic, and m2 muscarinic cholinergic receptors. J Biol Chem. 1995;270:720–731. doi: 10.1074/jbc.270.2.720. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Pals-Rylaarsdam R, Benovic JL, Hosey MM, Onorato JJ. Agonist-receptor-arrestin, an alternative ternary complex with high agonist affinity. J Biol Chem. 1997;272:28849–28852. doi: 10.1074/jbc.272.46.28849. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience. 2002;109:421–436. doi: 10.1016/s0306-4522(01)00511-5. [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Benovic JL, Gurevich VV. Arrestin2 expression selectively increases during neural differentiation. J Neurochem. 2004;91:1404–1416. doi: 10.1111/j.1471-4159.2004.02830.x. [DOI] [PubMed] [Google Scholar]
- Gurevich VV, Hanson SM, Song X, Vishnivetskiy SA, Gurevich EV. The functional cycle of visual arrestins in photoreceptor cells. Prog Retin Eye Res. 2011;30:405–430. doi: 10.1016/j.preteyeres.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane translocation. Structure. 2001;9:869–880. doi: 10.1016/s0969-2126(01)00644-x. [DOI] [PubMed] [Google Scholar]
- Hanson SM, Gurevich VV. The differential engagement of arrestin surface charges by the various functional forms of the receptor. J Biol Chem. 2006;281:3458–3462. doi: 10.1074/jbc.M512148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Klug CS, Gurevich VV. Visual arrestin binding to microtubules involves a distinct conformational change. J Biol Chem. 2006a;281:9765–9772. doi: 10.1074/jbc.M510738200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Francis DJ, Vishnivetskiy SA, Kolobova EA, Hubbell WL, Klug CS, Gurevich VV. Differential interaction of spin-labeled arrestin with inactive and active phosphor-hodopsin. Proc Natl Acad Sci USA. 2006b;103:4900–4905. doi: 10.1073/pnas.0600733103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Cleghorn WM, Francis DJ, Vishnivetskiy SA, Raman D, Song S, Nair KS, Slepak VZ, Klug CS, Gurevich VV. Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J Mol Biol. 2007a;368:375–387. doi: 10.1016/j.jmb.2007.02.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Gurevich EV, Vishnivetskiy SA, Ahmed MR, Song X, Gurevich VV. Each rhodopsin molecule binds its own arrestin. Proc Natl Acad Sci USA. 2007b;104:3125–3128. doi: 10.1073/pnas.0610886104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Van Eps N, Francis DJ, Altenbach C, Vishnivetskiy SA, Arshavsky VY, Klug CS, Hubbell WL, Gurevich VV. Structure and function of the visual arrestin oligomer. EMBO J. 2007c;26:1726–1736. doi: 10.1038/sj.emboj.7601614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Dawson ES, Francis DJ, Van Eps N, Klug CS, Hubbell WL, Meiler J, Gurevich VV. A model for the solution structure of the rod arrestin tetramer. Structure. 2008a;16:924–934. doi: 10.1016/j.str.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Vishnivetskiy SA, Hubbell WL, Gurevich VV. Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry. 2008b;47:1070–1075. doi: 10.1021/bi7021359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Schubert C, Gurevich VV, Sigler PB. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Huang SP, Brown BM, Craft CM. Visual Arrestin 1 acts as a modulator for N-ethylmaleimide-sensitive factor in the photoreceptor synapse. J Neurosci. 2010;30:9381–9391. doi: 10.1523/JNEUROSCI.1207-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamoto Y, Tamura C, Kamikubo H, Kataoka M. Concentration-dependent tetramerization of bovine visual arrestin. Biophys J. 2003;85:1186–1195. doi: 10.1016/S0006-3495(03)74554-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeschke G. Distance measurements in the nanometer range by pulse EPR. Chemphyschem. 2002;3:927–932. doi: 10.1002/1439-7641(20021115)3:11<927::AID-CPHC927>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- Kim M, Hanson SM, Vishnivetskiy SA, Song X, Cleghorn WM, Hubbell WL, Gurevich VV. Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry. 2011;50:2235–2242. doi: 10.1021/bi1018607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Vishnivetskiy SA, Van Eps N, Alexander NS, Cleghorn WM, Zhan X, Hanson SM, Morizumi T, Ernst OP, Meiler J, et al. Conformation of receptor-bound visual arrestin. Proc Natl Acad Sci USA. 2012;109:18407–18412. doi: 10.1073/pnas.1216304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984;176:473–478. doi: 10.1016/0014-5793(84)81221-1. [DOI] [PubMed] [Google Scholar]
- Laporte SA, Oakley RH, Zhang J, Holt JA, Ferguson SG, Caron MG, Barak LS. The 2-adrenergic receptor/arrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc Natl Acad Sci USA. 1999;96:3712–3717. doi: 10.1073/pnas.96.7.3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makino CL, Wen XH, Lem J. Piecing together the timetable for visual transduction with transgenic animals. Curr Opin Neurobiol. 2003;13:404–412. doi: 10.1016/s0959-4388(03)00091-6. [DOI] [PubMed] [Google Scholar]
- Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- Milano SK, Kim YM, Stefano FP, Benovic JL, Brenner C. Nonvisual arrestin oligomerization and cellular localization are regulated by inositol hexakisphosphate binding. J Biol Chem. 2006;281:9812–9823. doi: 10.1074/jbc.M512703200. [DOI] [PubMed] [Google Scholar]
- Moaven H, Koike Y, Jao CC, Gurevich VV, Langen R, Chen J. Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc Natl Acad Sci USA. 2013;110:9463–9468. doi: 10.1073/pnas.1301126110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogridge J. Using light scattering to determine the stoichiometry of protein complexes. Methods Mol Biol. 2004;261:113–118. doi: 10.1385/1-59259-762-9:113. [DOI] [PubMed] [Google Scholar]
- Nair KS, Hanson SM, Mendez A, Gurevich EV, Kennedy MJ, Shestopalov VI, Vishnivetskiy SA, Chen J, Hurley JB, Gurevich VV, Slepak VZ. Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron. 2005;46:555–567. doi: 10.1016/j.neuron.2005.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EN, Jr, Craft CM. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59:462–474. doi: 10.1016/j.neuron.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohguro H, Palczewski K, Walsh KA, Johnson RS. Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein Sci. 1994;3:2428–2434. doi: 10.1002/pro.5560031226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannier M, Veit S, Godt A, Jeschke G, Spiess HW. Dead-time free measurement of dipole-dipole interactions between electron spins. J Magn Reson. 2000;142:331–340. doi: 10.1006/jmre.1999.1944. [DOI] [PubMed] [Google Scholar]
- Peet JA, Bragin A, Calvert PD, Nikonov SS, Mani S, Zhao X, Besharse JC, Pierce EA, Knox BE, Pugh ENJ. Quantification of the cytoplasmic spaces of living cells with EGFP reveals arrestin-EGFP to be in disequilibrium in dark adapted rod photoreceptors. J Cell Sci. 2004;117:3049–3059. doi: 10.1242/jcs.01167. [DOI] [PubMed] [Google Scholar]
- Pugh EN, Jr, Lamb TD. Phototransduction in vertebrate rods and cones: molecular mechanisms of amplification, recovery and light adaptation. In: Stavenga DG, DeGrip WJ, Pugh EN Jr, editors. Handbook of biological physics molecular mechanisms in visual transduction. Elsevier; Amsterdam: 2000. pp. 183–255. [Google Scholar]
- Pulvermuller A, Schroder K, Fischer T, Hofmann KP. Interactions of metarhodopsin II. Arrestin peptides compete with arrestin and transducin. J Biol Chem. 2000;275:37679–37685. doi: 10.1074/jbc.M006776200. [DOI] [PubMed] [Google Scholar]
- Schubert C, Hirsch JA, Gurevich VV, Engelman DM, Sigler PB, Fleming KG. Visual arrestin activity may be regulated by self-association. J Biol Chem. 1999;274:21186–21190. doi: 10.1074/jbc.274.30.21186. [DOI] [PubMed] [Google Scholar]
- Schueler-Furman O, Wang C, Bradley P, Misura K, Baker D. Progress in modeling of protein structures and interactions. Science. 2005;310:638–642. doi: 10.1126/science.1112160. [DOI] [PubMed] [Google Scholar]
- Scott MG, Le Rouzic E, Perianin A, Pierotti V, Enslen H, Benichou S, Marullo S, Benmerah A. Differential nucleocytoplasmic shuttling of beta-arrestins. Characterization of a leucine-rich nuclear export signal in beta-arrestin2. J Biol Chem. 2002;277:37693–37701. doi: 10.1074/jbc.M207552200. [DOI] [PubMed] [Google Scholar]
- Shears SB. Assessing the omnipotence of inositol hexakisphosphate. Cell Signal. 2001;13:151–158. doi: 10.1016/s0898-6568(01)00129-2. [DOI] [PubMed] [Google Scholar]
- Shilton BH, McDowell JH, Smith WC, Hargrave PA. The solution structure and activation of visual arrestin studied by small-angle X-ray scattering. Eur J Biochem. 2002;269:3801–3809. doi: 10.1046/j.1432-1033.2002.03071.x. [DOI] [PubMed] [Google Scholar]
- Singhal A, Ostermaier MK, Vishnivetskiy SA, Panneels V, Homan KT, Tesmer JJ, Veprintsev D, Deupi X, Gurevich VV, Schertler GF, Standfuss J. Insights into congenital night blindness based on the structure of G90D rhodopsin. EMBO Rep. 2013;14:520–526. doi: 10.1038/embor.2013.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer ME, Hofmann KP, Heck M. Arrestin-rhodopsin binding stoichiometry in isolated rod outer segment membranes depends on the percentage of activated receptors. J Biol Chem. 2011;286:7359–7369. doi: 10.1074/jbc.M110.204941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer ME, Hofmann KP, Heck M. Distinct loops in arrestin differentially regulate ligand binding within the GPCR opsin. Nat Commun. 2012;3:995. doi: 10.1038/ncomms2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Raman D, Gurevich EV, Vishnivetskiy SA, Gurevich VV. Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J Biol Chem. 2006;281:21491–21499. doi: 10.1074/jbc.M603659200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Gurevich EV, Gurevich VV. Cone arrestin binding to JNK3 and Mdm2: conformational preference and localization of interaction sites. J Neurochem. 2007;103:1053–1062. doi: 10.1111/j.1471-4159.2007.04842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Gross OP, Emelianoff K, Mendez A, Chen J, Gurevich EV, Burns ME, Gurevich VV. Enhanced arrestin facilitates recovery and protects rod photoreceptors deficient in rhodopsin phosphorylation. Curr Biol. 2009;19:700–705. doi: 10.1016/j.cub.2009.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Vishnivetskiy SA, Seo J, Chen J, Gurevich EV, Gurevich VV. Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience. 2011;174:37–49. doi: 10.1016/j.neuroscience.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X, Seo J, Baameur F, Vishnivetskiy SA, Chen Q, Kook S, Kim M, Brooks EK, Altenbach C, Hong Y, et al. Rapid degeneration of rod photoreceptors expressing self-association-deficient arrestin-1 mutant. Cell Signal. 2013;25:2613–2624. doi: 10.1016/j.cellsig.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storez H, Scott MG, Issafras H, Burtey A, Benmerah A, Muntaner O, Piolot T, Tramier M, Coppey-Moisan M, Bouvier M, et al. Homo- and hetero-oligomerization of beta-arrestins in living cells. J Biol Chem. 2005;280:40210–40215. doi: 10.1074/jbc.M508001200. [DOI] [PubMed] [Google Scholar]
- Strissel KJ, Sokolov M, Trieu LH, Arshavsky VY. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Vishnivetskiy SA, Robert J, Hanson SM, Raman D, Knox BE, Kono M, Navarro J, Gurevich VV. Crystal structure of cone arrestin at 2.3Å: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- Tan E, Wang Q, Quiambao AB, Xu X, Qtaishat NM, Peachey NS, Lem J, Fliesler SJ, Pepperberg DR, Naash MI, Al-Ubaidi MR. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42:589–600. [PubMed] [Google Scholar]
- Vishnivetskiy SA, Hosey MM, Benovic JL, Gurevich VV. Mapping the arrestin-receptor interface: structural elements responsible for receptor specificity of arrestin proteins. J Biol Chem. 2004;279:1262–1268. doi: 10.1074/jbc.M308834200. [DOI] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Gimenez LE, Francis DJ, Hanson SM, Hubbell WL, Klug CS, Gurevich VV. Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J Biol Chem. 2011;286:24288–24299. doi: 10.1074/jbc.M110.213835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Chen Q, Palazzo MC, Brooks EK, Altenbach C, Iverson TM, Hubbell WL, Gurevich VV. Engineering visual arrestin-1 with special functional characteristics. J Biol Chem. 2013a;288:3394–3405. doi: 10.1074/jbc.M112.445437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vishnivetskiy SA, Ostermaierm MK, Singhal A, Panneels V, Homan KT, Glukhova A, Sligar SG, Tesmer JJ, Schertler GF, Standfuss J, Gurevich VV. Constitutively active rhodopsin mutants causing night blindness are effectively phosphorylated by GRKs but differ in arrestin-1 binding. Cell Signal. 2013b;25:2155–2162. doi: 10.1016/j.cellsig.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker WB, Donoso LA, Kalsow CM, Yankeelov JA, Jr, Organisciak DT. Experimental allergic uveitis. Isolation, characterization, and localization of a soluble uveitopathogenic antigen from bovine retina. J Immunol. 1977;119:1949–1958. [PubMed] [Google Scholar]
- Wang P, Wu Y, Ge X, Ma L, Pei G. Subcellular localization of beta-arrestins is determined by their intact N domain and the nuclear export signal at the C terminus. J Biol Chem. 2003;278:11648–11653. doi: 10.1074/jbc.M208109200. [DOI] [PubMed] [Google Scholar]
- Wang C, Schueler-Furman O, Baker D. Improved side-chain modeling for protein-protein docking. Protein Sci. 2005;14:1328–1339. doi: 10.1110/ps.041222905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U, Hall SW, Kühn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986a;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilden U, Wüst E, Weyand I, Kühn H. Rapid affinity purification of retinal arrestin (48 kDa protein) via its light-dependent binding to phosphorylated rhodopsin. FEBS Lett. 1986b;207:292–295. doi: 10.1016/0014-5793(86)81507-1. [DOI] [PubMed] [Google Scholar]
- Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual arrestins. J Mol Biol. 2011;406:467–478. doi: 10.1016/j.jmb.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Vishnivetskiy SA, Gurevich VV, Sanders CR. Elucidation of IP6 and heparin interaction sites and conformational changes in arrestin-1 by solution NMR. Biochemistry. 2010:10473–10485. doi: 10.1021/bi101596g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang T, Chen Q, Cho M-K, Vishnivetskiy SA, Iverson TI, Gurevich VV, Hubbell WL. Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc Natl Acad Sci USA. 2013;110:942–947. doi: 10.1073/pnas.1215176110. [DOI] [PMC free article] [PubMed] [Google Scholar]