

Abstract
The mammalian target of rapamycin (mTOR), a phosphoinositide 3-kinase related protein kinase, controls cell growth in response to nutrients and growth factors and is frequently deregulated in cancer. Here we report co-crystal structures of a truncated mTOR-mLST8 complex with an ATP transition state mimic and with ATP-site inhibitors. The structures reveal an intrinsically active kinase conformation, with catalytic residues and mechanism remarkably similar to canonical protein kinases. The active site is highly recessed due to the FKBP12-Rapamycin binding (FRB) domain and an inhibitory helix protruding from the catalytic cleft. mTOR activating mutations map to the structural framework that holds these elements in place, indicating the kinase is controlled by restricted access. In vitro biochemistry indicates that the FRB domain acts as a gatekeeper, with its rapamycin-binding site interacting with substrates to grant them access to the restricted active site. FKBP12-rapamycin inhibits by directly blocking substrate recruitment and by further restricting active site access. The structures also reveal active site residues and conformational changes that underlie inhibitor potency and specificity.
The mTOR pathway controls cell growth in response to energy, nutrients, growth factors and other environmental cues, and it figures prominently in cancer1,2. Central to the pathway is the mammalian target of rapamycin (mTOR) protein that belongs to the phosphoinositide 3-kinase (PI3K)-related protein kinase (PIKK) family3. mTOR assembles into two complexes with distinct inputs and downstream effects. mTOR Complex 1 (mTORC1) is defined by its RAPTOR subunit4-6, which is replaced by RICTOR in mTORC26,7. Both complexes also contain the requisite mLST8 subunit8,9, but they differ in a number of other subunits that interact with RAPTOR or RICTOR1.
mTORC1 regulates cell growth by promoting translation, ribosome biogenesis and autophagy1,4,5. Its activation requires nutrients and amino acids, which result in the RAPTOR-mediated recruitment of mTORC1 to lysosomes and late endosomes10,11, and co-localization with its activator, the small GTPase RHEB12,13. Proposed mechanisms of RHEB action include binding to and activating the mTOR kinase domain14, and the displacement of the mTORC1 inhibitor PRAS40 from RAPTOR15,16. RHEB in turn is negatively-regulated by the GTPase-activating domain of the TSC2 tumor suppressor, which relays signals from multiple growth factor and stress pathways17. mTORC1 substrates include the eIF4E-binding protein 1 (4EBP1) and ribosomal S6 kinases (S6K), which control cap-dependent translation initiation and elongation, respectively18. Phosphorylation of 4EBP1 and S6K1 is dependent on their Tor Signaling Sequence (TOS) motif19,20 that binds to RAPTOR, and which is also present in the negative regulator PRAS4021,22.
mTORC2 responds primarily to growth factors, promoting cell-cycle entry, cell survival, actin cytoskeleton polarization, and anabolic output6,7,23. Its substrates include the Ser/Thr protein kinases AKT, SGK and PKC, which share the hydrophobic motif (HM) phosphorylation site with S6K11,2.
Rapamycin, which forms a ternary complex with the FK506-binding protein12 (FKBP12) and the FRB (FKBP12-Rapamycin-Binding) domain of mTOR, is thought to be an allosteric inhibitor24,25. Rapamycin-FKBP12 inhibits mTORC1 to a variable extent that is substrate and phosphorylation-site dependent25, and it does not bind to mTORC223. To overcome these limitations, ATP-competitive inhibitors that potently and uniformly inhibit both mTORC1 and mTORC2 are being developed as anti-cancer agents26.
The six mammalian PIKKs regulate diverse cellular processes27. They share three regions of homology consisting of a ~600 residue FAT domain (FRAP, ATM, TRRAP), a ~300 residue PI3K-related protein kinase catalytic domain, and a ~35 residue FATC domain at the C-terminus28. In mTOR, the ~100 residue FRB domain is thought to occur in-between the FAT and catalytic domains, and the region N-terminal to the FAT domain is required for binding to RAPTOR and RICTOR1.
Here we present the 3.2 Å crystal structure of a ~1500 amino acid mTOR-mLST8 complex containing the FAT, FRB, kinase and FATC domains, as well as the structures of this complex bound to an ATP transition state analog and to ATP-competitive inhibitors. We discuss their implications for understanding mTOR function, regulation and inhibition by rapamycin and ATP-competitive compounds.
Overall structure of mTORΔN–mLST8
Crystals were grown using an N-terminally truncated human mTOR (residues 1376 to 2549; thereafter mTORΔN) bound to full-length human mLST8 (Supplementary Table 1). The complex was produced in an HEK293-F cell line that was stably-transfected sequentially by FLAG-tagged mLST8 and FLAG-tagged mTORΔN vectors. The kinase activity of mTORΔN-mLST8 is overall comparable to that of mTORC1 (Supplementary Fig. 1). mTORC1 is more active towards low-micromolar concentrations of S6K1ki (kinase-inactive mutant) and 4EBP1, consistent with RAPTOR recruiting these substrates through their TOS motifs, whereas mTORΔN-mLST8 is more active at higher substrate concentrations.
The mTORΔN-mLST8 structure has a compact shape (Fig. 1). The FAT domain, which consists of α–α helical repeats, forms a “C” shaped α solenoid that wraps halfway around the kinase domain and clamps onto it. mLST8 and the FRB domain protrude from the kinase domain, on opposite sides of the catalytic cleft. The FATC is integral to the kinase domain structure.
Figure 1. Structure of the mTORΔN-mLST8-ATPγS-Mg complex.
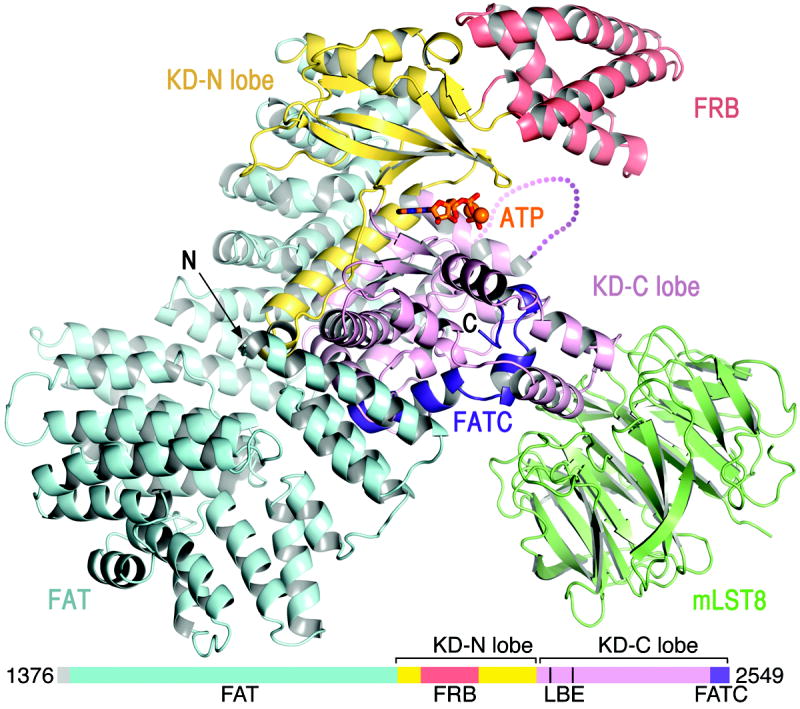
mTOR is coloured as indicated in the linear schematic, mLST8 is coloured green, ATP is shown as sticks, and Mg2+ ions as spheres.
The ~550-residue mTOR kinase domain (KD) adopts the two-lobe structure that is characteristic of both the PI3K and canonical protein kinase families29. It consists of an N-terminal lobe (N lobe), a larger C-terminal lobe (C lobe), and a cleft in-between the two that binds to ATP. The mTOR KD contains the core PI3K KD fold, but with substantial differences. While PI3K KD structures superimpose essentially across their entire ~350 residues, only ~250 residues of the mTOR KD superimpose on PI3Ks (Supplementary Table 2). In addition, the mTOR KD contains ~200 additional residues of insertions that decorate the common fold. The largest of these is the FRB domain (residues 2021 to 2118) that is inserted within the kinase N lobe (Fig. 1). There is also a ~40-residue insertion in the C lobe that forms the binding site for mLST8 (residues 2258 to 2296; thereafter LBE).
Kinase domain structure
The mTOR KD structure starts prior to the FRB domain, with the long kα1 helix that is present in PI3Ks as well (Fig. 2a and Supplementary Fig. 2a). The kα1 helix is integral to the structure of the N lobe, as it packs in the concave surface of the N lobe β sheet in both mTOR and PI3Ks29. The FRB insertion occurs immediately after kα1. It is followed by a β strand and two short helices that pack with the base of the FRB, and which replace the kα2 helix of the PI3K KD fold. Thereafter, the mTOR and PI3K N lobes share a similar structure.
Figure 2. mTOR kinase domain and active site conformation.
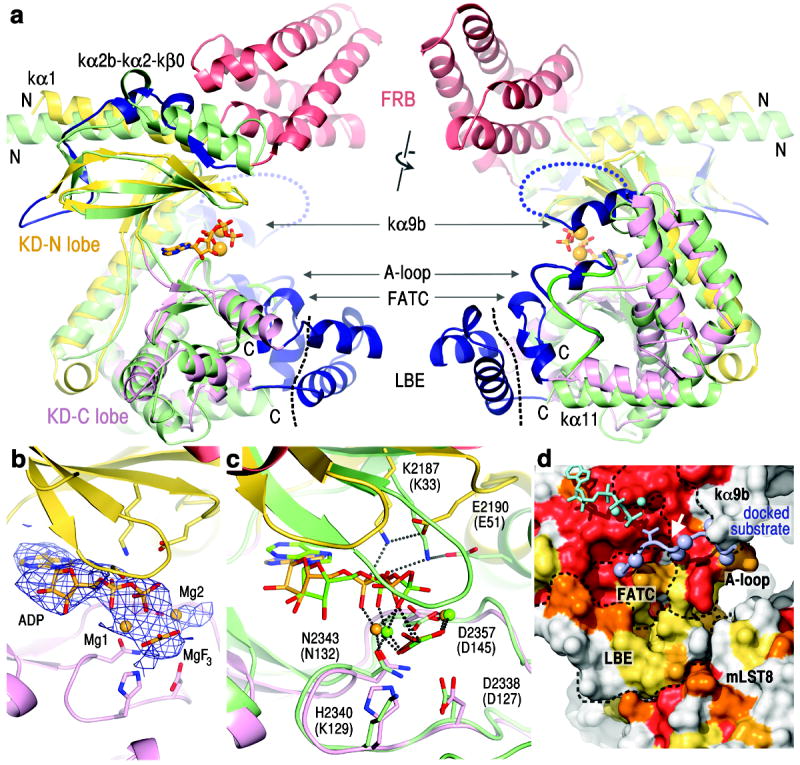
a, Superposition of the kinase domains of mTOR and PIK3C3 (PDB 3IHY; green) in two views rotated by ~180°. Left view is related to that of Figure 1. The FRB is colored red, and the remaining mTOR insertions dark blue. Black dashed line delineates the LBE, and the blue dotted loop indicates the disordered region between kα9b and kα10.
b, The 3.5 Å Fo-Fc electron density, contoured at 2.5 σ, of the mTOR TS complex before ADP-MgF3-Mg2 was built.
c, Superposition of the mTOR and CDK2 TS (PDB 3QHW) complexes. CDK2 and its nucleotide are coloured green (its residue labels in parentheses).
d, Molecular surface representation of the C lobe portion of the mTOR catalytic cleft, coloured according to conservation entropy (red invariant in 22 orthologs, orange in 21, yellow-orange in 20, and yellow in 18-19). Dashed lines delineate the boundaries of structural elements (labelled). The docked substrate peptide is coloured light blue, with its threonine phosphorylation site shown as sticks, the remaining side chains as spheres (Cβ atoms), and its +1 position indicated by a white arrow.
The mTOR structure indicates that the kα1 helix is present in all PIKKs (Supplementary Fig. 3a). The SMG1, DNAPKcs and TRRAP PIKKs also appear to have an FRB-like domain, as they contain insertions of 128, 95 and 128 residues, respectively, that are predicted to be alpha-helical. This is supported by the 6.6 Å x-ray diffraction data of DNAPKcs reported recently30. Although the deposited DNAPKcs model, which contains a PI3K-derived KD, lacks the FRB insertion, a reinterpretation of the data using the mTOR structure reveals electron density indicative of an FRB-like four-helix bundle (Supplementary Fig. 3b). ATM and ATR appear to lack a comparable insertion.
In the C lobe, the vicinity of the catalytic cleft contains four structural insertions compared to PI3Ks (LBE, kαAL, kα9b and FATC; Fig. 2a). These form a spine of interactions centered on the activation loop, which is a ~30 residue segment with a central role in the function and regulation of canonical protein kinases. It forms part of the polypeptide binding site, provides an active site residue, and also undergoes a conformational change, typically disorder-to-order transition, upon kinase activation31. The activation loop is thought to have an analogous role in PI3Ks, among which only in the class 3 PIK3C3/Vps34 structures it is ordered32. In the mTOR structure the entire activation loop is well ordered (Supplementary Fig. 2b), and its middle portion contains the kαAL helix insertion. One side of the activation loop packs with the kα9b insertion, and the other side with the FATC (Fig. 2a). The FATC is not unique to PIKKs in its entirety, as its N-terminal half forms a helix (kα11) that is also present in PI3K structures29,32. Its C-terminal half, which is absent from PI3Ks, forms three short helices that pack with the activation loop on one side and the LBE on the other (Fig. 2a).
The interactions the FATC makes with the activation loop suggest that it may have a role in stabilizing the activation loop structure, and the LBE, through its interactions with the FATC, may contribute to this indirectly (Supplementary Fig. 4a). The FATC and activation loop sequences are highly conserved among PIKKs. While the LBE is not conserved, all PIKK family members contain an LBE-like insertion that may similarly pack with FATC (Supplementary Fig. 3a).
The kα9b insertion (residues 2425 to 2436) plugs one end of the catalytic cleft (Fig. 2a). It partially overlaps with a segment, termed negative regulatory domain (residues 2430 to 2450), whose deletion activates mTOR in vitro33 and in vivo34,35. After kα9b, there is a 55 residue unstructured segment (2437 to 2491) that is not conserved and has variable length in mTOR orthologs (Supplementary Fig. 2a).
Active site conformation
To assess the activation state of the KD structure and investigate the mechanism of phosphotransfer, we co-crystallized mTORΔN-mLST8 with ADP, Mg2+ and MgF3−, a mimic of the γ-phosphate group of ATP in the transition state (TS)36 (Supplementary Table 1). The 3.5 Å Fo-Fc map of these crystals shows strong electron density extending from the β-phosphate group of ADP, consistent with a bound MgF3− group (Fig. 2b). The presence of two additional Mg2+ ions is supported by the anomalous dispersion maps of apo-crystals soaked in AMPPNP-Mn2+ (Supplementary Fig. 4b).
A superposition with the protein kinase CDK2 bound to the same ATP TS mimic36 reveals that the arrangement of key active site residues and MgF3− is remarkably conserved in mTOR (Fig. 2c and Supplementary Fig. 4c). These include the C lobe Asn2343 and Asp2357, which in both mTOR and protein kinases serve as metal ligands, and Asp2338, which in protein kinases plays a key catalytic role in orienting and activating the substrate hydroxyl group for nucleophilic attack36,37. The mTOR-CDK2 superposition also reveals a coincidence of the mTOR His2340 and CDK2 Lys129 side chains (Fig. 2c). In protein kinases, a basic residue at this position interacts with both the substrate hydroxyl group and the γ-phosphate TS mimic, and is postulated to stabilize the buildup of charge at the transition state36,37. Consistent with a similar role, we find that the kinase activity of the H2340A mTORΔN-mLST8 mutant is barely detectable, and comparable to the D2338A mutant previously shown to be inactive38 (Supplementary Fig. 4d).
These findings strongly suggest that the crystallized mTORΔN-mLST8 complex is intrinsically active in the absence of any additional regulatory subunits. They also indicate that PIKKs, and most likely PI3Ks, employ the same catalytic mechanism as canonical protein kinases, in contrast to a recently proposed PI3K mechanism32 (see legend of Supplementary Fig. 4c).
Substrate-binding site
The MgF3− γ-phosphate mimic points to an extended C lobe groove that is highly conserved and is the likely site of substrate binding. Figure 2d illustrates this with a CDK2-bound substrate peptide docked by superposing the mTOR and CDK2 TS36 complexes. The groove consists of the activation loop, as in protein kinases, but also of portions of the FATC and LBE.
In the (+) direction (C-terminal to phosphorylation site), the groove extends only to the +1 position, abruptly terminating with the kα9b helix that plugs this end of the cleft (Fig. 2d). Thereafter, the peptide can exit along the C lobe towards mLST8, or along the N lobe towards the FRB. The +1 portion of the groove has a pronounced pocket lined with three aromatic residues, consistent with the two classes of mTOR substrates, exemplified by 4EBP1 and S6K1, having a proline and tyrosine residue, respectively, at +1 (Supplementary Figs 5a and 5b). The groove extends substantially more in the (-) direction, and it may account for the low-level sequence preference at -4 and -5 suggested by a positional scanning peptide array39.
Restricted access to the active site
The four-helix FRB substantially extends the N lobe side of the catalytic cleft, while the LBE and mLST8 extend the C lobe side. This gives the cleft a deep, “V”-shaped cross-section, restricting access to the substrate-binding site at the bottom of the cleft (Fig. 3a). Substrate access is further hindered by one end of the cleft being plugged by kα9b and the following 55-residue unstructured segment. Portions of kα9b and the unstructured segment form the negative regulatory region33-35, suggesting that restricted access to the active site negatively regulates mTOR. Kα9b is likely the key element because deleting the bulk of the unstructured segment (residues 2443-2486) does not activate mTOR (Supplementary Fig. 6).
Figure 3. The kinase active site is recessed at the bottom of a deep cleft.
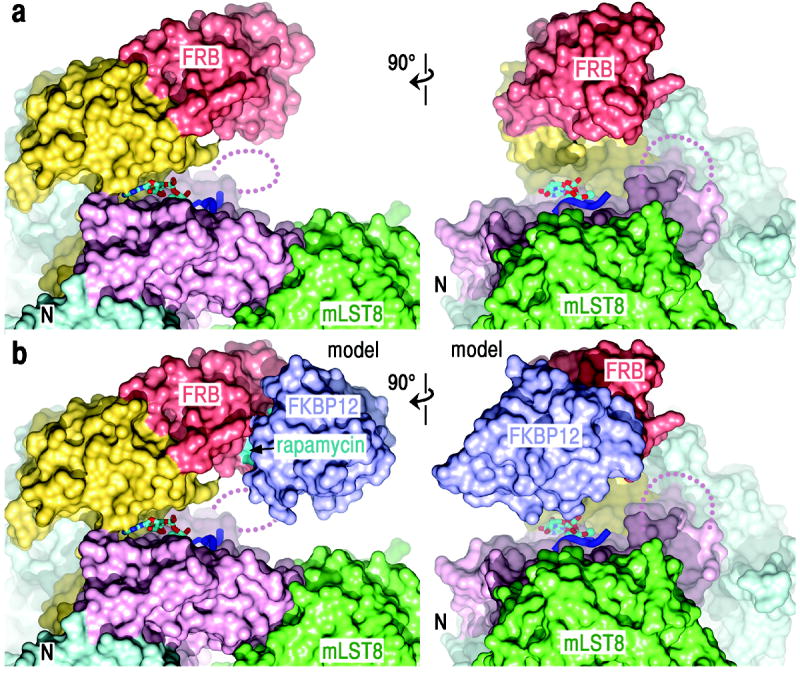
a, Surface representation of mTORΔN-mLST8-ADP-MgF3-Mg2 in two orthogonal views. Dark blue ribbon indicates the docked substrate peptide (positions -2 to +2) from Figure 2d.
b, Surface representation of the mTORΔN-mLST8-rapamycin-FKBP12 model. Rapamycin (cyan) and FKBP12 (blue) are labelled.
The end of the cleft opposite kα9b is unencumbered in the mTORΔN-mLST8 structure. However, it is near the FAT N-terminus, and the region deleted from the crystallized mTORΔN may restrict cleft access directly and/or through its associated RAPTOR or RICTOR proteins. In support, we find that towards TOS-mutant 4EBP1 and S6K1ki substrates, mTORΔN-mLST8 is more active than mTOR-mLST8, which in turn is more active than mTOR-mLST8-RAPTOR (Supplementary Fig. 6). A restricted active site would reduce the probability of substrates hitting the active site by random diffusion, and could thus be important in allowing for the regulation of phosphorylation by substrate-recruitment.
Inhibition by rapamycin-FKBP12
In a model constructed by superposing the FRB domains of the FRB-rapamycin-FKBP1224 and mTORΔN-mLST8 structures, FKBP12 extends from the FRB towards mLST8 on the C lobe, nearly capping the catalytic cleft (Fig. 3b). At their closest approach, FKBP12 and mLST8 are only 8 Å apart, with the catalytic center recessed by 37 Å. The model suggests that rapamycin-FKBP12 inhibits in part by drastically reducing the accessibility of the already constricted catalytic cleft. This is consistent with the extent of mTORC1 inhibition being substrate and phosphorylation site dependent25 (additional discussion in Supplementary Fig. 6 legend).
FRB recruits S6K1 to active site
The rapamycin-binding site maps to the FRB surface closest to the active site, in the midst of a hotspot of highly conserved residues (Fig. 4a). Four of these residues are invariant in 22 mTOR orthologs from yeast to man, and another four in 21 orthologs, in stark contrast to the FRB surface opposite from the active site (Fig. 4a and Supplementary Fig. 7). This raises the possibility that the rapamycin-binding site interacts with substrates to facilitate their entry to the recessed active site.
Figure 4. The rapamycin-binding site of the FRB recruits S6K1 into the catalytic cleft.
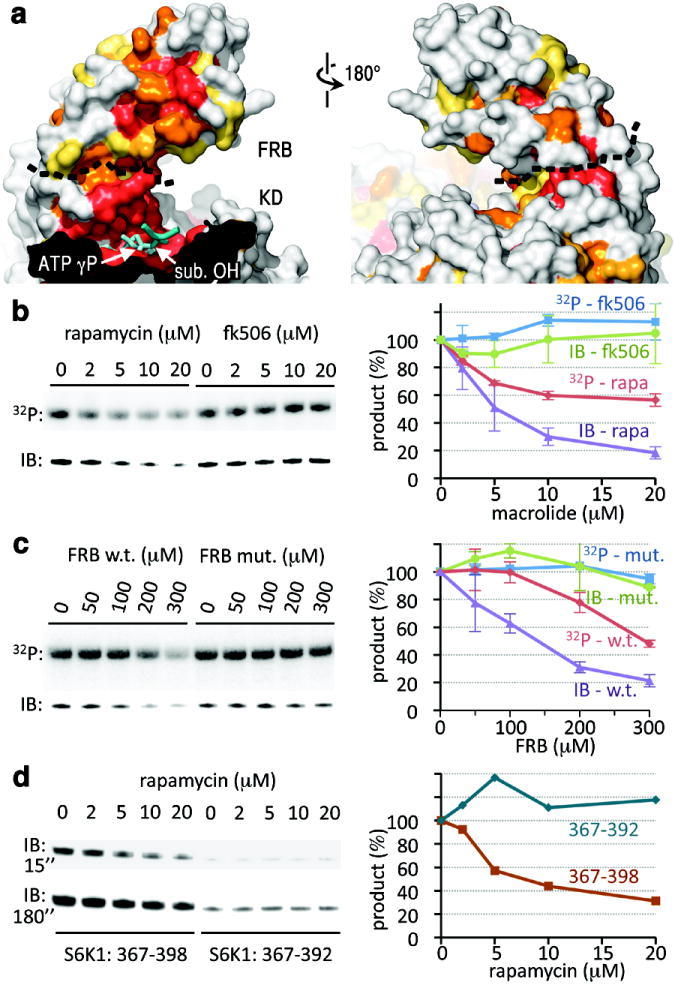
a, Surface representations of the FRB and portion of the catalytic cleft, coloured by sequence conservation as in Figure 2d. ADP-MgF3-Mg2 is in cyan with the TS mimic MgF3 group labelled “ATP γP”. The black dashed line delineates the FRB-KD boundary. The docked substrate peptide from Figure 2d is shown in blue (positions -1 to +1), with its phosphorylation site labelled “sub. OH”.
b, Phosphorylation of S6K1ki (10 μM) by mTORΔN-mLST8 (20 nM), measured by 32P incorporation (top panel) and by immunoblotting with a phosphoThr389-specific antibody (lower panel), in the presence of the indicated concentrations of rapamycin or FK506. The average and standard deviation of three independent repetitions is plotted as a percentage of the 0 μM macrolide reaction of each set.
c, Phosphorylation of S6K1ki (10 μM) by mTORΔN-mLST8 (20 nM) in the presence of the indicated concentrations of the wild type or S2035I mutant FRB. Reactions were repeated twice, and results plotted as in (b).
d, Phosphorylation of GST-tagged S6K1367-398 (10 μM) and S6K1367-392 (10 μM) by mTORΔN-mLST8 (20 nM) in the presence of the indicated concentrations of rapamycin. Two different exposures (15 and 180 sec.) of the phosphoThr389-specific immunoblot are shown. The quantitation of the 15 sec. immunoblot is plotted on the right.
This model is supported by the observation that mutation of Ser2035, a rapamycin contact38,40 at the centre of the conservation hotspot, reduces phosphorylation of S6K1 and 4EBP133. It can also explain inhibition of mTORC1 and mTORC2 by rapamycin in the absence of FKBP12, albeit at ~100-fold higher concentrations reflecting the weaker rapamycin-FRB binary interaction41. However, these observations can also be explained by the rapamycin-binding site interacting with other parts of mTOR, or with associated proteins, especially since rapamycin was thought to be an allosteric inhibitor25. To rule this out, we assayed mTORΔN-mLST8, and found that rapamycin but not the closely-related FK506 macrolide inhibits S6K1ki overall phosphorylation, as measured by 32P incorporation (Fig. 4b). Inhibition reaches ~45 % at the 20 μM solubility limit of rapamycin, close to the free rapamycin-FRB Kd. Phosphorylation of the key Thr389 site of the S6K1 HM motif, measured by a phosphoT389-specific antibody, is inhibited by ~80 % (Fig. 4b).
In a complementary approach, we tested whether the isolated FRB domain inhibits phosphorylation in trans. Figure 4c shows that wild type but not the S2035I FRB mutant38,40 inhibits overall S6K1ki phosphorylation by ~50 % and Thr389 phosphorylation by ~75 % at the highest concentration tested.
To map the region of S6K1 involved in FRB interactions, we reasoned that its deletion should reduce Thr389 phosphorylation and also render any residual phosphorylation insensitive to rapamycin. By testing successively truncated polypeptides from the S6K1 tail, we found that both criteria are met by residues 393 to 398 (Fig. 4d). In addition, residues 399 to 402 and 403 to 410 likely contribute to FRB interactions, as their truncation progressively reduces phosphorylation (Supplementary Fig. 8). N-terminal truncations up to residue 381 have no significant effect.
Together, these data indicate that the FRB provides a secondary substrate-recruitment site near the entrance of the catalytic cleft. We presume that while the TOS and possibly other motifs are the primary means of substrate recruitment, the secondary site may facilitate substrate entry into the otherwise restricted active site, and it may also provide an additional level of specificity, at least for a subset of substrates.
A bi-partite substrate-recruitment mechanism is analogous to what has been proposed for the calcineurin phosphatase, the target of FK506-FKBP12. Most calcineurin substrates require both a primary and a secondary recruitment motif for efficient dephosphorylation, and FK506-FKBP12 inhibits by binding to the secondary recruitment motif site42.
FAT domain
The FAT domain contains 28 α helices arranged as α–α helical repeats. Helices α1 to α22 belong to the TPR repeat family and form three discontinuous domains (TRD1, TRD2 and TRD3). Helices α23 to α28 belong to the HEAT family and form a single domain (HRD). The four domains pack sequentially to form a C-shaped α solenoid that clamps onto the KD (Supplementary Figs 9a and 9b). TRD1 interacts with the C lobe on one side of the KD, and after TRD2-TRD3 traverse to the other side, the HRD interacts with both the N and C lobes of the KD (Supplementary Figs 9c and 9d).
Both the TRD1-KD and HRD-KD contacts involve conserved residues, consistent with the two interfaces being important for the structure and function of mTOR. For example, a set of buried hydrogen bond contacts at the TRD1-KD interface (Glu1401-Arg2317) and at the HRD-KD interface (Arg1905-Glu2419 and Gln1941- Gln2200) involves residues invariant in 22 mTOR orthologs (Supplementary Figs 9c and 9d).
The TRD1 and HRD segments correspond to the FAT segments best conserved among PIKK family members28 as well, suggesting that the FAT domain clamping onto the KD is a common feature of this family. In addition, PI3Ks contain a HEAT-repeat domain analogous to the HRD in its structure and interactions with the KD, although they lack the rest of the FAT domain (Supplementary Fig. 9e).
mLST8
mLST8 consists of seven WD40 repeats. At the narrow end of the mLST8 β propeller structure, a surface that extends across six WD40 repeats binds to both helices and intervening loop of the LBE helix-loop-helix structure. The interface involves mostly polar LBE residues and polar/aromatic mLST8 residues, and it is dominated by hydrogen bonds (Supplementary Fig. 10).
mLST8 is thought to be a requisite activating subunit of mTOR complexes8,9. The structure suggests the extended interaction surface of mLST8 may directly stabilize the LBE structure, and indirectly influence the organization of the active site through the LBE-FATC-catalytic loop spine of interactions (Supplementary Fig. 4a). In support, we note that lack of mLST8 results in the association of mTOR with heat shock proteins9, and we find that the solubility of overexpressed mTOR is highly dependent on mLST8 co-expression (not shown). In addition, the growth-suppression phenotype of a temperature-sensitive yeast Lst8 allele can be rescued by mutations43 either at the LBE hydrophobic core (A2290V) or at the following kα5 helix (L2302Q), and the structure is consistent with these mutations stabilizing the local LBE structure.
Control of mTOR kinase activity
The model that active site restriction is a negative regulatory mechanism is supported by hyper-activating mutations43-45, most of which map to structural elements involved in limiting active site access. A large number of mutations cluster at the end of the catalytic cleft that is plugged by kα9b (Fig. 5 and Supplementary Fig. 11). They map to the kα3, kα9, kα9b and kα10 helices that pack extensively with each other, and to the FAT HRD portion that packs with kα3 and kα9. These mutations are likely to loosen the kα9b-centered structural framework that restricts access from this end of the cleft, presumably without destabilizing the entire active site structure. The role of the FAT clamp is highlighted by the widely studied E2419K activating mutation, which eliminates the Glu2419-Arg1905 salt bridge at the kα9-HRD interface (Supplementary Figs 9c and 11).
Figure 5. mTOR activating mutations map to structural elements involved in restricting active site access.
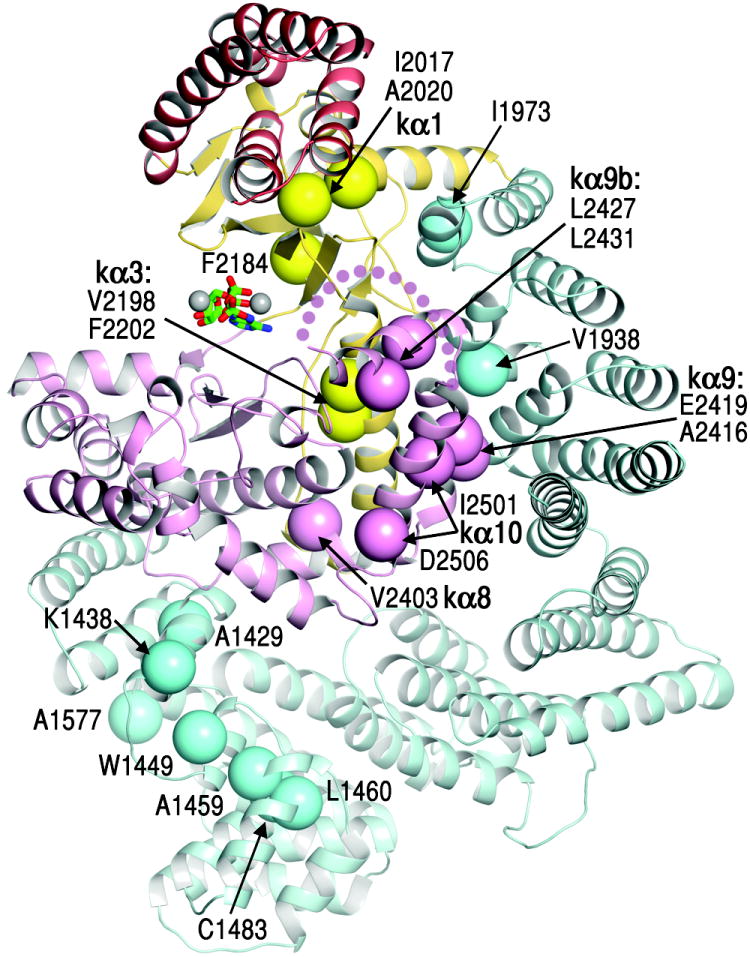
Activating mutations43-45 reported for mTOR, yeast Tor2 and Tor1p are shown as large spheres (labelled, with structural elements in bold), coloured as in Figure 1.
Two other hyper-activating mutations map to a portion of the kα1 helix that is sandwiched between the FRB base and the N lobe β sheet43,44. These mutations (I2017V and A2020V) are unlikely to unfold the FRB, because an FRB hydrophobic core mutation (W2027F) abolishes kinase activity40. The structure suggests that these mutations may loosen the rigid coupling of the FRB the catalytic cleft, increasing access to the active site.
A third cluster of mutations43 maps to the N-terminal portion of the FAT domain, to structural residues of TRD1 and of the TRD1-proximal portion of TRD2. These mutations would likely destabilize TRD1-KD interactions through loss of structural integrity. They may also destabilize the HRD-KD interactions, since the structural coupling of TRD1 and HRD would thermodynamically couple their KD contacts. In addition, the disposition of the N-terminal mTOR segment and its associated RAPTOR could change or become more flexible relative to the KD.
Inhibitors of the ATP-binding site
To explore the determinants of inhibitor potency and specificity, we determined the structures of mTORΔN-mLST8 bound to Torin2 and PP242, which are highly specific for mTOR, and to PI-103, which has dual specificity for mTOR and Class I PI3Ks (Supplementary Table 3).
The overall orientation of Torin2 in the ATP site is as predicted 46 (Fig. 6a and Supplementary Fig. 12a). The tricyclic benzonapththyridine ring binds to the adenine site and makes a hydrogen bond to the N-C lobe “hinge”, analogous to one of two backbone hydrogen bonds made by ATP and diverse PI3/protein kinase inhibitors. The amino-pyridine group reaches into the “inner hydrophobic pocket”, an area in the back of the cleft that many kinase inhibitors contact. However, it does not make the three predicted hydrogen bonds (to Asp2195, Asp2357 and Tyr2225)46. Rather, the structure reveals that a key interaction is the extensive stacking of the tricyclic benzonapththyridine ring with the indole group of Trp2239 from the “hinge” (Fig. 6a). In the ATPγS complex, Trp2239 stacks partially with a three-atom portion of the adenine. In the Torin2 complex, by contrast, Trp2239 stacks with a ten-atom portion of the tricyclic Troin2 ring. This likely makes a substantial contribution to the sub-nanomolar potency46 of Torin2. In addition, because Trp2239 is not present in canonical protein kinases or in PI3Ks, it would also contribute to Torin2’s ~800-fold specificity for mTOR over PI3Ks46. The structure also reveals that the Torin2 trifluoromethyl group packs into an N lobe pocket (Ile2163, Pro2169 and Leu2185; Fig. 6a). These contacts are less extensive than those made to Trp2239, and only Leu2185 is variable across the PIKK and PI3K families. They may contribute to Torin2-mTOR specificity, however, as this portion of the N lobe β sheet exhibits considerable variation in its relative orientation in PI3K structures.
Figure 6. Structures of the Torin2, PP242 and PI-103 inhibitors bound to the mTOR catalytic cleft.
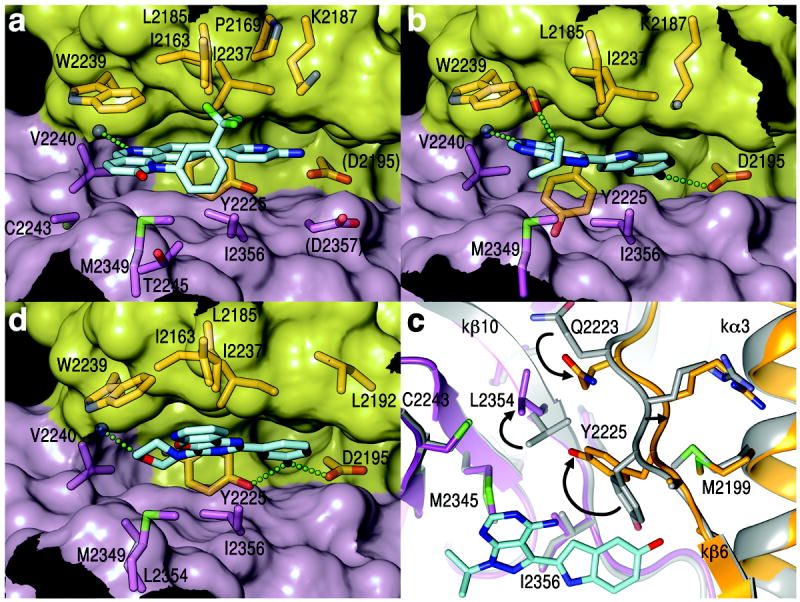
a Stick representation of Torin2 (C atoms coloured cyan, N blue, O red, F green) and of mTOR residues within 4 Å (except for D2195 and D2357). The mTOR cleft is shown in transparent surface representation, with the N lobe in yellow and C lobe in pink. Green dotted line indicates atoms within hydrogen-bonding distance and geometry.
b, PP242-mTOR structure, represented as in (a).
c, Conformational change in the inner hydrophobic pocket of mTOR on PP242 (cyan) binding. Arrows indicate side chain rotations and main chain shifts compared to ATPγS-bound mTOR (gray). View looking down the vertical axis of (b).
d, PI103-mTOR structure, represented as in (a).
PP242 consists of the adenine-mimetic pyrazolopyrimidine scaffold common to PI3/tyrosine kinase inhibitors, with a hydroxyindole substituent at a position that often points to the inner hydrophobic pocket47. In mTOR, this pocket is lined with residues conserved across the PI3K and PIKK families, and the selectivity of PP242 for mTOR was unexpected47. The structure reveals that while these two PP242 groups are generally positioned as predicted47, mTOR undergoes a conformational change that expands and deepens the inner hydrophobic pocket, with the PP242 hydroxyindole group reaching deep into the new space (Fig. 6b and Supplementary Fig. 12b). The conformational change involves the Tyr2225 side chain at the back of the pocket swinging (Δχ1 = 108°) out of the way of the hydroxyindole group (Fig. 6c and Supplementary Fig. 12c). Because Tyr2225 is part of the local hydrophobic core between the N and C lobes, this necessitates additional concerted changes. The Leu2354 side chain rotates (Δχ1 = 166°) away from the incoming Tyr2225, the Gln2223 side chain moves in to plug a resulting gap, and a 3-residue main chain stretch (residues 2223 to 2225) shifts by ~1 Å. In PI3Ks, Leu2354 is replaced by a conserved phenylalanine that cannot undergo a similar conformational change, at least without an even more extensive repacking of the local hydrophobic core. Leu2354 is thus likely to be a major, though indirect, determinant of the selectivity of PP242 for mTOR over PI3Ks and other kinases.
The multi-targeted PI-10348 does not take advantage of any of the aforementioned mTOR-specific features. The morpholine ring binds to the adenine pocket and makes two hydrogen bonds to the “hinge”, while the m-phenol group binds to the inner pocket and makes two hydrogen bonds to the Tyr2225 and Asp2195 side chains at the back of the cleft (Fig. 6d and Supplementary Fig. 12d). These hydrogen bonds are likely to be important for the high affinity of PI-103 for mTOR and, presumably, for Class I PI3Ks where the relevant protein groups are similarly positioned. In support, we note that the ~500-fold weaker affinity48 Class III PI3K–PI-103 complex lacks both m-phenol hydrogen bonds and one of the “hinge” hydrogen bonds32.
Conclusions
The mTORΔN structure reveals an intrinsically active but otherwise highly restricted catalytic centre, and together with activating mutations points to substrate recruitment as a major mechanism controlling the kinase activity. Our biochemical data on S6K1 indicates that in addition to a primary recruitment motif, a secondary recruitment motif proximal to the phosphorylation site is needed for efficient phosphorylation. The FRB acts as a gatekeeper by restricting access to the active site while also granting privileged substrates access through its binding site for the secondary motif. Co-crystal structures of ATP-competitive inhibitors reveal multiple determinants of mTOR specificity, including a conformational change deep inside the catalytic cleft.
Methods
Protein expression and purification
The boundary of the crystallized human mTORΔN (residues 1376-2549) was identified by screening successive N-terminal truncations of mTOR, produced by transient transfection in HEK293-F cells, for expression levels, solubility and size exclusion chromatography profile (not shown). For large-scale production of mTORΔN–mLST8, we generated an HEK 293-F cell line that was stably-transfected sequentially by modified pcDNA3.1 vectors encoding FLAG-tagged mLST8 and FLAG-tagged mTORΔN. In the absence of mLST8 co-expression, the level of soluble mTORΔN was substantially lower, and it co-purified with endogenous mLST8 (not shown). The mTORΔNΔ–2486-mLST8, full-length human mTOR-mLST8 and human mTORC1 (mTOR-mLST8-RAPTOR) complexes were similarly produced in HEK 293-F cell lines that were stably transfected with the FLAG-tagged versions of the proteins. Cells were grown as monolayers in 15 centimeter plates. They were lysed in 50mM Tris-Cl, pH 8.0, 100 mM KCl, 400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10 % (v/v) glycerol, 2 mM dithiothreitol (DTT) and protease inhibitors using French Press. After centrifugation, the FLAG-tagged proteins were affinity purified using anti-FLAG M2 agarose beads (Sigma). Following cleavage of the FLAG-tags by tobacco etch virus (TEV) protease, they were purified by ion exchange (MonoQ) and gel-filtration chromatography. The purified complex was concentrated to 5 mg ml-1 by ultrafiltration in 20 mM Tris-Cl, 500 mM NaCl, 10 mM DTT, pH 8.0 and was used for crystallization immediately. For kinase assays, 10% (v/v) glycerol was added to protein aliquots for storage at -80 °C.
Human S6K1αIIki protein, which contains the kinase-inactivating K100R mutation, was produced by infecting High Five insect cells with a pFastBac1 baculovirus expressing the FLAG-tagged protein. It was purified by affinity chromatography with anti-FLAG M2 agarose beads. The protein was concentrated to 8 mg ml-1 in 50 mM Tris-Cl, 400 mM NaCl, 1mM EDTA, 1mM EGTA, 5% (v/v) glycerol, 10 mM DTT, 1 mM Pefabloc, pH 8.0, and it was stored at -80 °C.
Human 4EBP1 was overexpressed in the E. Coli strain BL21(DE3) from a modified pGEX4T3 vector, and was purified by glutathione affinity chromatography, cleavage of the GST tag with TEV protease, fractionation on MonoQ column, removal of free GST by glutathione affinity, and finally gel filtration (Superdex75) chromatography. The peak fractions were concentrated to 20 mg ml-1 in 20 mM Tris-Cl, 200 mM NaCl, 5% (v/v) glycerol, 10 mM DTT, pH 8.0.
Crystallization and data collection
Crystals were grown by the hanging-drop vapour diffusion method at 4 °C. Apo-crystals of the mTORΔN–mLST8 complex were grown from 100 mM Tris-Cl, 6-8% (w/v) polyethylene glycol (PEG) 8000, 500 mM NaCl, 10% (v/v) glycerol, 10 mM DTT, pH 8.5. Crystals of the mTORΔN-mLST8 bound to ADP-MgF3-Mg2 were grown similarly, except the well-buffer contained 10 mM MgCl2, 3 mM ADP, and 20 mM NaF. The mTORΔN-mLST8-ATPγS-Mg2 complex was prepared by soaking apo-crystals for one hour in a stabilization buffer of 50 mM Tris-Cl, pH 8.5, 10 mM Tris-Cl, 8.0, 10% PEG8000, 0.1 M NaCl, 6% glycerol, supplemented with 5 mM MgCl2 and 1 mM ATPγS. The mTORΔN-mLST8-AMPPNP-Mn2 complex was prepared by soaking apo-crystals similarly, except the stabilization buffer had a pH of 7.5 and it was supplemented with 1 mM AMPPNP and 2 mM MnCl2, and the data was collected at the Manganese absorption edge. Crystals of mTORΔN-mLST8-Torin2 and mTORΔN-mLST8-PI-103 were prepared by mixing 1 mM of the inhibitors with the protein. Co-crystals appeared from the same condition as the apo-crystals. Crystals of mTORΔN-mLST8-PP242 were prepared by soaking apo-crystals for 2.5 hours in the stabilization buffer supplemented with 0.2 mM PP242. Apo-crystals were harvested in stabilization buffer, transferred to 50 mM Tris-Cl, pH 8.5, 10 mM Tris-Cl, pH 8.0, 0.1 M NaCl, 14% (w/v) PEG8000, 22% (v/v) glycerol, and were flash-frozen in liquid nitrogen. Crystals with ADP-MgF3-Mg2, ATPγS-Mg2, Torin2, PI103 and PP242 were flash-frozen similarly, except for the presence of the corresponding cofactors or inhibitors (0.1 mM) in the buffers. Diffraction data were collected at -170 °C at the ID24C and ID24E beamlines of the Advanced Photon Source, and they were processed with the HKL suite49.
Structure determination and refinement
Initial phases were obtained from isomorphous and anomalous differences of two heavy atom derivatives, prepared by soaking crystals in stabilization buffer lacking DTT and supplemented with 1 mM uranyl acetate (1 hour), or with 0.4 mM potassium-gold cyanide (75 min.) at at 4° C. Initial phases, calculated with the program SHARP50 had a mean figure of merit of 0.35 (35.0 to 4.5 Å). The uranyl derivative had a dispersive phasing power (Pp) of 0.89 and anomalous Pp of 0.60, with a dispersive Rcullis of 0.79 and anomalous Rcullis of 0.95. The gold derivative had a dispersive Pp of 0.80 and anomalous Pp of 0.29, with a dispersive Rcullis of 0.66 and anomalous Rcullis of 0.98. The phases were improved using solvent flattening and two-fold ncs averaging with multiple masks with the program DM51. The model was built using O52 and refined first with REFMAC551 and then with PHENIX53, using tight ncs restraints on atom positions. The final model contains residues 1385 to 2549 of human mTOR, and 8 to 324 of human mLST8. mTORΔN residues 1376-1384 at the N-terminus, residues 1815-1866 in the FAT domain, and residues 2437-2491 between kα9b and kα10 in the KD are disordered. mLST8 residues 1-7 and 325-326 from the N- and C-termini are disordered. The Ramachandran plot, calculated by PROCHECK, has 88.5, 11.0 and 0.5 % of the residues in the most favored, additionally allowed, and generously allowed regions, respectively. There are no residues in disallowed regions. The Rfree test set of the Native data contains 1699 reflections.
In vitro kinase assays
All kinase assays were performed in a buffer of 25 mM HEPES, pH 7.4, 100 mM NaCl, 10 mM MgCl2, 2 mM DTT, 3 % (v/v) glycerol, for 30 minutes at 30° C, in a final volume of 15 μl. For a typical reaction, the indicated amount of enzymes, substrates, and competitors were incubated together for 10 minutes on ice in the kinase buffer in a final volume of 13.5 μl. Reactions were started by the addition of 1.5 μl of cold ATP (0.5 mM final concentration) including 2 μCi of [γ-32P] ATP (6000 Ci/mmol, Perkin-Elmer). In assays with rapamycin, all reactions in a titration contained a constant level of 3.3 % DMSO. Reactions were stopped by the addition of 15 μl of 2X NuPAGE LDS sample buffer and boiling for 3 minutes, and were resolved in a 4-12 % NuPAGE Bis-Tris gel. Where applicable, each reaction was split into two for analysis by phosphorimaging and immunoblotting. All antibodies were obtained from Cell Signaling, except for the anti-FLAG antibody which was obtained from Sigma.
Supplementary Material
Acknowledgments
We thank D. King for mass spectroscopic analysis; H. Erdjument-Bromage for N-terminal sequencing; the staff of the Advanced Photon Source ID24 beamlines for help with data collection; Dorothea Robev for help with protein expression in HEK 293 cells. This work was supported by the Howard Hughes Medical Institute.
Footnotes
Author Contributions H.Y. and N.P.P. designed the experiments, solved the structures and wrote the manuscript. H.Y., D.G.R., J.D.K., B.M. and H.J.Y. carried out protein production, crystallization, and biochemical experiments. H.Y. performed all other experiments.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
The authors declare no competing financial interests.
The coordinates and structure factors have been deposited in the Protein Data Bank with accession numbers of 4JSN for the native structure, 4JSP for the ATPγS-Mg complex, 4JSV for the ADP-Mg-F complex, 4JSX for the Torin2 complex, 4JT5 for the PP242 complex, and 4JT6 for the PI-103 complex.
References
- 1.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 3.Keith CT, Schreiber SL. PIK-related kinases: DNA repair, recombination, and cell cycle checkpoints. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 4.Hara K, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 5.Kim DH, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 6.Loewith R, et al. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- 7.Sarbassov DD, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296–1302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 8.Chen EJ, Kaiser CA. LST8 negatively regulates amino acid biosynthesis as a component of the TOR pathway. J Cell Biol. 2003;161:333–347. doi: 10.1083/jcb.200210141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim DH, et al. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol Cell. 2003;11:895–904. doi: 10.1016/s1097-2765(03)00114-x. [DOI] [PubMed] [Google Scholar]
- 10.Sancak Y, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320:1496–1501. doi: 10.1126/science.1157535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim E, Goraksha-Hicks P, Li L, Neufeld TP, Guan KL. Regulation of TORC1 by Rag GTPases in nutrient response. Nat Cell Biol. 2008;10:935–945. doi: 10.1038/ncb1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saucedo LJ, et al. Rheb promotes cell growth as a component of the insulin/TOR signalling network. Nat Cell Biol. 2003;5:566–571. doi: 10.1038/ncb996. [DOI] [PubMed] [Google Scholar]
- 13.Stocker H, et al. Rheb is an essential regulator of S6K in controlling cell growth in Drosophila. Nat Cell Biol. 2003;5:559–565. doi: 10.1038/ncb995. [DOI] [PubMed] [Google Scholar]
- 14.Long X, Lin Y, Ortiz-Vega S, Yonezawa K, Avruch J. Rheb binds and regulates the mTOR kinase. Curr Biol. 2005;15:702–713. doi: 10.1016/j.cub.2005.02.053. [DOI] [PubMed] [Google Scholar]
- 15.Sato T, Nakashima A, Guo L, Tamanoi F. Specific activation of mTORC1 by Rheb G-protein in vitro involves enhanced recruitment of its substrate protein. J Biol Chem. 2009;284:12783–12791. doi: 10.1074/jbc.M809207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sancak Y, et al. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell. 2007;25:903–915. doi: 10.1016/j.molcel.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, et al. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
- 18.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 19.Schalm SS, Fingar DC, Sabatini DM, Blenis J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr Biol. 2003;13:797–806. doi: 10.1016/s0960-9822(03)00329-4. [DOI] [PubMed] [Google Scholar]
- 20.Nojima H, et al. The mammalian target of rapamycin (mTOR) partner, raptor, binds the mTOR substrates p70 S6 kinase and 4E-BP1 through their TOR signaling (TOS) motif. J Biol Chem. 2003;278:15461–15464. doi: 10.1074/jbc.C200665200. [DOI] [PubMed] [Google Scholar]
- 21.Oshiro N, et al. The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem. 2007;282:20329–20339. doi: 10.1074/jbc.M702636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fonseca BD, Smith EM, Lee VH, MacKintosh C, Proud CG. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J Biol Chem. 2007;282:24514–24524. doi: 10.1074/jbc.M704406200. [DOI] [PubMed] [Google Scholar]
- 23.Jacinto E, et al. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol. 2004;6:1122–1128. doi: 10.1038/ncb1183. [DOI] [PubMed] [Google Scholar]
- 24.Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273:239–242. doi: 10.1126/science.273.5272.239. [DOI] [PubMed] [Google Scholar]
- 25.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8:567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 26.Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. J Clin Invest. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lovejoy CA, Cortez D. Common mechanisms of PIKK regulation. DNA Repair (Amst) 2009;8:1004–1008. doi: 10.1016/j.dnarep.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosotti R, Isacchi A, Sonnhammer EL. FAT: a novel domain in PIK-related kinases. Trends Biochem Sci. 2000;25:225–227. doi: 10.1016/s0968-0004(00)01563-2. [DOI] [PubMed] [Google Scholar]
- 29.Walker EH, Perisic O, Ried C, Stephens L, Williams RL. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999;402:313–320. doi: 10.1038/46319. [DOI] [PubMed] [Google Scholar]
- 30.Sibanda BL, Chirgadze DY, Blundell TL. Crystal structure of DNA-PKcs reveals a large open-ring cradle comprised of HEAT repeats. Nature. 2010;463:118–121. doi: 10.1038/nature08648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nolen B, Taylor S, Ghosh G. Regulation of protein kinases; controlling activity through activation segment conformation. Mol Cell. 2004;15:661–675. doi: 10.1016/j.molcel.2004.08.024. [DOI] [PubMed] [Google Scholar]
- 32.Miller S, et al. Shaping development of autophagy inhibitors with the structure of the lipid kinase Vps34. Science. 2010;327:1638–1642. doi: 10.1126/science.1184429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMahon LP, Choi KM, Lin TA, Abraham RT, Lawrence JC., Jr The rapamycin-binding domain governs substrate selectivity by the mammalian target of rapamycin. Mol Cell Biol. 2002;22:7428–7438. doi: 10.1128/MCB.22.21.7428-7438.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sekulic A, et al. A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 2000;60:3504–3513. [PubMed] [Google Scholar]
- 35.Edinger AL, Thompson CB. An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene. 2004;23:5654–5663. doi: 10.1038/sj.onc.1207738. [DOI] [PubMed] [Google Scholar]
- 36.Bao ZQ, Jacobsen DM, Young MA. Briefly bound to activate: transient binding of a second catalytic magnesium activates the structure and dynamics of CDK2 kinase for catalysis. Structure. 2011;19:675–690. doi: 10.1016/j.str.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Madhusudan, Akamine P, Xuong NH, Taylor SS. Crystal structure of a transition state mimic of the catalytic subunit of cAMP-dependent protein kinase. Nat Struct Biol. 2002;9:273–277. doi: 10.1038/nsb780. [DOI] [PubMed] [Google Scholar]
- 38.Brown EJ, et al. Control of p70 s6 kinase by kinase activity of FRAP in vivo. Nature. 1995;377:441–446. doi: 10.1038/377441a0. [DOI] [PubMed] [Google Scholar]
- 39.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vilella-Bach M, Nuzzi P, Fang Y, Chen J. The FKBP12-rapamycin-binding domain is required for FKBP12-rapamycin-associated protein kinase activity and G1 progression. J Biol Chem. 1999;274:4266–4272. doi: 10.1074/jbc.274.7.4266. [DOI] [PubMed] [Google Scholar]
- 41.Shor B, et al. A new pharmacologic action of CCI-779 involves FKBP12-independent inhibition of mTOR kinase activity and profound repression of global protein synthesis. Cancer Res. 2008;68:2934–2943. doi: 10.1158/0008-5472.CAN-07-6487. [DOI] [PubMed] [Google Scholar]
- 42.Rodriguez A, et al. A conserved docking surface on calcineurin mediates interaction with substrates and immunosuppressants. Mol Cell. 2009;33:616–626. doi: 10.1016/j.molcel.2009.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ohne Y, et al. Isolation of hyperactive mutants of mammalian target of rapamycin. J Biol Chem. 2008;283:31861–31870. doi: 10.1074/jbc.M801546200. [DOI] [PubMed] [Google Scholar]
- 44.Reinke A, Chen JC, Aronova S, Powers T. Caffeine targets TOR complex I and provides evidence for a regulatory link between the FRB and kinase domains of Tor1p. J Biol Chem. 2006;281:31616–31626. doi: 10.1074/jbc.M603107200. [DOI] [PubMed] [Google Scholar]
- 45.Urano J, et al. Point mutations in TOR confer Rheb-independent growth in fission yeast and nutrient-independent mammalian TOR signaling in mammalian cells. Proc Natl Acad Sci U S A. 2007;104:3514–3519. doi: 10.1073/pnas.0608510104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu Q, et al. Discovery of 9-(6-aminopyridin-3-yl)-1-(3-(trifluoromethyl)phenyl)benzo[h][1,6]naphthyridin-2(1H)-one (Torin2) as a potent, selective, and orally available mammalian target of rapamycin (mTOR) inhibitor for treatment of cancer. J Med Chem. 2011;54:1473–1480. doi: 10.1021/jm101520v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Apsel B, et al. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol. 2008;4:691–699. doi: 10.1038/nchembio.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knight ZA, et al. A pharmacological map of the PI3-K family defines a role for p110alpha in insulin signaling. Cell. 2006;125:733–747. doi: 10.1016/j.cell.2006.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Otwinowski Z, Minor W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods in Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 50.Bricogne G, Vonrhein C, Flensburg C, Schiltz M, Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Cryst D. 2003;59 doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- 51.The CCP4 suite: programs for protein crystallography. Acta crystallographica. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 52.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta crystallographica. 1991;47(Pt 2):110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 53.Adams PD, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.