

Abstract
Objective
IBD is a group of complex, systemic disorders associated with intestinal inflammation and extraintestinal manifestations. Recent studies revealed Mendelian forms of IBD, which contributed significantly to our understanding of disease pathogenesis and the heritability of IBD.
Design
We performed exome sequencing in a family with Crohn’s disease (CD) and severe autoimmunity, analysed immune cell phenotype and function in affected and non-affected individuals, and performed in silico and in vitro analyses of cytotoxic T lymphocyte-associated protein 4 (CTLA-4) structure and function.
Results
A novel missense variant was identified in CTLA4 encoding CTLA-4, a coinhibitory protein expressed by T cells and required for regulation of T cell activation. The residue affected by the mutation, CTLA-4 Tyr60, is evolutionarily highly conserved, and the identified Y60C variant is predicted to affect protein folding and structural stability and demonstrated to cause impaired CTLA-4 dimerisation and CD80 binding. Intestinal inflammation and autoimmunity in carriers of CTLA-4 Y60C exhibit incomplete penetrance with a spectrum of clinical presentations ranging from asymptomatic carrier status to fatal autoimmunity and intestinal inflammation. In a clinically affected CTLA-4 Y60C carrier, T cell proliferation was increased in vitro and associated with an increased ratio of memory to naive T cells in vivo, consistent with impaired regulation of T cell activation.
Conclusions
Our results support the concept that variants in CTLA4 provide the basis for a novel Mendelian form of early-onset CD associated with systemic autoimmunity. Incomplete penetrance of autoimmunity further indicates the presence of other genetic and/or environmental modifiers.
INTRODUCTION
IBD is a group of complex immune-mediated disorders.1–3 While intestinal inflammation constitutes a hallmark of IBD, a wide variety of extraintestinal manifestations associated with IBD highlight the systemic character of immune dysfunction in IBD. The pathogenesis of intestinal and extraintestinal inflammation in IBD involves both host and environmental factors, but is incompletely understood. While little is known about the environmental factors involved in IBD pathogenesis, genome-wide association studies have provided significant insight into the genetic factors regulating disease susceptibility. These studies have so far revealed more than 163 genetic loci associated with IBD and have highlighted the complexity of genetic contributions to IBD.4 While intestinal inflammation in IBD is thus believed to result from complex gene–gene and gene–environment interactions, a number of rare, Mendelian forms of IBD have been described over the past several decades, which have demonstrated that alterations in single genes or pathways can elicit intestinal inflammation in a mono- or oligogenic manner.5,6 These studies have, in particular, highlighted an indispensable role of regulatory elements of the immune system in the control of intestinal and systemic inflammation. As such, mutations in FOXP3, a transcription factor critical for regulatory T cell (Treg) development, were found to be associated with autoimmunity, immunodeficiency and intestinal inflammation termed IPEX (immune dysregulation, polyendocri-nopathy, enteropathy, X linked).7,8 Similarly, mutations in IL10, a gene encoding a central immunoregulatory cytokine derived from Tregs and other T- and non-T cells,9–11 as well as variants in IL10RA and IL10RB encoding the interleukin (IL)-10 receptor subunits, are associated with severe intestinal inflammation and extraintestinal manifestations.12–16
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a coinhibitory molecule expressed by T cells, which is required for immunoregulation and prevention of inflammation.17 CTLA-4 exerts its actions through a variety of cell-autonomous and cell-non-autonomous roles. Thus, CTLA-4 shares sequence homology with the costimulatory protein CD28 and competes with CD28 for binding of its ligands CD80 and CD86 expressed by antigen-presenting cells (APCs), thereby contributing to suppression of T cell activation.17 In addition, CTLA-4 expressed by FoxP3+ Tregs inhibits interactions between conventional T cells and APCs through downmodulation of CD80 and CD86 expression by APCs.17 In accordance with a critical role of CTLA-4 in immune regulation, CTLA4 deficiency in mice is associated with massive lymphoproliferation, intestinal inflammation, autoimmunity and premature death.18–20 Similarly, antibody-mediated neutralisation of CTLA-4 in human cancer patients, while enhancing antitumour responses by T cells, is associated with colitis and autoimmunity in a significant subset of patients.21 Intriguingly, a recent report described CTLA4 variants associated with human autoimmunity and intestinal inflammation with incomplete penetrance, suggesting that genetic variants in CTLA4 may provide the basis for Mendelian forms of autoimmunity.22
Here, we report a family with a novel missense mutation in CTLA4 (CTLA-4 Y60C) associated with severe, early-onset Crohn’s disease (CD) as part of a complex autoimmune syndrome. We demonstrate that CTLA-4 Y60C affects an evolutionarily conserved residue critical for dimerisation of CTLA-4 and CD80 binding, which is associated with increased T cell activation and an increased ratio of memory to naive T cells in a clinically affected CTLA-4 Y60C carrier. Together, these results support the concept that mutations in CTLA4 can give rise to Mendelian forms of intestinal inflammation and autoimmunity.
METHODS
Patients and exome sequencing
The study was approved by the local ethics committee of Children’s Hospital Medical Center, Boston, Massachusetts, USA. All patients provided written informed consent. We performed whole exome enrichment and sequencing for the two clinically affected individuals (III.5, III.6), the mother (II.6) and the two fathers (II.5, II.9). All samples were enriched using Illumina’s TruSeq Exome Enrichment Kit and sequencing of 2×100 bp paired-end reads was performed on the Illumina HiSeq 2000. Low-stringency single nucleotide variant (SNV) calls were annotated using snpActs (http://www.snpacts.ikmb.uni-kiel.de). SNVs shared between the half-siblings were selected as SNVs of interest. The following SNVs were removed: Known single nucleotide polymorphisms (SNPs) in dbSNP130, known SNVs in our control exome data set,23 SNVs with a frequency greater than 1% in the 1000 Genomes Project data,24 SNVs with genotypes identical in the father of the respective affected child, non-coding non-splice site SNVs, synonymous coding SNVs, and SNVs not predicted to have a damaging effect by SIFT25 and PolyPhen2.26 Mutations in known artefact-prone genes27 were marked as potential artefacts. Our tool pibase V.1.4.9 was used with default settings to interrogate bam files of the two affected individuals (III.5, III.6) to generate a table of genotypes with quality annotations and further information as previously described.28 SNVs were kept only if the following quality criteria were fulfilled: stable genotype (no question mark in the ‘BestQual’ column), both alleles covered on the forward and reverse strand, and the SNV was not in regions of homology (no ‘H’ in the ‘Homology’ column). All final SNVs were manually inspected in Integrative Genomics Viewer (IGV).
To identify potential genetic modifiers, all unfiltered low-stringency SNVs with genotypes shared between the affected half-siblings (III.5, III.6) and present in the list of immunodeficiency genes29,30 were retained as potential modifying SNVs. High-quality genotypes of the half-siblings and their parents were obtained with pibase as described above. Genotypes with Mendelian errors between child and parents were manually proof-read and corrected or tagged. The sibling genotypes were annotated with our in house tool snpActs.
Sanger sequencing
Sanger sequencing was performed as described before31 using the primers and conditions described in online supplementary table S1. The SNP calling was performed using the semiautomated software novoSNP (http://www.molgen.ua.ac.be/bioinfo/novosnp/).
Flow cytometry
Peripheral blood mononuclear cell (PBMC) isolation and flow cytometry were performed as described previously.32 Briefly, PBMCs were isolated using Ficoll (BD Biosciences) and were staining with monoclonal antibodies for 20 min at 4°C. For intracellular staining, cells were first surface-stained, then permeabilised with Cytofix/Cytoperm (BD Biosciences) and washed with Perm/Wash buffer (BD Biosciences) according to the manufacturer’s instructions. Antibodies were added for 30 min at 4°C before washing with Perm/Wash buffer. Antibodies and reagents used for flow cytometry were obtained from BD Biosciences. Data were collected using a Miltenyi MACSQuant (Miltenyi Biotec) and analysed by Flowjo (Tree Star, Inc, Ashland, Oregon, USA).
Monocyte-derived dendritic cells
Monocytes were extracted from PBMCs by positive selection using CD14 magnetic beads (Miltenyi Biotec) and cultured at 1×106 cells/mL for 5 days in complete RPMI-1640 medium supplemented with 500 U/mL recombinant human IL-4 and 1000 U/mL recombinant human Granulocyte-macrophage-colony stimulating factor (GM-CSF) (PeproTech) as described before.31
Cell lines, CTLA-4 constructs, transfection and western blotting
HEK293 cells were cultured in RPMI-1640 containing 10% fetal bovine serum (FBS). wildtype (WT) CTLA-4 cDNA was obtained from Open Biosystems (GeneBank: BC074842). To obtain Y60C CTLA-4, the point mutation c.179A>G was introduced using the QuikChange II Site-Directed Mutagenesis Kit (Agilent, Santa Clara, California, USA). WT and Y60C cDNA were subcloned into pcDNA3.1 (Invitrogen) and the cDNA sequence was confirmed. Constructs were transfected into HEK293 cells using TansitLT1 (MirusBio, Madison, Wisconsin, USA) according to the manufacturer’s instruction. Western blotting was performed as described previously33 using the following antibodies: goat-anti-human CTLA-4 (R&D Biosystems, Cat: AF-386-PB) and mouse-anti-human β-actin (Sigma-Aldrich, clone AC-15).
CD80-Fc binding assay
HEK293 cells were transfected with WT CTLA, CTLA-4 Y60C or empty plasmid (pcDNA3.1). After 24 h, recombinant, HIS-tagged CD80-Fc fusion protein (R&D systems) was added at a final concentration of 2 μg/mL for 20 min at 4°C before detection by Penta-His Alexa fluor 488 (Qiagen).
Allele specific expression analysis
RNA was extracted from 5×106–1×107 PBMCs using the RNeasy Plus kit (Qiagen, Hilden, Germany). cDNA was generated using Superscript (Life Technologies, Carlsbad, California, USA). To generate standards for allele-specific gene expression, WT and Y60C CTLA-4 constructs (see above) were combined at predefined ratios. cDNA and standards were then subjected to genotyping using a custom Taqman genotyping assay (Applied Biosystems, Foster City, California, USA). The logarithm of the ratio of transcripts was plotted against the logarithm of the ratio between VIC and FAM,34 and the allele-specific expression of cDNA samples was determined (see online supplementary figure S1).
Functional Treg assay
CD4+ CD25+and CD4+CD25− T cells were separated using the CD4+CD25+ Regulatory T Cell Isolation Kit (Miltenyi Biotec) according to the manufacturer’s instructions. 3×103 CD4+CD25− T cells were cultured in 96-well round bottom plates in the presence of the indicated amount of autologous CD4+CD25+ Tregs, 3×104 T cell-depleted, irradiated, autologous feeders, and soluble anti-human CD3 (5 μg/mL; OKT3) and anti-human CD28 (5 μg/mL; CD28.2).35 After 5 days of culture, 3H-thymidine was added and incorporation was determined after 16 h as described previously.32
Statistical analysis
Statistical testing was performed using the unpaired Student’s t test. For multiple comparisons against the same control, one-way analysis of variance with Dunnett’s post hoc test was applied. p Values were calculated in a two-tailed manner.
RESULTS
A family with severe, early-onset CD and autoimmunity
The two index patients (III.5 and III.6), half-siblings from two unrelated and asymptomatic fathers (II.5, II.9) and an asymptomatic mother (II.6), both exhibited early-onset CD and severe autoimmunity (figure 1). Patient III.6 exhibited a severe multi-system autoimmune disorder associated with CD, type 1 diabetes mellitus (T1DM), autoimmune hypothyroidism, demyelinating encephalopathy, fibrosing interstitial lung disease, chronic sinusitis, suspected primary sclerosing cholangitis, hypogammaglobulinaemia and intermittent haematological cytopenias including red cell aplasia, for which she was intermittently transfusion-dependent. CD, classified A1b, L3, L4a, B1, G1 according to the Paris classification,36 was diagnosed at 13 years of age with gastric, small intestinal and colonic involvement. Her CD was characterised by chronic moderately active duodenitis with villous atrophy, crypt hyperplasia and absent Paneth and goblet cells, severe chronic gastritis, chronic severely active ileitis and chronic severely active colitis with increased enterocyte apoptosis and lymphocytic inflammation. Despite an aggressive treatment course that included mesalamine, oral and pulse steroids, 6-mercaptopurine, cyclophosphamide, rituximab, tacrolimus, infliximab and sirolimus, her enterocolitis was refractory and worsened significantly over time. Her symptoms consisted of growth failure, abdominal pain and high-output diarrhoea, ultimately leading to a dependence on parental nutrition for nutrition and fluid/electrolyte management. She had multiple extended hospitalisations for treatment and symptom management of her multisystem autoimmune disease and related complications including several episodes of bacterial sepsis, fungal sepsis, recurrent Clostridium difficile colitis and recurrent herpes zoster infection. At the age of 24 years, she developed methicillin-resistant Staphylococcus aureus bacteraemia with septic emboli, became critically ill with septic shock, acute respiratory distress syndrome and renal insufficiency. Her care was advanced to comfort measures only and she died. At autopsy, diffuse alveolar damage and multifocal lung pallor suggestive of pneumonia as well as acute tubular necrosis and pitted renal cortical surfaces, possibly representing emboli, were noted. Additional findings included fibrous pleural adhesions, hepatosplenomegaly, adrenal cortical atrophy, cardiomegaly and diffuse fatty replacement of the pancreas, consistent with the known diagnosis of T1DM.
Figure 1.
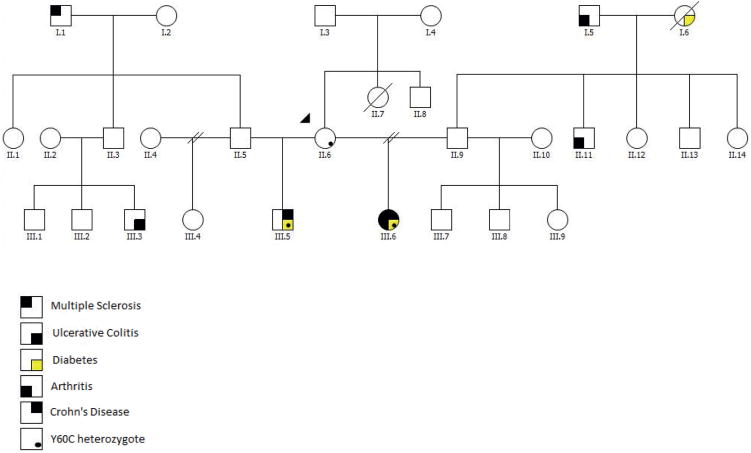
The pedigree of the family and disease manifestation.
Her half-brother, patient III.5, was diagnosed with CD at 7 years of age in the setting of early-onset T1DM (age of diagnosis 22 months). Shortly thereafter, he developed autoimmune haemolytic anaemia, neutropenia and thrombocytopenia. Pulmonary nodules and an inflammatory anterior mediastinal mass were discovered by CT scan. His CD was characterised by gastric, small intestinal and colonic involvement and was classified A1a, L3, L4a, B1, G1 according to the Paris classification.36 Despite aggressive treatment consisting of mesalamine, rituximab, oral and pulse dose steroids, 6-mercaptopurine, methotrexate, mycophenolate mofetil and tacrolimus, he developed severe proctocolitis refractory to treatment requiring partial colectomy at 14 years of age. Histopathological findings during this period were remarkable for severe chronic active gastritis with intestinal metaplasia, duodenal intraepithelial lymphocytosis and moderate villous atrophy, marked intraepithelial lymphocytosis and complete villous atrophy of the terminal ileum, moderate active colitis with lymphocytosis of the caecum, and ascending, transverse and descending colon with chronically severely active rectal colitis with ulcerations. Post-resection, he responded to infliximab and budesonide with improvement in his enterocolitis and haematological cytopenias. However, his longstanding treatment refractory atrophic gastritis progressed from intestinal metaplasia and recurrent polyps to multifocal gastric adenocarcinoma at 17 years of age. His treatment consisted of a total gastrectomy with extensive tumour-negative lymph node resection. Subsequent serial imaging has been negative for cancer recurrence. At 21 years of age, he is currently working and attending college.
The patients’ mother, II.6, has not reported any symptoms related to autoimmunity. A CT scan performed several years ago as part of an appendicitis evaluation revealed lung nodules as an incidental finding, but a further medical investigation was not performed and she remains well.
Exome sequencing identifies a novel missense variant in CTLA4
We performed whole exome sequencing for the two clinically affected individuals (III.5, III.6), the mother (II.6) and the two fathers (II.5, II.9). The average sequencing coverage ranged between 39-fold and 52-fold with over 87% of the target covered by more than eightfold in all samples (see online supplementary table S2). Overall, 25 481 low-stringency variant-calls with the same genotype in both half-siblings were identified. After deleting low-stringency SNV calls listed in dbSNP130, identified in our control exome data set23 or with a frequency greater than 1% in the 1000 Genomes Project data,24 2029 low-stringency rare variants remained. After removing alleles that were present in the respective fathers (II.5, II.9) of the clinically affected individuals (III.5, III.6), 1388 low-stringency SNV calls remained, of which 195 were non-synonymous and remained after applying SIFT and PolyPhen2 filtering as described in Methods section. After quality-filtering with pibase,28 14 SNVs present in both affected individuals (III.5, III.6) remained, all of which were inherited from the mother (II.6) (table 1). Among these candidate genes, we did not further investigate SNVs in genes with poorly characterised or unknown function (ARRDC2, ARMC6, CPXM1, DHRS11, KLHDC4, LHPP, POFUT2, RBMS2), restricted tissue expression unlikely to contribute to the observed clinical phenotype (LGALS12 in adipocytes, PRND in testis, LECT2 in liver) or known associations with clinical phenotypes not observed here (SLC26A2 and diastrophic dysplasia37). Among the remaining two candidate genes, CTLA4 and FURIN (see Discussion section for further information), we focused on CTLA4 since the encoded protein acts as a coinhibitory protein expressed by T cells, whose antibody-mediated neutralisation in humans and deletion in mice are associated with intestinal inflammation and autoimmunity.18–21 CTLA4 c.179A>G (Tyr60Cys, Y60C) was detected as a novel, de novo, heterozygous missense mutation in the mother (II.6), which was inherited to both clinically affected individuals (III.5, III.6) and not shared with the fathers (II.5, II.9), as confirmed by Sanger sequencing (see online supplementary figure S2). In two CTLA-4 Y60C carriers with available cDNA from PBMCs (II.6, III.5), we confirmed equal expression of WT and Y60C CTLA-4 by an allele-specific assay34 in accordance with bi-allelic expression and a heterozygous state of the mutation (see online supplementary table S3). While these assays were performed with total PBMCs and not with sorted T cells or Tregs, they suggest that epigenetic processes such as imprinting are unlikely to contribute to incomplete disease penetrance in the mother. Further, several individuals in the family also harboured common SNPs in CTLA4 (see online supplementary table S4), which included, in particular, CTLA4 c.49A>G, an SNP previously demonstrated to be associated with T1DM, Graves’ disease and Hashimoto thyroiditis.38–40 However, these common CTLA4 SNPs were shared between the clinically affected individuals (III.5, III.6), the unaffected mother (II.6) and partially with other unaffected family members (II.5 and II.9), and are thus unlikely to explain incomplete disease penetrance in the mother (see online supplementary table S4).
Table 1.
Potentially deleterious single nucleotide variants (SNVs) in the affected individuals (III.5, III.6) that are not present in the unaffected fathers (II.5, II.9)
| Chr | Position | Ref. Base | Var. Base | Mut type | Gene symbol | AA Subst | PhyloP44 | Grant-ham | pph2 Prob. | SIFT score |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 204 443 623 | A | G | het | CTLA4 | Y60C | 0.67 | 194 | 1.00 | 0.00 |
| 5 | 135 314 912 | C | T | het | LECT2 | G63E | 4.06 | 98 | 1.00 | 0.00 |
| 5 | 149 340 131 | C | G | het | SLC26A2 | S261C | 5.42 | 112 | 1.00 | 0.01 |
| 10 | 126 162 711 | C | T | het | LHPP | R47W | 1.09 | 101 | 0.94 | 0.03 |
| 11 | 63 039 655 | C | T | het | LGALS12 | T253I | −0.37 | 89 | 0.88 | 0.02 |
| 12 | 55 268 352 | C | T | het | RBMS2 | P357L | 3.32 | 98 | 0.92 | 0.00 |
| 15 | 89 221 193 | G | A | het | FURIN | G146S | 5.00 | 56 | 0.98 | 0.01 |
| 16 | 86 321 735 | A | C | het | KLHDC4 | S175A | 2.83 | 99 | 0.61 | 0.01 |
| 17 | 32 025 571 | T | A | het | DHRS11 | C69S | 4.50 | 112 | 1.00 | 0.01 |
| 19 | 17 981 660 | G | A | het | ARRDC2 | V216M | 0.75 | 21 | 0.99 | 0.01 |
| 19 | 19 029 430 | C | T | het | ARMC6 | A475V | 1.40 | 64 | 0.99 | 0.00 |
| 20 | 2 725 075 | G | A | het | CPXM1 | R354C | 6.47 | 180 | 1.00 | 0.00 |
| 20 | 4 653 387 | C | T | het | PRND | R64C | 0.56 | 180 | 1.00 | 0.00 |
| 21 | 45 521 464 | C | T | het | POFUT2 | A243T | 4.91 | 58 | 0.99 | 0.01 |
Only novel SNVs with in silico predicted damaging effects (PolyPhen probability >0.50 and SIFT score ≤0.05 etc) are shown. No homozygous or compound heterozygous variants applying to these criteria were identified. For further details on SNV filtering, see the Methods section. Positions are according to hg18.
Highlighted in bold is the CTLA4 variant discussed in the manuscript.
Chr, chromosome; Ref., reference; Var., variant; Mut., mutation; AA subst., amino acid substitution; pph2 Prob., PolyPhen 2 probability.
CTLA-4 Y60C affects an evolutionarily conserved residue critical for CTLA-4 dimerisation and binding of its ligand CD80
CTLA-4 Tyr60 is an evolutionarily highly conserved, aromatic and hydrophobic residue in the core of the extracellular Ig-like domain of CTLA-4 (figure 2A, B). The Tyr60 residue packs tightly against the side-chains of Val69, Val131 and Leu133 (figure 2B), thus contributing to the hydrophobic core of the Ig-domain and presumably stabilising the protein conformation. The mutation Y60C replaces aromatic Tyr with the much smaller and polar Cys residue and is thus likely to affect the structure of CTLA-4. Accordingly, CTLA-4 Y60C, compared with WT CTLA-4, exhibited impaired dimerisation under non-reducing conditions, while total protein expression as determined under reducing conditions showed only a minor decrease (figure 2C). Upon ectopic expression in HEK293 cells, CTLA-4 Y60C, in the absence of WT CTLA-4, exhibited considerably decreased cell surface expression, while intracellular CTLA-4 Y60C expression was less affected in accordance with western blot results (figure 3). Importantly, despite residual cell surface expression of CTLA-4 Y60C, binding of its ligand CD80 was abolished. These data are consistent with structural in silico predictions and confirm that CTLA-4 Y60C exhibits impaired cell surface expression and is unable to bind its ligand CD80.
Figure 2.
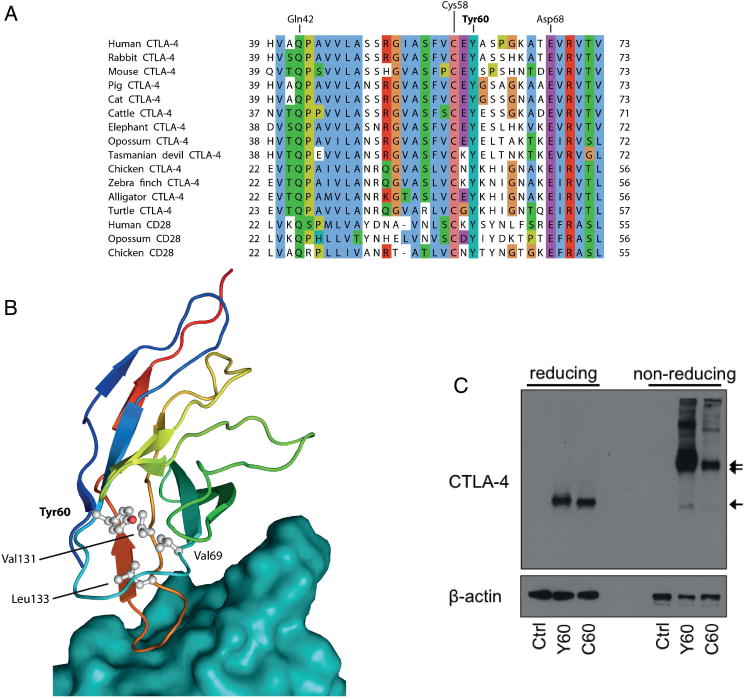
CTLA-4 Tyr60 is an evolutionarily conserved residue critical for CTLA-4 structural stability and dimerisation. (A) A multiple sequence alignment of human CTLA-4 (residues 39–73) and CD28 with homologues from other mammals, birds and reptiles shows Tyr60 to be conserved in both paralogs in amniotes. Residue numbering is shown at the line ends and, for selected conserved human CTLA-4 residues, above the alignment. All sequences are from RefSeq and identifiers are listed in online supplementary table S6. (B) The protein structure of CTLA-4 in complex with CD80 (model based on53) shows Tyr60 (grey) to be located at the core of CTLA-4 (rainbow colouring) not far from the surface that binds CD80 (teal). Tyr60 packs tightly against the side-chains of Val69, Val131 and Leu133 (ball and stick rendering), contributing to the hydrophobic core of the Ig-like domain and stabilising the protein fold. (C) Western blot of CTLA-4 and β-actin under reducing and non-reducing conditions, as indicated, in HEK293 cells after transfection of WT (Tyr60) or mutant (Cys60) CTLA-4 or empty control plasmid. The CTLA-4 monomer is indicated by one arrow, the dimer by two arrows.
Figure 3.
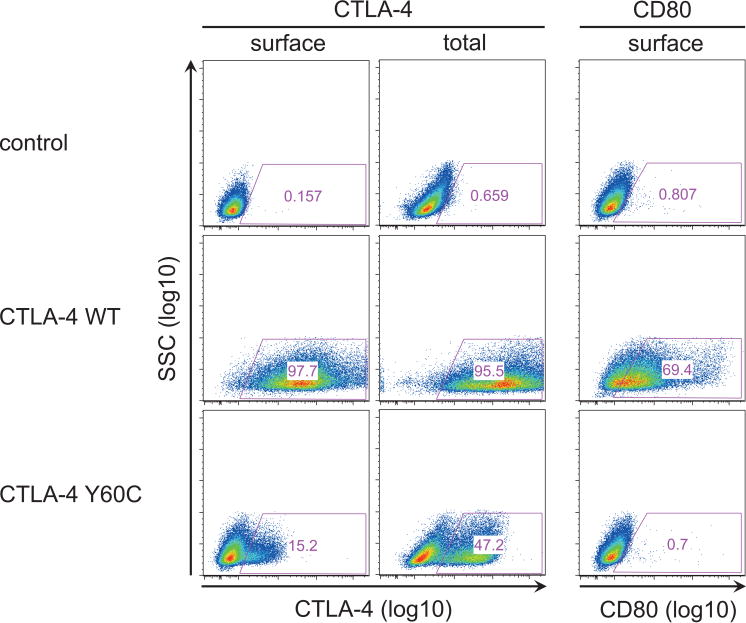
CTLA-4 Y60C is associated with impaired binding of its ligand CD80. CTLA-4 expression and CD80 binding as determined by flow cytometry in HEK293 cells after transfection of WT CTLA-4, Y60C CTLA-4 and empty plasmid (control). Total expression of CTLA-4 reflects combined surface and intracellular staining after cell permeabilisation.
Surface and intracellular expression of CTLA-4 by primary CD4+CD25+FoxP3+ and CD4+CD25− FoxP3− T cells were indistinguishable between homozygous carriers of WT CTLA-4 and heterozygous carriers of Y60C (see online supplementary figure S3), suggesting that an increased state of T cell activation (see below), an increase in WT CTLA-4 protein expression and/or a chaperoning function of WT CTLA-4 lead to unimpaired overall CTLA-4 expression in primary T cells of heterozygous CTLA-4 Y60C carriers.
Increased T cell proliferation in the clinically affected CTLA-4 Y60C carrier
A comprehensive phenotypic analysis of PBMCs was performed in patient III.5, his father (II.5) and his mother (II.6), while patient III.6 died early during the course of this investigation with only limited phenotypic data available. Both clinically affected carriers of CTLA-4 Y60C (III.5, III.6) exhibited a decrease in B cell and natural killer (NK) cell numbers (see online supplementary figure S4A and table S5). In patient III.5 (data from patient III.6 are not available), CD4+ T cells, but not CD8+ T cells, contained an increased percentage of CD45RO+ memory T cells and a corresponding decrease in naive T cells expressing CD62L (figure 4A, B). Interestingly, the patient’s clinically unaffected mother, expressing the CTLA-4 Y60C variant, exhibited a less pronounced phenotype with an increased ratio of memory to naive T cells but no alterations in relative and absolute numbers of B cells and NK cells (figure 4A, B, online supplementary figure S4A, and data not shown). The expression of costimulatory molecules by T cells, monocytes, monocyte-derived dendritic cells and B cells was not affected in CTLA-4 Y60C carriers (see online supplementary figure S4C–E). Similarly, the number of Foxp3+ CD25+CD4+ Tregs and the expression of other Treg markers such as latency-associated peptide (LAP) and glucocorticoid-induced TNFR family related gene (GITR) were indistinguishable between CTLA-4 WT and Y60C carriers. However, T cells obtained from the clinically affected carrier of CTLA-4 Y60C (III.5) showed hyperproliferation of naive T cells in response to antibody-mediated T cell receptor crosslinking in vitro, both in the absence and the presence of autologous Tregs (figure 4D), suggesting a defect which extends beyond Tregs to naive T cells. Of note, despite sharing the CTLA-4 Y60C variant, the patient’s mother (II.6) exhibited unimpaired T cell proliferation in vitro in accordance with the absence of clinically overt autoimmunity.
Figure 4.
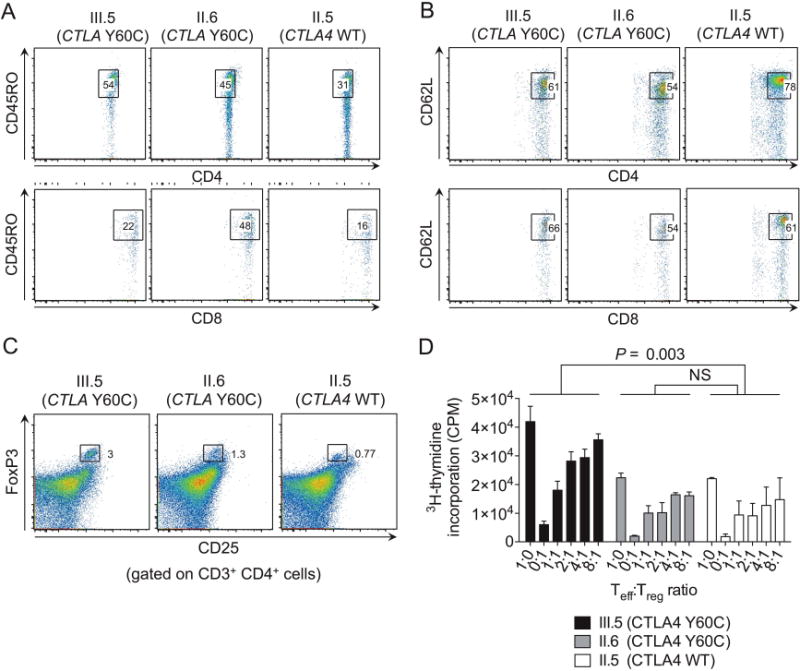
T cell phenotype and function in carriers of WT and Y60C CTLA-4. (A and B) Percentage of CD45RO+ (A) and CD62L+ (B) cells among CD4+ (upper panel) and CD8+ T cells (lower panel). (C) Percentage of Tregs among CD3+ CD4+ T cells as determined by flow cytometry. (D) T cell proliferation as determined by 3H-thymidine incorporation. In (D), mean±SEM of triplicates are shown. Statistical analysis was performed using paired, one-way analysis of variance followed by Dunnett’s post hoc test.
Investigation of potential genetic modifiers responsible for incomplete disease penetrance
To identify potential genetic modifiers responsible for incomplete disease penetrance in the mother carrying CTLA4 Y60C, we filtered, in the exomes of the kindred, variants with a genotype shared between both affected children in 207 genes with known association with immunodeficiency (see Methods section). This revealed 208 low-stringency candidate SNVs, of which 113 were validated as high-quality SNVs. After exclusion of synonymous variants and filtering for variants with a genotype shared between the affected children but not with the unaffected mother, 18 SNVs were detected in 13 genes (see online supplementary table S7). This included missense variants in eight genes (BLM, C6, CD8B, DOCK8, IL7R, SERPING1, RNF168, STXBP2), none of which were predicted in silico to be damaging (pph2 >0.50 and SIFT score ≤0.05). In addition, one intronic SNV was found in CD8B and variants in the 3′ untranslated region were found in five genes (GFI1, IL10, IL10RB, TMC8, GTF2H5). While SNVs in TMC8 and GTF2H5 are associated with phenotypically unrelated diseases (epidermodysplasia verruciformis and trichothiodystrophy, respectively) and are thus unlikely to act as modifiers, mutations in IL10 and IL10RB are associated with early-onset IBD12–16 and the observed variants in these genes may thus influence the susceptibility to IBD in the described kindred. Although all four variants in IL10 and IL10RB are common, allele G of marker rs3024496 in IL10 was reported to positively correlate with blood IL-10 concentrations41 and was homozygous (GG) in the unaffected mother and heterozygous (AG) in the affected children. As such, it is possible that rs3024496 contributes to incomplete disease penetrance in the mother.
DISCUSSION
Rare Mendelian forms of IBD have provided unique insight into the pathogenesis of IBD and have revealed that alterations in single genes or pathways critical for epithelial barrier function as well as innate and adaptive immunity are associated with intestinal inflammation, which in these cases typically manifests in association with systemic autoimmunity and immunodeficiency.5,6 Here, we report a family with severe, early-onset CD and autoimmunity associated with a missense mutation in CTLA4. These data suggest that variants in CTLA4, similar to those in FOXP3, IL10, IL10RA and IL10RB, promote intestinal and systemic inflammation through dysfunction of a central axis of regulatory immunity.
Clinical and immunological findings in CTLA-4 Y60C carriers reported here resemble those recently reported for patients with CTLA4 mutations,22 which further supports the concept of a causal relationship between CTLA-4 variants and autoimmunity. Patients in both cohorts showed autoimmunity of incomplete penetrance with predominant inflammatory affection of the intestine, lung and brain, autoimmune anaemia and thrombocytopenia, and immunodeficiency associated with opportunistic, and often fatal, infections. Similarly, an overlapping immunological phenotype was described with T cell hyperproliferation, an expansion of memory Tcells, and deficiency in B cells and NK cells. Furthermore, in both reports, asymptomatic carriers of CTLA4 mutations showed less pronounced phenotypic and functional alterations in cellular immunity compared with clinically affected individuals, which suggests a potential mechanistic involvement of these factors in prevention of overt autoimmunity. Alternatively, differences in T cell phenotype and function between affected and unaffected carriers of CTLA4 variants may represent a consequence, rather than cause, of the presence or absence of inflammation.
Fusion proteins of CTLA-4 including abatacept and belatacept are approved for the treatment of rheumatoid arthritis and the prevention of kidney transplant rejection, respectively. While abatacept failed to show efficacy in moderate-to-severe CD and UC,42 it is possible that CTLA-4 fusion proteins could provide beneficial effects in patients with autoimmunity and CTLA4 variants through provision of functional CTLA-4. This could serve as an approach of personalised medicine to inhibit CD28 signalling in the presence of genetic CTLA4 deficiency. It is noteworthy that the patients identified here were heterozygous carriers of CTLA4 variants suggesting that expression of the unaffected CTLA4 allele was not sufficient to prevent autoimmunity and intestinal inflammation. It is therefore possible that dominant negative effects of CTLA4 variants and/or the presence of genetic or environmental modifiers limit the efficacy of the functional allele to operate, making CTLA-4 fusion proteins of potential interest to study in patients with similar types of genetic mutations.
Our data highlight a number of remarkable features of autoimmunity related to CTLA-4 variants, which also illustrates the challenges of linking exome data to clinical phenotypes. Thus, CTLA-4 Y60C carriers exhibited incomplete penetrance of autoimmunity and a broad spectrum of clinical manifestations. In addition, proliferative T cell responses varied among carriers of CTLA-4 Y60C. While both findings may be surprising at first glance, they are consistent with observations made by Kuehn et al22 who reported several asymptomatic carriers of CTLA4 variants as well as affected individuals with unimpaired Treg function in vitro. The latter is consistent with unimpaired immunoregulatory function of Ctla4-deficient murine Tregs in vitro43–45 and supports the concept of compensatory pathways, which contribute to unaltered function of CTLA-4-deficient Tregs in vitro and may provide the basis for incomplete penetrance of autoimmunity in vivo. Of note, incomplete penetrance is not uncommon in autosomal-dominant disorders.46 In addition, incomplete disease penetrance is representative of the vast majority of Mendelian forms of IBD pointing towards the presence of genetic and/or environmental modifiers in individual regulation of disease susceptibility.5,6 To identify potential genetic modifiers, we first examined the CTLA4 gene for additional SNPs given that common CTLA4 variants were previously shown to be associated with IBD and autoimmunity.38–40,40a,40b Several individuals in the family harboured common CTLA4 SNPs including CTLA4 c.49A>G, a SNP previously demonstrated to be associated with T1DM, Graves’ disease and Hashimoto thyroiditis.38–40 However, the contribution of CTLA4 c.49A>G to T1DM and autoimmune hypothyroidism in the affected family members remains unknown as this SNV was previously rejected as causal SNP and yielded inconsistent results in terms of its effect on T cell proliferation.22,47–49 Furthermore, common CTLA4 SNPs were shared between clinically affected individuals, the unaffected mother and partially with other unaffected family members, and are thus unlikely to explain incomplete disease penetrance in the mother. In another approach to identify potential genetic modifiers, we filtered SNVs in genes known to be associated with immunodeficiency and with genotypes shared between the affected half-siblings but not with the unaffected mother. This approach identified four SNVs in IL10 and IL10RB, two genes in which mutations are associated with early-onset IBD.12–16 Allele G of marker rs3024496 in IL10, which was found to be homozygous in the unaffected mother and heterozygous in the affected children, was reported to positively correlate with IL-10 concentrations in blood.41 It is thus possible that this variant may contribute to protection from autoimmunity and intestinal inflammation in the mother, which warrants further investigation. Finally, while shared between the affected half-siblings (III.5, III.6) and their mother (II.6), a SNV located in FURIN may contribute to the observed clinical phenotype. Furin is a proprotein convertase involved in the processing of regulatory transforming growth factor β 1 (TGF-β1) whose genetic deletion in murine CD4+ T cells is associated with impaired TGF-β1 production, altered Treg function and spontaneous intestinal inflammation.50 As such, it is possible that the observed SNV in FURIN contributes to the pathogenesis of intestinal inflammation in the affected individuals, particularly in the presence of CTLA-4 Y60C.
Recent technical developments such as next generation sequencing have considerably facilitated the discovery of Mendelian forms of immune-mediated disorders. However, despite significant advances in the filtering of genetic results, a wealth of SNVs remaining after filtering often prevents the unambiguous identification of variants linked to disease pathogenesis. It is thus important to note that studies in genetically modified mice have largely facilitated the identification of disease-related genetic variants in approaches of exome sequencing as they provided mechanistic evidence for a link between individual genetic defects and disease manifestation. It is remarkable in this context that the description of intestinal inflammation and autoimmunity in mice deficient for Il10, Il10rb and Ctla4 preceded the identification of similar human defects by years, which highlights the critical role of animal studies in approaches to delineate the mechanistic basis of human diseases.18–20,51,52
Together, our data support the burgeoning data that variants in CTLA-4 provide the basis for autosomal-dominant Mendelian forms of early-onset CD and autoimmunity. Future work is required to delineate environmental and genetic modifiers responsible for incomplete disease penetrance in this disorder.
Supplementary Material
Significance of this study.
What is already known on this subject?
► Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a coinhibitory protein required for regulation of T cell activation.
► CTLA4 deficiency in mice is associated with fatal lymphoproliferation, intestinal inflammation and autoimmunity.
► Neutralisation of CTLA-4 by monoclonal antibodies in human cancer is associated with intestinal inflammation and autoimmunity.
What are the new findings?
► We provide evidence for a primary role of CTLA4 mutations in the pathogenesis of Crohn’s disease (CD) and autoimmunity through the demonstration of a Mendelian form of severe multi-system inflammation associated with a missense mutation in CTLA4.
► The identified, novel CTLA-4 variant affects an evolutionarily conserved residue of CTLA-4, is predicted to affect protein folding, and is demonstrated to cause impaired CTLA-4 dimerisation and CD80 binding.
► Autoimmunity associated with CTLA-4 Y60C exhibits incomplete disease penetrance.
How might it impact on clinical practice in the foreseeable future?
► Screening for CTLA-4 variants should be offered to individuals with early-onset CD and autoimmunity.
Acknowledgments
We are deeply indebted to and grateful for the help and support of the family members who contributed to this study. The authors thank Sandra May (Kiel, Germany) for help with Sanger sequencing. This work was supported by: NIH grants DK044319, DK051362, DK053056 and DK088199; the Harvard Digestive Diseases Center (HDDC) (DK0034854) (RSB); the Deutsche Forschungsgemeinschaft (DFG) (ZE 814/4-1, ZE 814/5-1, ZE 814/6-1); and the European Research Council (ERC Starting Grant agreement n° 336528) (SZ). Infrastructure support was received from the PopGen biobank and the DFG Cluster of Excellence 306 ‘Inflammation at Interfaces’.
Footnotes
Additional material is published online only. To view please visit the journal online (http://dx.doi.org/10.1136/gutjnl-2014-308541).
Contributors SZ, MT, EM, EH-C and SKD performed functional experiments. B-SP, BS and MF performed genetic analyses. JKL performed in silico analyses of CTLA-4 Y60C. SS contributed to study coordination. DW and AML contributed clinical information and provided patient samples. SZ, AF and RSB designed the study, coordinated the experimental work and wrote the manuscript with input from coauthors. All authors discussed the results and commented on the manuscript.
Competing interests None.
Ethics approval Ethics Committee of Children’s Hospital Boston.
Provenance and peer review Not commissioned; externally peer reviewed.
References
- 1.Khor B, Gardet A, Xavier RJ. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–17. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- 4.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uhlig HH. Monogenic diseases associated with intestinal inflammation: implications for the understanding of inflammatory bowel disease. Gut. 2013;62:1795–805. doi: 10.1136/gutjnl-2012-303956. [DOI] [PubMed] [Google Scholar]
- 6.Uhlig HH, Schwerd T, Koletzko S, et al. COLORS in IBD Study Group and NEOPICS The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology. 2014 doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bennett CL, Christie J, Ramsdell F, et al. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet. 2001;27:20–1. doi: 10.1038/83713. [DOI] [PubMed] [Google Scholar]
- 8.Wildin RS, Ramsdell F, Peake J, et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet. 2001;27:18–20. doi: 10.1038/83707. [DOI] [PubMed] [Google Scholar]
- 9.Chaudhry A, Samstein RM, Treuting P, et al. Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity. 2011;34:566–78. doi: 10.1016/j.immuni.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rubtsov YP, Rasmussen JP, Chi EY, et al. Regulatory T cell-derived interleukin-10 limits inflammation at environmental interfaces. Immunity. 2008;28:546–58. doi: 10.1016/j.immuni.2008.02.017. [DOI] [PubMed] [Google Scholar]
- 11.Olszak T, Neves JF, Dowds CM, et al. Protective mucosal immunity mediated by epithelial CD1d and IL-10. Nature. 2014;509:497–502. doi: 10.1038/nature13150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glocker EO, Frede N, Perro M, et al. Infant colitis—it’s in the genes. Lancet. 2010;376:1272. doi: 10.1016/S0140-6736(10)61008-2. [DOI] [PubMed] [Google Scholar]
- 14.Kotlarz D, Beier R, Murugan D, et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: implications for diagnosis and therapy. Gastroenterology. 2012;143:347–55. doi: 10.1053/j.gastro.2012.04.045. [DOI] [PubMed] [Google Scholar]
- 15.Moran CJ, Walters TD, Guo CH, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–23. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Begue B, Verdier J, Rieux-Laucat F, et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am J Gastroenterol. 2011;106:1544–55. doi: 10.1038/ajg.2011.112. [DOI] [PubMed] [Google Scholar]
- 17.Wing K, Yamaguchi T, Sakaguchi S. Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol. 2011;32:428–33. doi: 10.1016/j.it.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 18.Chambers CA, Cado D, Truong T, et al. Thymocyte development is normal in CTLA-4-deficient mice. Proc Natl Acad Sci USA. 1997;94:9296–301. doi: 10.1073/pnas.94.17.9296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tivol EA, Borriello F, Schweitzer AN, et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 20.Waterhouse P, Penninger JM, Timms E, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 21.Beck KE, Blansfield JA, Tran KQ, et al. Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol. 2006;24:2283–9. doi: 10.1200/JCO.2005.04.5716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuehn HS, Ouyang W, Lo B, et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science. 2014;345:1623–7. doi: 10.1126/science.1255904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stade B, Seelow D, Thomsen I, et al. GrabBlur—a framework to facilitate the secure exchange of whole-exome and -genome SNV data using VCF files. BMC Genomics. 2014;15(Suppl 4):S8. doi: 10.1186/1471-2164-15-S4-S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abecasis GR, Altshuler D, Auton A, et al. 1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–73. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–14. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuentes Fajardo KV, Adams D, Program NCS, et al. Detecting false-positive signals in exome sequencing. Hum Mutat. 2012;33:609–13. doi: 10.1002/humu.22033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forster M, Forster P, Elsharawy A, et al. From next-generation sequencing alignments to accurate comparison and validation of single-nucleotide variants: the pibase software. Nucleic Acids Res. 2013;41:e16. doi: 10.1093/nar/gks836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Notarangelo LD, Fischer A, Geha RS, et al. International Union of Immunological Societies Expert Committee on Primary Immunodeficiencies Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeissig Y, Petersen BS, Milutinovic S, et al. XIAP variants in male Crohn’s disease. Gut. 2014 doi: 10.1136/gutjnl-2013-306520. [DOI] [PubMed] [Google Scholar]
- 32.Zeissig S, Dougan SK, Barral DC, et al. Primary deficiency of microsomal triglyceride transfer protein in human abetalipoproteinemia is associated with loss of CD1 function. J Clin Invest. 2010;120:2889–99. doi: 10.1172/JCI42703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA. 2009;106:14524–9. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo HS, Wang Z, Hu Y, et al. Allelic variation in gene expression is common in the human genome. Genome Res. 2003;13:1855–62. doi: 10.1101/gr.1006603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Walker MR, Kasprowicz DJ, Gersuk VH, et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+CD25− T cells. J Clin Invest. 2003;112:1437–43. doi: 10.1172/JCI19441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levine A, Griffiths A, Markowitz J, et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: the Paris classification. Inflamm Bowel Dis. 2011;17:1314–21. doi: 10.1002/ibd.21493. [DOI] [PubMed] [Google Scholar]
- 37.Hastbacka J, de la Chapelle A, Mahtani MM, et al. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell. 1994;78:1073–87. doi: 10.1016/0092-8674(94)90281-x. [DOI] [PubMed] [Google Scholar]
- 38.Donner H, Braun J, Seidl C, et al. Codon 17 polymorphism of the cytotoxic T lymphocyte antigen 4 gene in Hashimoto’s thyroiditis and Addison’s disease. J Clin Endocrinol Metab. 1997;82:4130–2. doi: 10.1210/jcem.82.12.4406. [DOI] [PubMed] [Google Scholar]
- 39.Marron MP, Raffel LJ, Garchon HJ, et al. Insulin-dependent diabetes mellitus (IDDM) is associated with CTLA4 polymorphisms in multiple ethnic groups. Hum Mol Genet. 1997;6:1275–82. doi: 10.1093/hmg/6.8.1275. [DOI] [PubMed] [Google Scholar]
- 40.Nistico L, Buzzetti R, Pritchard LE, et al. The CTLA-4 gene region of chromosome 2q33 is linked to, and associated with, type 1 diabetes. Belgian Diabetes Registry. Human molecular genetics. 1996;5:1075–80. doi: 10.1093/hmg/5.7.1075. [DOI] [PubMed] [Google Scholar]
- 40a.Lee YH, Kim JH, Seo YH, et al. CTLA-4 polymorphisms and susceptibility to inflammatory bowel disease: a meta-analysis. Hum Immunol. 2014;75:414–21. doi: 10.1016/j.humimm.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 40b.Chen Z, Brant SR, Li C, et al. CTLA4-1661A/G and 3’UTR long repeat polymorphisms are associated with ulcerative colitis and influence CTLA4 mRNA and protein expression. Genes and immunity. 2010;11:573–83. doi: 10.1038/gene.2010.16. [DOI] [PubMed] [Google Scholar]
- 41.Assis S, Marques CR, Silva TM, et al. IL10 single nucleotide polymorphisms are related to upregulation of constitutive IL-10 production and susceptibility to Helicobacter pylori infection. Helicobacter. 2014;19:168–73. doi: 10.1111/hel.12119. [DOI] [PubMed] [Google Scholar]
- 42.Sandborn WJ, Colombel JF, Sands BE, et al. Abatacept for Crohn’s Disease and Ulcerative Colitis. Gastroenterology. 2012;143:62–9.e4. doi: 10.1053/j.gastro.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 43.Kataoka H, Takahashi S, Takase K, et al. CD25(+)CD4(+) regulatory T cells exert in vitro suppressive activity independent of CTLA-4. Int Immunol. 2005;17:421–7. doi: 10.1093/intimm/dxh221. [DOI] [PubMed] [Google Scholar]
- 44.Takahashi T, Tagami T, Yamazaki S, et al. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J Exp Med. 2000;192:303–10. doi: 10.1084/jem.192.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang Q, Boden EK, Henriksen KJ, et al. Distinct roles of CTLA-4 and TGF-beta in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34:2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 46.Price S, Shaw PA, Seitz A, et al. Natural history of autoimmune lymphoproliferative syndrome associated with FAS gene mutations. Blood. 2014;123:1989–99. doi: 10.1182/blood-2013-10-535393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kouki T, Sawai Y, Gardine CA, et al. CTLA-4 gene polymorphism at position 49 in exon 1 reduces the inhibitory function of CTLA-4 and contributes to the pathogenesis of Graves’ disease. J Immunol. 2000;165:6606–11. doi: 10.4049/jimmunol.165.11.6606. [DOI] [PubMed] [Google Scholar]
- 48.Maurer M, Loserth S, Kolb-Maurer A, et al. A polymorphism in the human cytotoxic T-lymphocyte antigen 4 (CTLA4) gene (exon 1+49) alters T-cell activation. Immunogenetics. 2002;54:1–8. doi: 10.1007/s00251-002-0429-9. [DOI] [PubMed] [Google Scholar]
- 49.Ueda H, Howson JM, Esposito L, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature. 2003;423:506–11. doi: 10.1038/nature01621. [DOI] [PubMed] [Google Scholar]
- 50.Pesu M, Watford WT, Wei L, et al. T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature. 2008;455:246–50. doi: 10.1038/nature07210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kuhn R, Lohler J, Rennick D, et al. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–74. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 52.Spencer SD, Di Marco F, Hooley J, et al. The orphan receptor CRF2-4 is an essential subunit of the interleukin 10 receptor. J Exp Med. 1998;187:571–8. doi: 10.1084/jem.187.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stamper CC, Zhang Y, Tobin JF, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature. 2001;410:608–11. doi: 10.1038/35069118. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.