

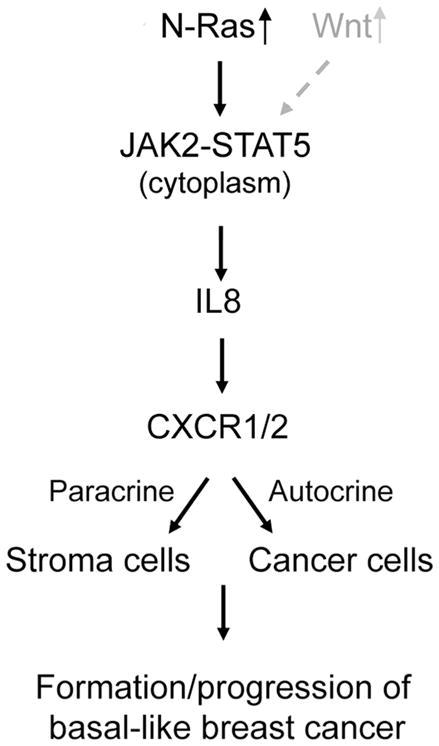
SUMMARY
Basal-like breast cancers (BLBCs) are aggressive, and their drivers are unclear. We have found that wild-type N-RAS is overexpressed in BLBCs, but not in other breast cancer subtypes. Repressing N-RAS inhibits transformation and tumor growth, while overexpression enhances these processes even in preinvasive BLBC cells. We identified N-Ras-responsive genes, most of which encode chemokines, e.g., IL8. Expression levels of these chemokines and N-RAS in tumors correlate with outcome. N-Ras, but not K-Ras, induces IL8 by binding and activating the cytoplasmic pool of JAK2; IL8 then acts on both the cancer cells and stromal fibroblasts. Thus BLBC progression is promoted by increasing activities of wild-type N-Ras, which mediates autocrine/paracrine signaling that can influence both cancer and stroma cells.
Graphical Abstract
INTRODUCTION
Approximately 20% of breast cancers belong to the “basal-like” subtype. Basal-like breast cancers (BLBCs) are of great clinical importance because they are the most aggressive breast cancers, with very poor prognosis (Sorlie et al., 2003). These tumors are commonly “triple negative,” lacking estrogen receptor (ER), HER2, and progesterone receptor; as such, they cannot be treated by current targeted therapies, which are largely directed against ER or HER2. Thus it is urgent to identify drivers for BLBC that can be targeted in order to treat this aggressive form of breast cancer.
The basal-like tumors are so named because they express markers typical of the cells in the basal layer in the mammary duct (Perou et al., 2000), approximately 1% of whose cells are postulated to have stem/progenitor cell properties. BLBC cells and human embryonic stem (ES) cells have been found to express a common set of genes (Ben-Porath et al., 2008), suggesting that the BLBC cells are enriched with cells having stem cell properties. We have thus sought to identify a common growth mechanism in these cells that may lead to the discovery of a driver for BLBC.
Humans have three RAS genes, H-, N-, and K-RAS, and the latter can undergo alternative splicing to produce two isoforms, K-Ras-4A and K-Ras-4B. As a result, human cells have a total of 4 Ras proteins. Oncogenic RAS mutations are among the most frequent genetic alterations in human tumors — over 30% of all human tumors contain an oncogenic RAS mutation (Pylayeva-Gupta et al., 2011). In breast cancers, however, oncogenic RAS mutations are rare (Bos, 1989). But different wild-type RAS genes are selectively overexpressed in different sub-types of human breast cancer cells (Hoadley et al., 2007) — BLBC cells selectively overexpress N-RAS, while luminal breast cancer cells selectively overexpress H-RAS, suggesting that the gain of activity of a particular wild-type Ras protein might play a key role in promoting the development of these subsets of breast cancers.
In this study we present evidence that N-Ras is a driver for BLBCs. By analyzing genes whose expression is N-Ras dependent, we illustrate a key mechanism by which N-Ras can promote BLBCs, namely it activates Janus kinase 2 (JAK2), leading to interleukin 8 (IL8/CXCL8) induction, which stimulates not only cancer cells themselves but possibly also stromal fibroblasts to create a proinvasive microenvironment.
RESULTS
N-RAS is selectively overexpressed in BLBCs
As mentioned above, N-RAS has been shown to be selectively overexpressed in BLBCs cell lines. In this study, we first determined whether in human breast cancers N-RAS is also selectively overexpressed. Upon examining The Cancer Genome Atlas (TCGA) RNA-seq data (TCGA, 2012), we found that N-RAS expression levels are highest in BLBCs, and lowest in normal adjacent tissues (Figure 1A). N-RAS expression seems to negatively correlate with expression of ESR1 (encoding the ER-α, ρ = − 0.27, P = 8.4×10−7 Spearman’s rank correlation), ERBB2/HER2 (ρ = − 0.19, P = 3×10−4) and PGR (encoding the progesterone receptor, ρ = − 0.23, P = 2.4×10−5). Other microarray data sets (Prat et al., 2010) reinforce these observations (Figure S1A). Furthermore, we found that N-RAS mRNA levels inversely correlate with N-RAS promoter methylation (Figure 1B), supporting the possibility that N-Ras overexpression may be partly mediated by epigenetic demethylation at the N-RAS promoter.
Figure 1. N-RAS is selectively overexpressed in BLBCs and its expression levels associate with poor clinical outcome.
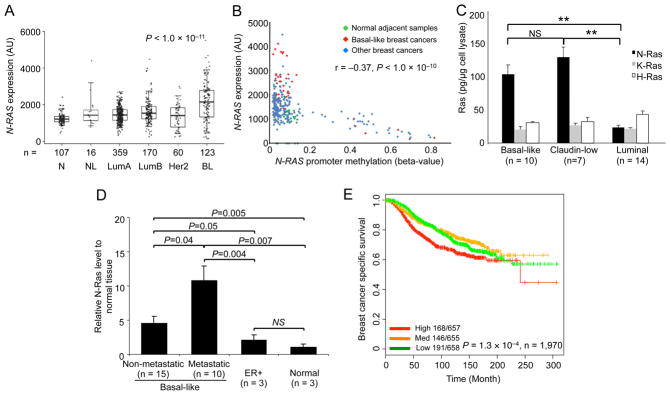
(A) Box-whisker plots of N-RAS expression levels in different breast cancer subtypes from RNAseq data in TCGA. (N=normal adjacent tissues, NL=normal like, LumA=luminal A, LumB=luminal B, Her2=Her2 positive, BL=basal-like). AU, arbitrary unit. P-value by ANOVA. (B) N-RAS mRNA in the indicated breast cancers or tissues (panel A) correlated with N-RAS promoter methylation in the same TCGA data sets. P-value by Spearman’s correlation. (C) N-Ras protein levels in different subtypes of breast cancer cell lines (n = number of cell lines, see Supplemental Information) were measured by Western blot. (D) N-Ras in tumors from indicated PDXs, relative to normal mouse mammary tissues, analyzed by Western blot as above. (E) Kaplan-Meier plots of breast cancer-specific survival in patients (n = 1,970) by tertiles (high, medium, and low levels) of N-RAS mRNA levels over time in the METABRIC dataset. P-value by log-rank test. See also Figure S1.
To determine whether N-Ras is overexpressed in BLBCs at the protein level, we used Western blot to analyze a large collection of human breast cancer cell lines that recapitulates the molecular complexity seen in human breast tumors (Neve et al., 2006). As shown in Figure 1C, BLBC cells express approximately 4 times more N-Ras protein than do luminal breast cancer cells, leading to more GTP-N-Ras in the former (Figure S1B). In addition, the N-Ras levels in BLBC cells are higher than those of H- and K-Ras. In contrast, luminal breast cancer cells have more H-Ras than N- and K-Ras proteins, agreeing with our previous study examining the mRNA levels (Hoadley et al., 2007). We also examined several patient-derived xenograft (PDX) lines, the tumors in which preserve key pathological features and biomarkers of the original tumors (Zhang et al., 2013), and found that basal-like PDXs have more N-Ras than ER+ PDXs (Figure 1D). Finally, in support of the concept that human ES cells and BLBCs express a similar set of genes, we examined two human ES cell lines and found that they too express more N-Ras than K- and H-Ras (Figure S1C).
N-Ras levels and outcome
To determine whether N-Ras expression levels are important for breast cancer, we searched databases with patient outcome information. As shown in Figure 1E, we found that high N-RAS expression levels associate with poor breast cancer-specific survival in the METABRIC data sets (Curtis et al., 2012). We also examined another database (Prat et al., 2010) and came to the same conclusion (Figure S1D). We note that in all our bioinformatics analyses, high N-RAS mRNA levels are inseparable from the basal-like subtype, which supports the concept that high N-Ras levels drive the formation of this subtype of breast cancer. To further examine whether N-Ras levels can influence outcome within BLBCs, we analyzed the aforementioned PDXs (Figure 1D), and found that basal-like PDXs that are metastatic in mice have higher levels of N-Ras protein than those basal-like PDXs that are not metastatic. We conclude that BLBCs selectively overexpress primarily one of the Ras proteins, N-Ras, and that high levels of N-Ras associate with poor outcome.
N-Ras, but not K-Ras, is required for efficient growth of just BLBC cells
To determine whether selective N-Ras overexpression in BLBCs is functionally relevant, we repressed N-RAS by shRNA (clone #1) by 70–90% in all the examined cell lines (Figure 2A), without affecting levels of H-Ras, K-Ras, and GAPDH. When N-Ras was so suppressed, only the growth of BLBC cells was greatly inhibited (50–80% reduction, Figure 2B), while the growth of luminal breast cancer cells and untransformed “normal” breast mammary epithelial cells was not significantly impacted (Figure 2B). Like BLBCs, claudin-low cells are also frequently triple-negative (Prat et al., 2010), and their N-Ras protein levels are not statistically different from those of BLBC cells (Figure 1C). Surprisingly, however, N-RAS repression did not affect the growth of the claudin-low cell lines (Figure 2B and Figure S2A). To further assess any possible off-target effects of gene silencing in BLBC, we overexpressed an N-RAS cDNA refractory to this shRNA (N-RAS*) and found that it rescued the growth inhibition in N-RAS-repressed basal-like cells (Figure 2C). Using two additional shRNA clones (#2 and #3), we came to the same conclusion (Figure S2 A and B).
Figure 2. N-Ras is required for efficient growth, transformation, and tumor formation of BLBC cells only.

(A) Cells expressing control or shRNAs targeting N-RAS or K-RAS4B were analyzed by Western blot for Ras proteins using isoform-specific antibodies. GAPDH was the loading control. The numbers below the blot indicate protein levels relative to the non-silenced control. (B) Cells were transduced by the shRNAs as in panel A and the growth rates were measured. (C) An N-RAS cDNA refractory to shRNA (N-RAS*) or a K-RAS4B cDNA was overexpressed in N-RAS-silenced SUM102PT cells. The growth rates of these cells were measured (top), and indicated proteins were analyzed by Western blot (bottom). The numbers below indicate protein levels relative to the controls. (D) Cells were transduced by shRNAs as in panel A and seeded either alone or together with HMFs (1:1) in soft agar. (E) N-RAS in indicated cells was similarly silenced as above with or without N-RAS* and examined for invasiveness (left). N-Ras in these cells was analyzed by Western blot (right). (F) Control or N-RAS-repressed SUM102PT cells were injected with HMFs into nude mice (n = 6). The control cells were also injected alone (n = 6) as an additional control. Kaplan-Meier curves of the portion of tumor-free animals are shown on the left while pictures of mice are shown in the middle. N-RAS silencing in the tissue was confirmed by RT-PCR (right). P-value (Gehan-Breslow-Wilcoxon test) was calculated between the green and red curves. (G) Control or N-RAS-repressed MDA-MB-468 cells carrying luciferase were injected into nude mice (2 mammary glands per mouse and 5 mice in each group, n = 10 tumors). Their luciferase activity (Figure S2I) was measured weekly (left) to calculate the tumor growth rates (between week-8 and week-13, middle). N-RAS silencing in the tissue was confirmed by RT-PCR (right). (H) SUM102PT cells carrying the inducible N-RAS shRNA were co-injected with HMFs (1:1) into nude mice (n = 9). When tumors became detectable (arrow), these mice were treated with doxycycline (DOX) or the vehicle control, and the tumor size was measured twice per week. See also Figure S2.
We also examined K-RAS and found that K-RAS repression (Figure 2A) did not affect cell growth (Figure 2B) and that K-RAS overexpression did not rescue the growth inhibition of N-RAS-repressed basal-like cells (Figure 2C). These data suggest that most BLBC cell lines not only selectively overexpress N-Ras, but also depend on N-Ras, but not K-Ras, for efficient cell growth. Finally, we found that human ES cells are also dependent on N-Ras for proliferation (our unpublished observation).
Efficient transformation of BLBC cells is dependent on N-Ras
We first examined colony formation in soft agar (anchorage independent growth) and found that this activity was inhibited by N-RAS repression in two malignant BLBC cell lines (MDA-MB-468, Figure 2D, and SUM149PT, Figure S2C), but not in claudin-low cell lines (MDA-MB-231, Figure 2D, and SUM159PT, Figure S2C). We also examined a preinvasive BLBC cell line, SUM102PT, which was derived from intraductal carcinoma and shows ductal carcinoma in situ (DCIS) features in 3D culture (Jedeszko et al., 2009; Kaur et al., 2012). SUM102PT cells are only weakly transformed and do not efficiently form colonies in soft agar. Colony formation is induced, however, when these cells are co-cultured with human mammary fibroblasts (HMFs), a key component of the stroma (Figure 2D). When N-RAS was silenced in SUM102PT cells, their ability to form colonies in soft agar even in co-cultured with HMFs was also greatly inhibited. Similar results were observed using two other shRNAs (Figure S2 D and E). We also examined cell invasiveness and found that it was decreased by N-RAS knock-down only in the basal-like MDA-MB-468 cells, but not in the claudin-low MDA-MB-231 cells (Figure 2E). This defect was similarly rescued by the shRNA-resistant N-RAS*, suggesting that the gene silencing is also selective when cell invasion is measured.
While N-Ras is not essential for growth and transformation of claudin-low cells, N-RAS-repressed claudin-low cells, but not BLBC cells, appeared morphologically smaller after prolonged culturing in vitro (our unpublished results). Thus, although claudin-low and BLBC cells are both usually triple-negative, increasing evidence supports their classification into two different subtypes (Herschkowitz et al., 2012), and it seems that one key difference between them is how the cells react to N-Ras.
N-Ras is required for efficient tumor formation and/or tumor growth of BLBC cells
To determine whether N-Ras is required for BLBC cells to form tumors in vivo, we first examined SUM102PT cells, which can form tumors when co-transplanted with HMFs into mice (Figure 2F). However, when N-RAS was silenced in SUM102PT cells, tumor latency increased, and the tumor number (Figure 2F), as well as tumor volume (Figure S2F), decreased. Similar results were obtained using shRNA-3 (Figure S2G), which was made into an inducible vector (Figure S2B). The effect of shRNA-3 on tumor formation can be rescued by wild-type N-RAS (shRNA-3 targets a non-coding region, Figure S2G), suggesting that the observed growth inhibition is caused by N-RAS repression with a high degree of selectivity. A similar growth inhibition was observed when N-RAS-repressed malignant MDA-MB-468 cells were transplanted into mice (Figure 2G and Figure S2H). To further examine N-Ras’s role in BLBC, we switched off N-RAS expression after tumors were detectable and found that their growth was substantially decreased (Figure 2H).
N-Ras overexpression promotes transformation of BLBC cells
To ascertain whether N-Ras can promote the development of BLBCs, we first overexpressed it in the malignant basal-like MDA-MB-468 and BT20 cells and found that their ability to form colonies in soft agar was readily enhanced (Figure 3A); in contrast, N-Ras overexpression did not enhance colony formation of the claudin-low MDA-MB-231 and SUM159PT cells. To more stringently test the concept that N-Ras can drive BLBC, we overexpressed it in the preinvasive SUM102PT cells and found that colony formation in soft agar was increased even without HMFs (Figure 3B). Finally, we overexpressed N-Ras in the untransformed basal-like MCF10A cells and found that they too were transformed after a relatively long latency (Figure 3C). We also examined whether N-Ras overexpression can promote cell migration and invasion in the preinvasive SUM102PT and normal MCF10A cells. As shown in Figure 3D, N-Ras did enhance migration of these two cell lines, but not invasion (Figure S3A).
Figure 3. N-Ras overexpression promotes transforming activities and tumor growth of BLBC cells.

(A) Control or N-Ras-overexpressing cells were seeded in soft agar for colony formation. (B) Control or N-Ras overexpressing SUM102PT cells were seeded in soft agar alone or with HMFs (2:1) for the indicated durations (left). N-Ras overexpression was confirmed by Western blot (right). (C) N-Ras was similarly overexpressed in MCF10A cells and seeded in soft agar. Colony number was measured 2 months later (left). Western blot validated N-Ras overexpression (right). (D) Control or N-Ras overexpressing cells were measured for their migration abilities. (E) Control or N-Ras overexpressing SUM102PT cells were co-injected with HMFs (2:1) into nude mice (n = 10 mice). A Kaplan-Meier plot of the portion of tumor-free mice over time is shown on the left (P-value by Gehan-Breslow-Wilcoxon test), while a plot of tumor volumes over time is shown in the middle. Overexpression of the human N-RAS in the tissue was confirmed by RT-PCR (right). (F) An inducible N-RAS vector was injected into the mammary glands of 26 MMTV-Wnt1 mice, which were then randomized to receive DOX or water, and the portion of tumor-free mice was plotted over time. P-value by Gehan-Breslow-Wilcoxon test (left). Levels of N-Ras (middle), JAK2, and phospho-JAK2 (right) in 7 tumors from the +DOX group (n = 7) and 4 tumors untreated by DOX (n = 4) were examined by Western blot. See also Figure S3.
N-Ras overexpression promotes tumor formation in mouse models of BLBC
To determine whether N-Ras overexpression can promote tumor formation, we first investigated SUM102PT cells, which weakly form tumors in mice when co-transplanted with HMFs. Overexpressing N-Ras in SUM102PT cells shortened the latency for tumor formation, which correlated with an increase in tumor growth rate (Figure 3E).
Like N-Ras, the canonical Wnt pathway in BLBC is not frequently mutated, but its activity is up-regulated, which predicts poor outcome (Khramtsov et al., 2010). In mice, MMTV-Wnt1 efficiently induces hyperplastic early lesions, and tumors do eventually emerge after a long latency (Tsukamoto et al., 1988). These tumors resemble human BLBC (Herschkowitz et al., 2007). Intriguingly, it has been known for some time that ≈50% of the mammary tumors arising in MMTV-Wnt1 transgenic mice carry oncogenic mutations affecting ras (Podsypanina et al., 2004). Examining the other half of the tumors, those without oncogenic ras mutations, we found that they have very high levels of N-Ras (Figure S3B). We then went on to construct an inducible vector to overexpress N-Ras in MMTV-Wnt1 mice and found that N-Ras overexpression accelerated tumor formation (Figure 3F).
Identifying N-Ras-dependent genes
To uncover the role of N-Ras specifically in BLBC cells, we repressed N-RAS expression and then profiled gene expression changes in two different basal-like cell lines (SUM102PT and SUM149PT). We also similarly analyzed a claudin-low cell line (SUM159PT) as control because its growth is not sensitive to N-RAS-repression. As shown in Table S1, despite the fact that the two BLBC cell lines are different, N-RAS repression readily induced changes of expression of a similar set of genes, while such changes were not detected in the claudin-low cells. We then used this gene set to search against the Molecular Signature Database (MSigDB) to reveal the biological pathways in which they are involved (Table S2). Strikingly, those genes whose expression is down-regulated when N-RAS is repressed appear to be enriched for genes involved in immune/inflammatory responses, e. g., cytokines and chemokines. Conversely, genes that are up-regulated during N-RAS repression are enriched for markers of differentiation or development (e.g., keratins, collagens, and cytoskeletal proteins).
N-RAS-controlled genes are clinically relevant and associate with clinical outcome
To ascertain whether the genes identified above using cell lines may also be controlled by N-Ras in human tumors, we asked whether there was any correlation in the expression levels between N-RAS and the identified genes in several databases of human tumors. As shown in Table 1, in the METABRIC data set, we found a good correlation in expression levels between N-RAS and genes that were down-regulated, but not with those that were up-regulated, when N-RAS expression was suppressed. Expression of neither set of genes correlated with those of K-RAS. These conclusions were supported by examining another database (Prat et al., 2010). For the rest of the study, we thus focused on those genes that are down-regulated when N-RAS expression itself is also down-regulated. We will call these the “Down” genes hereafter.
Table 1.
Correlation of gene expression levels in human tumors1.
| N-Ras sensitive genes2 |
N-RAS
|
K-RAS
|
||
|---|---|---|---|---|
| r | P | r | P | |
|
|
|
|||
| Down | 0.4 | 2.2 × 10−16 | 0.0 | 0.1 |
| Up | 0.0 | 0.9 | −0.1 | 1.5 × 10−6 |
Notes:
n=1,992 tumors (METABRIC database).
Median-weighted expression levels of all the genes that are down- or up-regulated when N-RAS expression is repressed (Table S1) were separately analyzed by Pearson correlation to determine whether these correlated with expression of N-RAS and K-RAS. In addition, we similarly analyzed just those six genes validated to be down-regulated upon N-RAS repression (Figure 4A), and found that their expression levels also correlated with those of N-RAS (r = 0.3, P = 2.2 × 10−16), but not with K-RAS.
We selected those Down genes whose levels were changed ≥ 2-fold for validation by qPCR. As summarized in Figure 4A, the expression of these 6 genes is dependent on N-RAS, but not on K-RAS, in the basal-like cell lines, but not in the control claudin-low cell lines. Finally, expression of these validated genes returned nearly the same correlation with N-RAS expression levels in human tumors as shown in Table 1. We will call these 6 genes in Figure 4A “N-Ras responsive genes” hereafter.
Figure 4. N-RAS-controlled genes are clinically relevant and associate with outcome.

(A) The mRNAs from basal-like cell lines (SUM102PT and SUM149PT) and claudin-low cell lines (SUM159PT and MDA-MB-231), transduced by indicated shRNAs as in Figure 2A, were analyzed with qPCR. The analysis of IL8 is shown (left) as an example. The table on the right shows the expression changes of indicated genes in BLBC cells after being normalized to those in claudin-low cells. See Table S1 for the list of all the identified genes and Table S2 for MSigDB analysis. (B) The expression levels of the 6 N-Ras responsive genes were added up into a single score, which was analyzed in the indicated breast tissues. (C) Kaplan-Meier curves of breast cancer-specific survival in patients by tertiles (high, medium, and low levels) of the sum of expression levels of the 6 N-Ras-responsive genes in the METABRIC dataset. P-values by log-rank test. See also Figure S4.
To determine whether these N-Ras responsive genes are also clinically relevant, we first analyzed a published database in which gene expression in normal, DCIS, and invasive ductal carcinoma (IDC) was profiled (Knudsen et al., 2012) and found that the sums of the expression levels of these 6 N-Ras responsive genes are much higher in DCIS or IDC than in normal tissues (Figure 4B). Consistent with the observation that metastatic PDXs have higher levels of N-Ras (Figure 1D), there is an intriguing trend here that the expression levels of these genes are higher in IDC than DCIS (Figure 4B), but the differences are not statistically significant. We obtained the same results after examining another database (Lee et al., 2012).
In addition, we interrogated several data sets with clinical outcomes as described earlier. Upon analyzing the METABRIC database, we found that expression levels of these N-Ras responsive genes, like those of N-RAS itself, significantly associate with breast cancer-specific survival (Figure 4C). We obtained the same results using the list of all the Down genes (Table S1), suggesting that these validated genes represent the key signaling output of just N-Ras in BLBCs. We have also performed Principal Component Analysis on another data set (Prat et al., 2010) and come to the same conclusion (Figure S4A). These data support the concept that activity from this N-Ras pathway can promote progression of BLBCs, leading to poor outcome.
IL8, a key N-Ras responsive gene, stimulates cancer cells in an autocrine fashion
The N-Ras responsive genes encode mostly cytokines and chemokines, which are secreted signaling molecules that may stimulate growth of cancer cells in an autocrine fashion. To test this, we prepared conditioned media from control or N-RAS-repressed BLBC cells and found that the latter could not efficiently support the growth of parental cells (Figure 5A).
Figure 5. Functional validation of N-Ras controlled genes.

(A) The growth media from control and N-RAS-repressed SUM102PT cells were mixed with fresh medium (1:1) and then added to parental SUM102PT cells for measuring growth rate. (B) The media collected from control or N-RAS-repressed cells were examined by ELISA for IL8 (n = 3 separate experiments). (C) Control or IL8-silenced SUM102PT cells, with or without IL8 overexpression, were seeded for growth rate measurement. (D) Growth rates of cells treated with the IL8 receptor antagonist repertaxin relative to those of vehicle (DMSO)-treated cells were measured. (E) SUM102PT cells were silenced as in Figure 2A, and then treated with recombinant IL8 and/or CCL3 at concentrations that did not enhance the growth of non-silenced cells. The growth rate of the non-silenced vehicle-treated cells was set to 1. (F) Left, the cells expressing control or IL8 shRNAs were seeded in soft agar for colony formation. Right, the cells were seeded into soft agar and then treated with repertaxin. (G) Migration of HMFs in response to the control or N-RAS-repressed cancer cells. (H) Left, IL8 was similarly repressed as in panel F, and the cells were seeded into soft agar with HMFs. Right, the cells were co-seeded with HMFs in soft agar, and treated with repertaxin. (I) SUM102PT cells were seeded in soft agar with IL8RA or IL8RB-silenced HMFs (right). The silencing was confirmed by Western blot with IL8 receptor levels in the non-silenced controls set to 1 (left). (J) HMFs were co-cultured with control or N-RAS-repressed cells (left) or treated with IL8 (right) for 7 days. HMF lysates were analyzed by Western blot for α-SMA. GAPDH was the loading control. The numbers below indicate protein levels relative to the controls. (K) SUM102PT cells were co-injected with HMFs into nude mice (2 mammary glands injected per mouse and 10 mice for each group, n = 20 tumors). When tumor formation occurred (arrow), repertaxin was administered daily. Tumor size was measured every 5 days (left). Human N-RAS expression in tumors was examined by RT-PCR (right). See also Figure S5.
Among the identified chemokines, we noticed that IL8, like N-RAS, is overexpressed in BLBCs and correlates with poor outcome (Hartman et al., 2013; Rody et al., 2011), Figure S5A). Furthermore, IL8 has been shown to be a Ras-induced gene elsewhere (O’Hayer et al., 2009; Sparmann and Bar-Sagi, 2004), suggesting that activating IL8 induction may be a core Ras function. We first performed ELISA to measure IL8 in the culture media of breast cancer cell lines of various subtypes (Figure 5B). In agreement with the aforementioned microarray and qPCR results, IL8 levels were greatly reduced when N-RAS was repressed in BLBC cells, but not in luminal and claudin-low cells. We thus repressed IL8 using several shRNAs targeting the non-coding region (Figure S5B), and found that the growth of BLBC cells was readily inhibited, which can be rescued by overexpressing IL8 (Figure 5C). To determine whether IL8 acts on known IL8 receptors in cancer cells, we treated cells with the IL8 receptor antagonist repertaxin, which recapitulated the selective growth inhibition of BLBC cells caused by shRNAs against IL8 and N-RAS (Figure 5D). Conversely, adding recombinant IL8 (Figure 5E) or overexpressing IL8 (Figure S5C) partially rescued the growth defect caused by N-RAS-repression. Finally, as shown in Figure 5F, repressing IL8 by either shRNAs or repertaxin blocked soft agar colony formation selectively in BLBC cells (but not in claudin-low cells). We note that adding back IL8 at the reported dosage only partially rescues the growth defect caused by N-RAS repression. During the course of analyzing the UNC337 database, we found that high mRNA levels of CCL3, another N-Ras responsive chemokine (Figure 4A), also associate with poor outcome (Figure S5D). When CCL3 was added together with IL8, the growth defect of N-RAS-repressed cells was fully rescued (Figure 5E). These data suggest that N-Ras controls the growth and transforming activity of BLBC cells in an autocrine fashion by inducing chemokines such as IL8 and perhaps CCL3 as well.
N-Ras-induced IL8 mediates paracrine signaling from cancer cells to mammary fibroblasts
It is possible that N-Ras-induced cytokines/chemokines from the cancer cells can also influence the tumor microenvironment by a paracrine mechanism to impact tumorigenesis. In support of this, we found that when N-RAS in cancer cells was repressed, migration of HMFs toward BLBC cells, but not toward claudin-low cells, was inhibited (Figure 5G). To determine whether IL8 is a key paracrine signaling molecule, we performed co-culture experiments with HMFs and measured soft agar colony formation. Our data show that while soft agar colony formation of both basal-like and claudin-low cells was enhanced by HMFs (Figure 2D), IL8 silencing or repertaxin treatment (Figure 5H), like N-RAS silencing (Figure 2D), inhibited this activity only in BLBC cells. To directly test that IL8, produced in the cancer cells, acts on the HMFs, we repressed the IL8 receptor A or B in HMF cells and found that soft agar colony formation of co-cultured BLBC cells was greatly suppressed (Figure 5I and Figure S5E).
Tumor fibroblasts, called carcinoma associated fibroblasts (CAFs), are emerging as a key stromal component that can promote tumor progression (Kharaishvili et al., 2014). The cell of origin of CAFs remains unclear. A key type of CAFs display myofibroblast properties, e. g., expression of α-SMA. To define how the N-Ras-IL8 pathway regulates HMFs, we investigated the possibility that IL8 secreted by BLBC cells can signal to HMFs to induce myofibroblast-like properties by measuring α-SMA induction. Indeed, as shown in Figure 5J, repressing N-RAS only in BLBC cells led to a decrease of α-SMA level in the co-cultured HMF cells, while directly adding recombinant IL8 to HMF increased it. These data are consistent with the possibility that IL8 induced by N-Ras in BLBC cells can promote myofibroblast/CAF-like properties in the fibroblasts.
Blocking IL8 suppresses tumor growth of BLBC cells
IL8 inhibitors are widely available and in clinical trials for treating a wide range of diseases including cancers. We thus investigated whether repertaxin as a single agent can efficiently block tumor growth using the SUM102PT mouse model. As shown in Figure 5K, repertaxin substantially reduced the rate of tumor growth without affecting N-RAS expression levels. Thus, our results support the concept that repertaxin administered alone can block the pathway downstream from N-Ras to impede growth of BLBCs early in development.
N-Ras controls IL8 induction by activating JAK2
As shown earlier, repressing N-RAS expression inhibited IL8 induction in BLBC cells. Conversely, when N-Ras was overexpressed, IL8 levels increased (Figure 6A). IL8 induction is known to be driven by JAK2 (Britschgi et al., 2012); thus, we asked whether N-Ras induces IL8 via this known pathway. As shown in Figure 3F, N-Ras overexpression can induce JAK2 activation (as measured by an increase in its auto-phosphorylation) in mice. Conversely, when N-RAS was silenced in BLBC cells (but not in claudin-low cells) in vitro, JAK2 activity was suppressed (Figure 6B). JAK2 is known to activate STAT5 (Britschgi et al., 2012; Wagner and Rui, 2008), and as shown in Figure 6C, N-RAS repression also inhibited activation of STAT5 (but not STAT3). In contrast, silencing K-RAS had no effect on JAK2/STAT5 activation (Figure S6A). We also examined known Ras effector pathways and found that N-RAS repression did not affect Erk1/2 activation in BLBC cells (Figure S6B). N-Ras repression did inhibit Akt phosphorylation (Figure S6B). However, inhibiting Akt by MK2206 did not affect JAK2 and STAT5 phosphorylation (Figure S6C), and N-Ras-overexpressed cells are still sensitive to this compound (Figure S6D). These results suggest that N-Ras mainly signals down JAK2 to activate STAT5 in BLBC.
Figure 6. N-Ras controls IL8 induction by activating JAK2.

(A) The media harvested from control and N-RAS-overexpressing SUM102PT cells (n = 3 separate experiments) were analyzed for IL8 by ELISA. (B) The lysates of control or N-RAS-repressed cells (n=3 triplicates) were analyzed by ELISA for total and phosphorylated JAK2. (C) The control and N-RAS-repressed cells were examined by Western blot for indicated proteins. (D) The control and N-RAS-overexpressed cells, which were treated with or without TG101348, were seeded in soft agar for colony formation. (E) N-Ras was overexpressed as above, and the cells were treated with TG101348 to measure IC50. (F) N-RAS-silenced cells were also transduced to express JAK2-(V617F) before the growth rates (left) and colony formation in soft agar (middle) relative to the non-silenced control were measured. Protein expression levels were examined by Western blot (right). (G) HT1080 cells were transfected to express indicated Yn and Yc-tagged proteins and seeded into 96-well plates in triplicate (n = 3). The reconstituted YFP signal was detected by FACS (gated by untransfected cells as control), and an example of this is shown on the left. The percent YFP-positive cells was quantified and shown in the middle. Cells were also examined by Western blot to show that tagged N-Ras and JAK2 are expressed at comparable levels in various samples (right). (H) HT1080 cells carrying Yn and Yc-tagged proteins were examined by FACS. (I) HT1080 cells expressing Yn- and Yc-tagged proteins, as well as CFP-tagged cell compartment markers (GalT for Golgi, Rab5A for early endosomes, and Rab7A for late endosomes), were examined by confocal microscopy. (J) Cells overexpressing wild type or palmitoylation-deficient N-Ras were seeded in soft agar for colony formation (left). The right panel shows that both N-Ras proteins were expressed at comparable levels determined by Western blot. (K) MDA-MB-468 cells expressing indicated N-Ras proteins were treated with TG101348 for 16 hours, and then examined by Western blot. See also Figure S6.
To determine whether activities induced by N-Ras overexpression are dependent on JAK2, we sought to block such activities by a JAK2 inhibitor (TG101348). N-Ras overexpression in BLBC cells can either induce (SUM102PT cells) or enhance (MDA-MB-468 cells) colony formation in soft agar. As shown in Figure 6D, this activity was readily inhibited by TG101348. Moreover, MDA-MB-468 cells carrying just the vector control could barely form colonies in soft agar when treated with TG101348 (50 colonies or so); however, when N-Ras was overexpressed in these cells, close to 200 colonies could be seen with TG101348. Thus N-Ras overexpression apparently activates more JAK2, making the cells 4 times more resistant to TG101348. To further examine the impact of N-Ras on JAK2 activation, we measured IC50s of TG101348 in these BLBC cells with or without N-Ras overexpression. As shown in Figure 6E, N-Ras overexpressed cells returned IC50s that are approximately 3 times greater than the cells with no N-Ras overexpression. Conversely, when a constitutively active JAK2 (JAK2-(V617F)) was overexpressed, inhibition of cell growth and soft agar colony formation by N-RAS-repression were rescued (Figure 6F). These data strengthen the concept that N-Ras signals down JAK2 to control BLBC.
Physical interactions between JAK2 and N-Ras
While JAK2 activity is critical for many cancers, including breast cancer, the mechanisms controlling this activity are not well defined. We investigated the possibility that N-Ras can directly activate JAK2 by physically binding to JAK2, using the BiFC (bimolecular fluorescence complementation) method, which we have employed to study Ras-effector interactions (Cheng et al., 2011; Zheng and Chang, 2014; Zheng et al., 2012a) due to its greater sensitivity to detect weak and transient protein-protein interactions. In this system, one protein is fused to an N-terminal fragment of YFP (Yn), while its binding partner is fused to a C-terminal fragment of YFP (Yc). Binding of these two proteins can bring Yn and Yc to close proximity to facilitate refolding of a functional YFP detectable by FACS and microscopy. As shown in Figure 6G, FACS analysis showed that a YFP signal could be readily detected when Yc-JAK2 was paired with Yn-N-Ras but not with Yn-GST or just Yn. Likewise, Yn-N-Ras and just Yc barely induced any YFP signals. These data suggest that JAK2 and N-Ras interact with considerable specificity in this assay. Next we investigated whether the binding between N-Ras and JAK2 is dependent on the effector-binding loop in N-Ras. We have previously created a mutant Ras, RasΔE, in which all amino acid residues in the effector-binding loop were mutated to alanine, and its binding to Raf-RBD was only half of that of wild type N-Ras (Figure 6G). Likewise, we found that JAK2 binding to N-Ras was also reduced by half with N-RasΔE. To assess whether the binding between N-Ras and JAK2 is GTP-dependent, we compared the binding between wild type N-Ras and a constitutively GTP-bound N-Ras, N-Ras-(G12V), as well as the N-Ras-(S17N) mutant, which is mostly GDP-bound. As shown in Figure 6H, JAK2, like Raf, bound most strongly to N-Ras-(G12V) and most weakly to N-Ras-S17N, suggesting that the binding between N-Ras and JAK2 is largely GTP-dependent.
JAK2, as well as the STAT proteins, is mostly cytoplasmic. In fact, endogenous JAK2 is found almost exclusively in the cytoplasm (including the nucleus) but is not detectable at the plasma membrane (Dawson et al., 2009). This mode of subcellular localization has been later confirmed independently elsewhere including in mammary cells (Dawson et al., 2011). While N-Ras and K-Ras-4B have an identical effector binding loop, we and others have shown that they localize to and signal from different cell compartments —whereas a large portion of N-Ras is cytoplasmic, K-Ras-4B is almost exclusively on the plasma membrane (see Discussion). We thus analyzed the BiFC binding by confocal microscopy to uncover where N-Ras and JAK2 bind in the cell. As shown in Figure 6I, the binding between N-Ras and JAK2 takes place mostly in the cytoplasm. These binding sites appear as dots that are outside the nucleus and do not overlap with Golgi or endosome markers. In contrast, N-Ras binds Raf on the plasma membrane. As a control, we also examined K-Ras-4B but quickly learned that it was more difficult to detect its binding to JAK2. To boost the signal, instead of using the wild type version of the protein (as with N-Ras in Figure 6I), we used the oncogenic K-Ras-4B-(G12V), and acquired the image using up to 3 times more laser power. As shown in Figure 6I, K-Ras-4B-(G12V) bound JAK2 almost exclusively at the plasma membrane.
H- and N-Ras proteins must be palmitoylated at C-terminal cysteine residues to be targeted to the plasma membrane. We and others have shown that when these proteins are restricted to the cytoplasm by mutating these cysteines, the resulting mutant Ras proteins can still transform cells and interact with cytoplasmic effectors (Cheng et al., 2011; Chiu et al., 2002; Zheng et al., 2012a). We expressed such a cytoplasm-restricted N-Ras (N-Ras-(C181S)) and found that it can induce colony formation in soft agar of BLBC cells as efficiently as wild type Ras (Figure 6J). Note that N-Ras-(C181S) can more efficiently induce STAT5 activation, which is inhibited by TG101348 (Figure 6K). These data strongly suggest that N-Ras is directly responsible for activating the large cytoplasmic pool of JAK2 to promote IL8 induction in BLBCs.
DISCUSSION
BLBCs are aggressive, and their drivers are mostly unknown. Our data suggest that the formation and/or progression of BLBCs are driven by overexpressed wild type N-Ras, a key function of which is to activate JAK2, either alone or together with the Wnt pathway. The activated JAK2 in turn induces expression of a subset of cytokines and chemokines, such as IL8 and CCL3, in the cancer cells to stimulate tumorigenesis activity in an autocrine fashion. These cytokines and chemokines may also signal to stromal cells to create a proinvasive tumor microenvironment in a paracrine fashion. Many components in this N-Ras-chemokine and Wnt pathway have commercially available inhibitors; thus targeting them singularly or in combination may increase our chance to find an effective targeted therapy for this devastating form of breast cancer.
Our data demonstrating that JAK2 is a potential N-Ras effector in BLBC agrees with previous findings linking N-Ras and JAK2 in cancers. For example, the JAKs can be activated by the IL6 receptor (Mihara et al., 2012), and in a multiple myeloma cell line whose growth is IL6-dependent, activated N-Ras can fully replace IL6 to promote growth and prevent apoptosis (Billadeau et al., 1995). Similarly, oncogenic N-Ras requires JAKs to be fully functional in leukemia (Kong et al., 2014). This apparent convergence between N-Ras and JAK2 is intriguing considering they are thought to be activated by different receptor families — Ras by growth factor receptors and JAK2 by cytokine receptors. Whether this N-Ras-JAK2 pathway is turned on by growth factors or cytokines or both is not resolved in this study, however. Furthermore, while JAK2 can bind N-Ras in the BiFC assay, it remains an open question whether such binding alone is sufficient to activate JAK2 in BLBC. It is possible that after binding to Ras, JAK2 activation may be further mediated by cofactors. These cofactors, as well as the upstream receptors, may control the JAK2 pathway in a cell-type dependent manner to partially explain why JAK2 activation in claudin-low cells is not N-Ras dependent.
While K-RAS is the most frequently mutated RAS gene in cancers, our data suggest that K-Ras is not required for growth in BLBC cells. This selectivity can be partially explained by the fact that there is approximately 5 times more N-Ras than K-Ras in these cells (Figure 1C). However, we and others have demonstrated that Ras proteins are not created equal, and that there may be real isoform-specific Ras activities in cancers. For example, in a colon cancer mouse model, no colon cancer can be seen when oncogenic K-RAS is replaced by knocked-in oncogenic N-RAS (Haigis et al., 2008). One model to explain the apparent Ras isoform-specific functions centers on the fact that Ras protein structures diverge greatly at the C-terminus, which controls Ras subcellular localization. This has led to the compartmentalized signaling model, whereby different Ras proteins can signal to different pools of effectors that are spatially compartmentalized in the cells (Chang and Philips, 2006; Cheng and Chang, 2011; Fehrenbacher et al., 2009). While K-Ras-4B is found almost exclusively on the plasma membrane, a substantial amount of N-Ras is in the cytoplasm. We have previously identified several cytoplasmic effectors that interact with N-Ras but not with K-Ras-4B (Zheng et al., 2012b). In this study, we show that only N-Ras, but not K-Ras-4B, can efficiently activate the cytoplasmic pool of JAK2. Considering that JAK2, as well its effectors such as STAT5, is mostly cytoplasmic, it can be most efficiently activated by the cytoplasmic pool of N-Ras without having to be first translocated to the plasma membrane. We must caution that it is inadequate to categorically suggest that K-Ras is not important for BLBCs. During the course of identifying N-Ras-dependent genes, we found that expression of CXCL1, which encodes a critical factor for linking chemotherapy resistance and metastasis (Acharyya et al., 2012), is also influenced by K-Ras, suggesting that K- and N-Ras may coordinately regulate another set of genes that are important for breast cancers.
If breast cancer can be driven by wild-type N-Ras, it is baffling why oncogenic N-RAS mutations are rare in breast cancers. Our gene expression data show that an oncogenic N-Ras influences very few genes in common with those controlled by wild-type N-Ras, such as IL8 (Table S3 and Figure S4B). Thus it is possible that an oncogenic N-Ras is not needed during the early phase of breast tumorigenesis. Alternatively, blockage in proliferation caused by oncogenic Ras-induced stresses, such as DNA damage (Grabocka et al., 2014) and MAP kinase pathway desensitization (Young et al., 2014), can be overcome by wild type Ras present in the same cell. We thus speculate that an oncogenic N-RAS may occur early during breast tumorigenesis but is later lost once tumorigenesis begins to depend on wild type Ras and other oncogenic agents.
EXPERIMENTAL PROCEDURES
The following procedures can be found in Supplemental information on line: Cell lines and cell culture method, plasmid construction, transfection, microarrays, clinical data analyses, and measurement of proteins, cell transformation, cell migration, and GTP-Ras.
Tumor formation in mice
Half a million cancer cells, either alone or mixed with HMFs, were re-suspended in Growth Factor-Reduced Matrigel (BD Biosciences) and PBS, and injected into the No. 4 and 9 mammary glands of 5–6 week old female nude mice (Harlan Laboratories). Tumor volume (mm3) was calculated by the equation: 0.5 × (length) × (width)2. Tumor formation was recorded when it was ≥30 mm3. To measure tumor growth by luciferase activity, MDA-MB-468 cells were transduced with pWPT-FLuc-RFP, and the RFP+-cells were isolated by FACS, and their luciferase activity was measured as described (Wang et al., 2015). “Total counts” were used to estimate tumor size. To block tumor growth using repertaxin, tumor-bearing mice were randomized and given repertaxin (Britschgi et al., 2012) when average tumor volume reached ≈30 mm3. DOX (0.2 mg/ml) or water was given to mice when inducible expression vectors were used. The transgenic MMTV-Wnt1 mouse experiments were performed as described (Bu et al., 2009; Li et al., 2000). Tumors were detected by palpation 3 times a week. The initial detectable size is around 2 mm in diameter, and the presence of tumor was further confirmed by monitoring its growth. The presence of all tumors was later confirmed by Harris Hematoxylin and Eosin staining.
Protein binding measured by BiFC
HT1080 cells, chosen for low autofluorescence, were co-transfected to express Yn and Yc fusion proteins, and 24 hours later, the protein binding was detected as described (Zheng and Chang, 2014). While we typically set the argon laser power at 30% when acquiring images, we needed to use up to 80% laser power when examining the weak binding between Yn-K-Ras-4B-(G12V) and Yc-JAK2.
Statistical analysis
Results from multiple measurements are usually presented as mean ± s.e.m. Data were examined by Student’s t-test unless otherwise mentioned. Human tumor databases were analyzed using the R statistical packages. All reported P values were two-sided, unless otherwise indicated. P values for the IC50 measurement were determined by Software R with the DRC package. We note that throughout this paper, *, P < 0.05; **, P < 0.01; NS, Not Significant.
Supplementary Material
HIGHLIGHTS.
N-RAS is more highly expressed in BLBCs than in other breast cancer subtypes.
N-Ras promotes tumorigenesis even in preinvasive and untransformed cells.
N-Ras binds and activates cytoplasmic JAK2, leading to efficient IL8 induction.
IL8 not only stimulates BLBC cells but may also influence stromal fibroblasts.
Acknowledgments
We thank Mark Philips and David Rowley for kindly providing research materials; Cuijuan Yu, Jun Liu, Jun Xu, Xiaomei Zhang, Frances S. Kittrell, and David G. Edwards for technical support; Gary Chamness for thoughtfully editing the manuscript; and Xiaosong Wang, Jeffrey M. Rosen, and C. Kent Osborne for discussion. The use of FACS for BiFC and shRNA was assisted by the C-BASS core, supervised by Dan Liu and supported by the NIH (P30 CA125123). General cell sorting was performed with assistance by Joel M. Sederstrom at the Cytometry and Cell Sorting Core with funding from the NIH (P30 AI036211, P30 CA125123, and S10 RR024574). We thank Margaret Goodell for general advice on the study of ES cells, and technical support by Robert Milczarek, Ping Zhang, and Anna Crane in the Pluripotential Stem Cell Core, supported by a grant from NIH (P01 GM81627). The pathology work was performed with assistance from Rena Mao and Joy Guo in the Pathology Core in the Breast Center supervised by Carolina Gutierrez. This work was also made possible by the iPlant Collaborative, funded by the NSF (#DBI-0735191). ZZ was supported by a postdoctoral fellowship from the Susan G. Komen for the Cure Foundation (PDF0707860). YL is supported by U54CA149196 and R01 CA124820. CJC and SGH are supported in part by NIH grant P30 CA125123. ECC was supported by grants from the NIH (P50-CA58183 and P50-CA186784), the Nancy Owens Memorial Foundation, the Mary Kay Ash Foundation, and the Cancer Prevention and Research Institute of Texas (CPRIT, RP130135).
C.M.P is an equity stock holder, consultant, and Board of Director Member, of BioClassifier LLC and GeneCentric Diagnostics. C.M.P is also listed an inventor on a patent application on the PAM50 assay. M.T.L is a scientific founder of, and limited partner in StemMed LTD. and a manager of StemMed Holdings LLC.
Footnotes
Author Contributions
Z.Y.Z., L.T., W.B., C.F., X.G., H.W., Y.H.L, C.J.C., Y.L., M.T.L., D.E., T.P.Z. S.G.H., C.M.P., D.M., X.H.F.Z, and E.C.C. conducted and/or designed the experiments, and Z.Y.Z. and E.C.C. wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165–178. doi: 10.1016/j.cell.2012.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Porath I, Thomson MW, Carey VJ, Ge R, Bell GW, Regev A, Weinberg RA. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat Genet. 2008;40:499–507. doi: 10.1038/ng.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billadeau D, Jelinek DF, Shah N, LeBien TW, Van Ness B. Introduction of an activated N-ras oncogene alters the growth characteristics of the interleukin 6-dependent myeloma cell line ANBL6. Cancer Res. 1995;55:3640–3646. [PubMed] [Google Scholar]
- Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- Britschgi A, Andraos R, Brinkhaus H, Klebba I, Romanet V, Muller U, Murakami M, Radimerski T, Bentires-Alj M. JAK2/STAT5 inhibition circumvents resistance to PI3K/mTOR blockade: a rationale for cotargeting these pathways in metastatic breast cancer. Cancer Cell. 2012;22:796–811. doi: 10.1016/j.ccr.2012.10.023. [DOI] [PubMed] [Google Scholar]
- Bu W, Xin L, Toneff M, Li L, Li Y. Lentivirus vectors for stably introducing genes into mammary epithelial cells in vivo. Journal of mammary gland biology and neoplasia. 2009;14:401–404. doi: 10.1007/s10911-009-9154-4. [DOI] [PubMed] [Google Scholar]
- Chang EC, Philips MR. Spatial segregation of Ras signaling: new evidence from fission yeast. Cell Cycle. 2006;5:1936–1939. doi: 10.4161/cc.5.17.3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Chang EC. Busy traveling Ras. Cell Cycle. 2011;10:1180–1181. doi: 10.4161/cc.10.8.15259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng CM, Li H, Gasman S, Huang J, Schiff R, Chang EC. Compartmentalized Ras proteins transform NIH 3T3 cells with different efficiencies. Mol Cell Biol. 2011;31:983–997. doi: 10.1128/MCB.00137-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu VK, Bivona T, Hach A, Sajous JB, Silletti J, Wiener H, Johnson RL, 2nd, Cox AD, Philips MR. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4:343–350. doi: 10.1038/ncb783. [DOI] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–352. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature. 2009;461:819–822. doi: 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MA, Bannister AJ, Saunders L, Wahab OA, Liu F, Nimer SD, Levine RL, Gottgens B, Kouzarides T, Green AR. Nuclear JAK2. Blood. 2011;118:6987–6988. doi: 10.1182/blood-2011-10-385278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher N, Bar-Sagi D, Philips M. Ras/MAPK signaling from endomembranes. Mol Oncol. 2009;3:297–307. doi: 10.1016/j.molonc.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabocka E, Pylayeva-Gupta Y, Jones MJ, Lubkov V, Yemanaberhan E, Taylor L, Jeng HH, Bar-Sagi D. Wild-type H- and N-Ras promote mutant K-Ras-driven tumorigenesis by modulating the DNA damage response. Cancer Cell. 2014;25:243–256. doi: 10.1016/j.ccr.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, et al. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman ZC, Poage GM, den Hollander P, Tsimelzon A, Hill J, Panupinthu N, Zhang Y, Mazumdar A, Hilsenbeck SG, Mills GB, Brown PH. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines IL-6 and IL-8. Cancer Res. 2013;73:3470–3480. doi: 10.1158/0008-5472.CAN-12-4524-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, Simin K, Weigman VJ, Mikaelian I, Usary J, Hu Z, Rasmussen KE, Jones LP, Assefnia S, Chandrasekharan S, et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007;8:R76. doi: 10.1186/gb-2007-8-5-r76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschkowitz JI, Zhao W, Zhang M, Usary J, Murrow G, Edwards D, Knezevic J, Greene SB, Darr D, Troester MA, et al. Comparative oncogenomics identifies breast tumors enriched in functional tumor-initiating cells. Proc Natl Aca Sci USA. 2012;109:2778–2783. doi: 10.1073/pnas.1018862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Weigman VJ, Fan C, Sawyer LR, He X, Troester MA, Sartor CI, Rieger-House T, Bernard PS, Carey LA, Perou CM. EGFR associated expression profiles vary with breast tumor subtype. BMC Genomics. 2007;8:258. doi: 10.1186/1471-2164-8-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedeszko C, Victor BC, Podgorski I, Sloane BF. Fibroblast hepatocyte growth factor promotes invasion of human mammary ductal carcinoma in situ. Cancer Res. 2009;69:9148–9155. doi: 10.1158/0008-5472.CAN-09-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, Mao S, Li Q, Sameni M, Krawetz SA, Sloane BF, Mattingly RR. RNA-Seq of human breast ductal carcinoma in situ models reveals aldehyde dehydrogenase isoform 5A1 as a novel potential target. PLoS One. 2012;7:e50249. doi: 10.1371/journal.pone.0050249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharaishvili G, Simkova D, Bouchalova K, Gachechiladze M, Narsia N, Bouchal J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer cell international. 2014;14:41. doi: 10.1186/1475-2867-14-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khramtsov AI, Khramtsova GF, Tretiakova M, Huo D, Olopade OI, Goss KH. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am J Pathol. 2010;176:2911–2920. doi: 10.2353/ajpath.2010.091125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen ES, Ertel A, Davicioni E, Kline J, Schwartz GF, Witkiewicz AK. Progression of ductal carcinoma in situ to invasive breast cancer is associated with gene expression programs of EMT and myoepithelia. Breast cancer research and treatment. 2012;133:1009–1024. doi: 10.1007/s10549-011-1894-3. [DOI] [PubMed] [Google Scholar]
- Kong G, Wunderlich M, Yang D, Ranheim EA, Young KH, Wang J, Chang YI, Du J, Liu Y, Tey SR, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. The Journal of clinical investigation. 2014;124:2762–2773. doi: 10.1172/JCI74182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Stewart S, Nagtegaal I, Luo J, Wu Y, Colditz G, Medina D, Allred DC. Differentially expressed genes regulating the progression of ductal carcinoma in situ to invasive breast cancer. Cancer Res. 2012;72:4574–4586. doi: 10.1158/0008-5472.CAN-12-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Hively WP, Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene. 2000;19:1002–1009. doi: 10.1038/sj.onc.1203273. [DOI] [PubMed] [Google Scholar]
- Mihara M, Hashizume M, Yoshida H, Suzuki M, Shiina M. IL-6/IL-6 receptor system and its role in physiological and pathological conditions. Clinical science. 2012;122:143–159. doi: 10.1042/CS20110340. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hayer KM, Brady DC, Counter CM. ELR+ CXC chemokines and oncogenic Ras-mediated tumorigenesis. Carcinogenesis. 2009;30:1841–1847. doi: 10.1093/carcin/bgp198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Podsypanina K, Li Y, Varmus HE. Evolution of somatic mutations in mammary tumors in transgenic mice is influenced by the inherited genotype. BMC Med. 2004;2:24. doi: 10.1186/1741-7015-2-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat A, Parker JS, Karginova O, Fan C, Livasy C, Herschkowitz JI, He X, Perou CM. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast cancer research: BCR. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rody A, Karn T, Liedtke C, Pusztai L, Ruckhaeberle E, Hanker L, Gaetje R, Solbach C, Ahr A, Metzler D, et al. A clinically relevant gene signature in triple negative and basal-like breast cancer. Breast Cancer Res. 2011;13:R97. doi: 10.1186/bcr3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci U S A. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparmann A, Bar-Sagi D. Ras-induced interleukin-8 expression plays a critical role in tumor growth and angiogenesis. Cancer Cell. 2004;6:447–458. doi: 10.1016/j.ccr.2004.09.028. [DOI] [PubMed] [Google Scholar]
- TCGA. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto AS, Grosschedl R, Guzman RC, Parslow T, Varmus HE. Expression of the int-1 gene in transgenic mice is associated with mammary gland hyperplasia and adenocarcinomas in male and female mice. Cell. 1988;55:619–625. doi: 10.1016/0092-8674(88)90220-6. [DOI] [PubMed] [Google Scholar]
- Wagner KU, Rui H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. Journal of mammary gland biology and neoplasia. 2008;13:93–103. doi: 10.1007/s10911-008-9062-z. [DOI] [PubMed] [Google Scholar]
- Wang H, Yu C, Gao X, Welte T, Muscarella AM, Tian L, Zhao H, Zhao Z, Du S, Tao J, et al. The osteogenic niche promotes early-stage bone colonization of disseminated breast cancer cells. Cancer cell. 2015;27:193–210. doi: 10.1016/j.ccell.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A, Lou D, McCormick F. Oncogenic and wild-type Ras play divergent roles in the regulation of mitogen-activated protein kinase signaling. Cancer Discov. 2014;3:112–123. doi: 10.1158/2159-8290.CD-12-0231. [DOI] [PubMed] [Google Scholar]
- Zhang X, Claerhout S, Prat A, Dobrolecki LE, Petrovic I, Lai Q, Landis MD, Wiechmann L, Schiff R, Giuliano M, et al. A renewable tissue resource of phenotypically stable, biologically and ethnically diverse, patient-derived human breast cancer xenograft models. Cancer Res. 2013;73:4885–4897. doi: 10.1158/0008-5472.CAN-12-4081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZY, Chang EC. A bimolecular fluorescent complementation screen reveals complex roles of endosomes in ras-mediated signaling. Methods Enzymol. 2014;535:25–38. doi: 10.1016/B978-0-12-397925-4.00002-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZY, Cheng CM, Fu XR, Chen LY, Xu L, Terrillon S, Wong ST, Bar-Sagi D, Songyang Z, Chang EC. CHMP6 and VPS4A mediate the recycling of Ras to the plasma membrane to promote growth factor signaling. Oncogene. 2012a;31:4630–4638. doi: 10.1038/onc.2011.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng ZY, Xu L, Bar-Sagi D, Chang EC. Escorting Ras. Small GTPases. 2012b;3:236–239. doi: 10.4161/sgtp.20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.