

Background: Cellobionic acid phosphorylase (CBAP) catalyzes the reversible phosphorolysis of cellobionic acid into glucose 1-phosphate and gluconic acid.
Results: Crystal structures of CBAP complexed with various ligands were determined.
Conclusion: CBAP has a unique substrate recognition site for aldonic acids that contains positively charged residues.
Significance: This study provided the first insight into the mechanism of sugar catabolism after oxidative cellulose degradation.
Keywords: cellulase, glycoside hydrolase, phosphorylase, site-directed mutagenesis, x-ray crystallography
Abstract
The microbial oxidative cellulose degradation system is attracting significant research attention after the recent discovery of lytic polysaccharide mono-oxygenases. A primary product of the oxidative and hydrolytic cellulose degradation system is cellobionic acid (CbA), the aldonic acid form of cellobiose. We previously demonstrated that the intracellular enzyme belonging to glycoside hydrolase family 94 from cellulolytic fungus and bacterium is cellobionic acid phosphorylase (CBAP), which catalyzes reversible phosphorolysis of CbA into glucose 1-phosphate and gluconic acid (GlcA). In this report, we describe the biochemical characterization and the three-dimensional structure of CBAP from the marine cellulolytic bacterium Saccharophagus degradans. Structures of ligand-free and complex forms with CbA, GlcA, and a synthetic disaccharide product from glucuronic acid were determined at resolutions of up to 1.6 Å. The active site is located near the dimer interface. At subsite +1, the carboxylate group of GlcA and CbA is recognized by Arg-609 and Lys-613. Additionally, one residue from the neighboring protomer (Gln-190) is involved in the carboxylate recognition of GlcA. A mutational analysis indicated that these residues are critical for the binding and catalysis of the aldonic and uronic acid acceptors GlcA and glucuronic acid. Structural and sequence comparisons with other glycoside hydrolase family 94 phosphorylases revealed that CBAPs have a unique subsite +1 with a distinct amino acid residue conservation pattern at this site. This study provides molecular insight into the energetically efficient metabolic pathway of oxidized sugars that links the oxidative cellulolytic pathway to the glycolytic and pentose phosphate pathways in cellulolytic microbes.
Introduction
The establishment of cost-efficient degradation systems of cellulosic biomass is a challenging task as cellulose is the most abundant renewable biopolymer on Earth; however, it is intrinsically recalcitrant (1, 2). Hydrolytic cellulase systems involving cellobiohydrolases, endoglucanases, and β-glucosidases have been extensively studied. The recent discovery of lytic polysaccharide mono-oxygenases (3–5), which cleave glycosidic bonds by oxidization at C1 and/or C4 positions to form aldonolactone or 4-ketoaldose, shifted the paradigm of research on cellulases and chitinases toward oxidative degradation mechanisms (6–9). Cellobiose dehydrogenase has also been demonstrated to catalyze the oxidation of cellobiose at the C1 position to produce cellobiono-1,5-lactone (CbL)3 (10–12). Extracellular β-glucosidases do not efficiently hydrolyze CbL due to their narrow substrate preference and strong product inhibition (13, 14). Therefore, CbL is a major end product by a combination of the extracellular oxidative (lytic polysaccharide mono-oxygenase and cellobiose dehydrogenase) and hydrolytic (e.g. cellobiohydrolase and endoglucanase) enzymes (15). In our previous study, a novel enzyme, cellobionic acid phosphorylase (CBAP; EC 2.4.1.321), was discovered from two cellulolytic microbes, the plant pathogenic bacterium Xanthomonas campestris (XCC4077; XcCBAP) and the red bread mold Neurospora crassa (NCU09425; NcCBAP) of the phylum Ascomycota (16). CbL is spontaneously hydrolyzed to form cellobionic acid (Glc-β1,4-glconic acid; CbA), and CBAP catalyzes phosphorolysis of this compound to produce α-d-glucose 1-phosphate (G1P) and d-gluconic acid (GlcA). The function of CBAP was also confirmed in vivo. Deletion of the NcCBAP gene (ndvB) in N. crassa resulted in the accumulation of CbA during cultivation on cellulose (17), and the NcCBAP gene complemented the ascB gene in Escherichia coli that is responsible for growth on CbA (18). The reaction of CBAP is reversible, and the synthetic reaction using G1P and GlcA as the donor and acceptor substrates produces CbA and inorganic phosphate (Pi). In the synthetic reaction, XcCBAP can also utilize d-glucuronic acid (GlcUA) as the acceptor. The reaction with GlcUA exclusively produced a β1,3-linked disaccharide 3-O-β-d-glucopyranosyl-d-glucuronic acid (Glc-β1,3-GlcUA) (16).
In the Carbohydrate-Active enZyme (CAZy) database (19), CBAP belongs to glycoside hydrolase (GH) family 94, which mainly consists of inverting phosphorylases acting on β-linked di- or oligosaccharides such as cellobiose phosphorylase (CBP; EC 2.4.1.20), cellodextrin phosphorylase (EC 2.4.1.49), laminaribiose phosphorylase (EC 2.4.1.31), and N,N′-diacetylchitobiose phosphorylase (ChBP; EC 2.4.1.280). Within this family, crystal structures of CBPs from Cellvibrio gilvus (CgCBP) (20), Cellulomonas uda (21), and Clostridium thermocellum (22) and ChBP from Vibrio proteolyticus (VpChBP) (23) have been determined. However, a phylogenetic analysis of GH94 phosphorylases indicated that CABP is distantly related to members that are characterized and have known structures with very low amino acid sequence identity (21–24%) (16). In this study, we characterized another CBAP enzyme (Sde_0906; SdCBAP) from the genome of the marine bacterium Saccharophagus degradans that can degrade various polysaccharides, including cellulose (24). Biochemical and structural characterization of SdCBAP revealed the detailed molecular mechanism of the key metabolic enzyme that connects the oxidative cellulose degradation and downstream pathways.
Experimental Procedures
Chemicals
G1P (disodium salt hydrate), GlcA (sodium salt), and GlcUA (sodium salt) were purchased from Sigma-Aldrich, Nacalai Tesque (Kyoto, Japan), and Wako Pure Chemicals (Osaka, Japan), respectively. CbA and Glc-β1,3-GlcUA were produced by the synthetic reaction of XcCBAP and purified as described previously (16).
Cloning, Expression, and Purification
A gene encoding Sde_0906 (GenBankTM accession number AAM43298.1) was amplified by PCR from genomic DNA of S. degradans 2-40 using KOD-plus DNA polymerase (Toyobo, Osaka, Japan) with the following oligonucleotides based on the genome sequence (GenBank accession number CP000282) (25): 5′-aaaccatgggcttaaaagccattaacaac-3′ and 5′-tttctcgaggtgtgtggcaggtaatag-3′ (restriction enzyme sites underlined). The amplified gene was purified using a FastGene Gel/PCR Extraction kit (Nippon Genetics Co., Tokyo, Japan), digested by NcoI and XhoI (New England Biolabs, Beverly, MA), and inserted into pET28a(+) (Novagen, Madison, WI) to encode a His6 tag fusion at the C terminus of the recombinant protein. The expression plasmid was introduced into E. coli DH5α (Toyobo) and verified by sequencing (Operon Biotechnologies, Tokyo, Japan). For native and selenomethionine (SeMet)-labeled protein expression, the plasmid was introduced into E. coli BL21 CodonPlus (DE3)-RIL (Stratagene, La Jolla, CA) and E. coli B834 (DE3) strains, respectively. The transformants were cultured in Luria-Bertani medium (native protein) or LeMaster medium (SeMet-labeled protein) containing 100 mg/liter kanamycin at 37 °C until the absorbance reached 0.6 at 600 nm. The protein expression was induced by 0.1 mm isopropyl β-d-thiogalactopyranoside and continued at 25 °C for 18 h. The cell-free extract was first purified by nickel affinity chromatography (HisTrap FF crude, GE Healthcare), and the enzyme was eluted with a stepwise increase in the imidazole concentration (20 and 250 mm) in 50 mm HEPES-NaOH (pH 7.0). Next, the enzyme was further purified on a Mono Q column (GE Healthcare) with an increasing linear gradient of 0–100% saturation of NaCl using ÄKTA (GE Healthcare). The protein concentration was determined spectrophotometrically at 280 nm using a theoretical extinction coefficient of ϵ = 146,110 m−1 cm−1 based on the amino acid sequence. The molecular mass of purified SdCBAP was estimated by SDS-PAGE and by gel filtration column chromatography using HiLoad 16/60 Superdex 200 prep grade (GE Healthcare) equilibrated with 50 mm HEPES-NaOH (pH 7.0) and 150 mm NaCl at a flow rate of 1.0 ml/min. A Gel Filtration Markers kit for molecular weights (Sigma-Aldrich) was used for standards.
Measurement of Enzymatic Activity
The catalytic activities of SdCBAP were measured as described previously (16). The phosphorolytic activity was determined by quantifying the G1P released during the reaction in 40 mm MES-NaOH (pH 6.5) containing 10 mm CbA and 10 mm Pi at 30 °C using the phosphoglucomutase-glucose 6-phosphate dehydrogenase method. The synthetic activity was determined by measuring the increase in Pi using a reaction mixture containing 10 mm G1P and 10 mm GlcA in 40 mm MES-NaOH (pH 6.5) at 30 °C.
Structural Determination of Reaction Products
Structures of reaction products were determined by NMR spectra as described previously (16). Reaction products were generated in 1 ml of reaction mixture (pH 6.5) containing 450 mm G1P and 450 mm GlcA or GlcUA with 22 μm SdCBAP at 30 °C for 24 h. After treatment with α-d-glucose-1-phosphatase from E. coli at 30 °C for 24 h, the reaction products were separated on a Toyopearl HW-40S column and then lyophilized. The amount of product obtained was 43 and 42 mg from GlcA or GlcUA, respectively. The structures of the products were identified by comparing their 1H and 13C NMR spectra acquired in D2O with those of authentic data (16) using a Bruker Avance 500 or Bruker Avance 800 spectrometer (Bruker Biospin, Rheinstetten, Germany).
Kinetic Analysis
The initial velocities of the phosphorolytic reactions were determined under standard conditions with SdCBAP (7 nm–17 μm for the wild-type and mutant enzymes) and a combination of initial concentrations of cellobionic acid (0.5–3.0 mm) and Pi (0.1–2.0 mm). The kinetic parameters were calculated by curve fitting the experimental data to the theoretical Equation 1 for a sequential bi-bi mechanism using GraFit version 7.0.2 (Erithacus Software Ltd., London, UK).
 |
Kinetic analysis of the synthetic reactions was performed under the standard conditions with SdCBAP (9.0 nm–17 μm for the wild-type and mutant enzymes) and various concentrations of GlcA or GlcUA (0.5–20 mm) as the acceptor or G1P (0.3–5.0 mm) as the donor with 10 mm for each opposite substrate. The kinetic parameters were calculated by curve fitting the experimental data to the Michaelis-Menten equation v = kcat [E]0 [S]/(Km + [S]) using GraFit version 7.0.2.
Temperature and pH Profiles
The effects of pH on the phosphorolytic and synthetic activities using 20 nm SdCBAP were measured under the standard conditions described above using the following 40 mm buffers: sodium citrate (pH 3.0–5.5), BisTris-HCl (pH 5.5–7.0), HEPES-NaOH (pH 7.0–8.5), and glycine-NaOH (pH 8.5–10.5). The thermal and pH stabilities were evaluated by measuring the residual synthetic activity under the standard conditions after incubation of 9.9 μm SdCBAP at a temperature range of 30 to 90 °C for 15 min and at various pH values at 4 °C for 24 h, respectively.
Site-directed Mutagenesis
Q190A, R609A, and K613A mutants were constructed using a PrimeSTAR Mutagenesis basal kit (Takara) in accordance with the manufacturer's protocol. The following primers and their complementary strands were used (mutation sites are underlined): 5′-cgccttatgctaaagtcgccgactacttt-3′ (Q190A), 5′-gacgtaggcgcagttacccaaaaattccca-3′ (R609A), and 5′-acccaagcattcccaggctctgcagaa-3′ (K613A). The mutations were confirmed by DNA sequencing. The mutant enzymes were expressed and purified using the same procedure as described above.
Crystallography
All crystals were obtained at 25 °C using the sitting drop vapor diffusion method by mixing 1 μl of protein solution containing 15–30 mg/ml protein with an equal volume of reservoir solution containing 0.1 m sodium citrate (pH 5.6–6.0), 0.1 m Li2SO4, 0.6 m (NH3)2SO4, and 5% (v/v) glycerol. Crystals completely grew in 3 weeks. Crystals were cryoprotected in the reservoir solutions supplemented with 20% (w/v) glycerol and flash cooled at 100 K in a stream of nitrogen gas. Each complex crystal was obtained by soaking in the cryoprotectant solution containing a ligand (50 mm GlcA, 50 mm CbA, or 20 mm Glc-β 1,3-GlcUA) for 2 min. Diffraction data were collected using a charge-coupled device camera on beamlines BL17A (Photon Factory) and NW12A (Photon Factory-Advanced Ring) at the High Energy Accelerator Research Organization (KEK, Tsukuba, Japan) and processed using HKL2000 (26). The initial phase calculation, phase improvement, and automated model building were performed using PHENIX (27). Manual model rebuilding and refinement were achieved using Coot (28) and Refmac5 (29). Initial coordinates and parameters of CbA and Glc-β1,3-GlcUA molecules were built with the JLigand program (30). The molecular interfaces were analyzed using the PDBe PISA server (31). Molecular graphic images were prepared using PyMOL (Schrödinger, LLC, New York, NY).
Results and Discussion
Biochemical Characterization of Sde_0906 (SdCBAP)
Before our identification of CBAPs from X. campestris and N. crassa, the sde_0906 (cep94B) gene of S. degradans was predicted as cytoplasmic cellobiose/cellodextrin phosphorylase (25). A previous study on recombinant Sde_0906 protein (Cep94B) showed that the gene product did not exhibit phosphorylase activity on cellobiose or cellodextrin (32). Because we noticed that Sde_0906 is closely related to XcCBAP and NcCBAP with high amino acid sequence identities (60.4 and 61.9%, respectively), we prepared its purified recombinant protein and measured the CBAP activities. We subsequently designated the enzyme as SdCBAP. The molecular masses of SdCBAP as deduced from the amino acid sequence and estimated by SDS-PAGE and calibrated gel filtration chromatography were 88.7, 87, and 147 kDa, respectively, suggesting that it is dimeric in solution. The phosphorolytic reaction followed a sequential bi-bi mechanism, and the kinetic parameters were kcat = 92 ± 2 s−1, KmA = 0.12 ± 0.03 mm, KiA = 0.44 ± 0.2 mm, and KmB = 0.061 ± 0.009 mm (Fig. 1). The kinetic parameters of the synthetic reactions using GlcA and GlcUA as the acceptor were also determined (Table 1). SdCBAP exhibited significant activity to GlcUA, although it was 9-fold lower than that of GlcA due to decreased kcat and increased Km values. Reaction products synthesized from GlcA and GlcUA were determined by NMR spectra measurements, confirming that they were CbA and Glc-β1,3-GlcUA, respectively, and no other products were detected (data not shown). The Km value for G1P was 0.41 ± 0.1 mm when GlcA was used as the acceptor. SdCBAP was stable up to 35 °C during 15 min of incubation and in the pH range of 5.5–10.5. The optimum pH for the synthetic reaction was pH 6.5–7.0 (data not shown). The characteristics of SdCBAP were similar to those of XcCBAP and NcCBAP (16).
FIGURE 1.
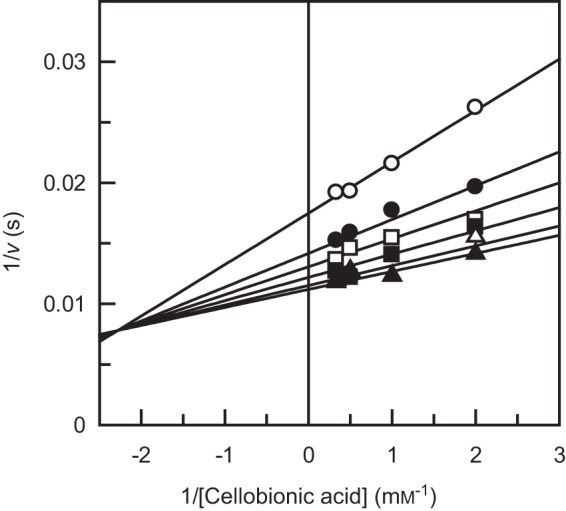
Double reciprocal plots of the phosphorolysis of cellobionic acid catalyzed by SdCBAP. The initial velocities of the phosphorolytic reaction were determined with a combination of initial concentrations of CbA (0.5, 1.0, 2.0, and 3.0 mm) and Pi (open circles, 0.1 mm; filled circles, 0.2 mm; open squares, 0.3 mm; filled squares, 0.5 mm; open triangles, 1.0 mm; filled triangles, 2.0 mm).
TABLE 1.
Specific activities and kinetic parameters for the synthetic and phosphorolytic reactions of wild-type SdCBAP and its mutants
| Enzyme | Synthesis acceptor |
Phosphorolysis substrate CbA specific activitya | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
GlcA |
GlcUA |
||||||||
| Specific activitya | kcat | Km | kcat/Km | Specific activitya | kcat | Km | kcat/Km | ||
| units/mg | s−1 | mm | s−1mm−1 | units/mg | s−1 | mm | s−1mm−1 | units/mg | |
| WT | 93.2 | 130 ± 6 | 1.7 ± 0.2 | 76 | 10 | 34.6 ± 0.7 | 13.4 ± 0.5 | 2.58 | 62 |
| Q190A | 3.3 | 24 ± 3 | 41 ± 7 | 0.58 | 1.1 | NDb | ND | 0.16 | 9.04 |
| R609A | <0.005 | ND | ND | ND | <0.001 | ND | ND | ND | <0.001 |
| K613A | <0.01 | ND | ND | ND | <0.01 | ND | ND | ND | <0.002 |
a The specific activities were measured at 10 mm substrates.
b ND, not determined.
Crystal Structure of SdCBAP
The crystal structure of SdCBAP was solved by the single wavelength anomalous dispersion method using an SeMet derivative. A ligand-free structure and complex structures with GlcA, CbA, and Glc-β1,3-GlcUA were determined (Table 2). The crystals contained one molecule in the asymmetric unit. The monomer structure consists of four distinct parts (Fig. 2A): an N-terminal β sandwich domain (residues 1–277; blue), a helical linker region (278–308; green), a catalytic (α/α)6 barrel domain (319–709; yellow), and a β sheet domain (red). The β sheet domain consists of a middle segment (residues 309–318) and the C-terminal segment (710–785). The protomers in the asymmetric units are related by 2-fold crystallographic symmetry to form a dimer (Fig. 2B), which corresponds to the state in solution as confirmed by the biochemical analysis described above. The dimer is tightly connected with an interface area of 2,700 Å. There are 44 hydrogen bonds and 18 salt bridges, and the estimated ΔiG value is −13.2 kcal/mol. The major contact area at the dimer interface comprises the N-terminal and catalytic barrel domains. The overall monomer and dimer structure of SdCBAP was basically similar to those of GH94 CgCBP and VpChBP (20, 23) with some variations at peripheral loop regions. The most prominent difference from other GH94 structures is the presence of a long loop region with a short α helix (186–203; Fig. 2, magenta) in the N-terminal domain. This region partly forms the active site in the neighboring protomer (described below).
TABLE 2.
Data collection and refinement statistics
| Peak | Ligand-free | CbA | GlcA | Glc-β1,3-GlcUA | |
|---|---|---|---|---|---|
| Data collection | |||||
| Protein Data Bank code | 4ZLE | 4ZLF | 4ZLG | 4ZLI | |
| Beamline | BL17A | BL17A | NW12A | BL17A | BL17A |
| Wavelength (Å) | 0.9790 | 0.9988 | 1.0000 | 0.9702 | 0.9732 |
| Space group | P3121 | P3121 | P3121 | P3121 | P3121 |
| Unit cell (Å) | a = b = 107.2, c = 186.6 | a = b = 106.9, c = 185.3 | a = b = 107.2, c = 186.8 | a = b = 107.0, c = 186.5 | a = b = 107.1, c = 185.8 |
| Resolution (Å)a | 50.00–2.40 (2.44–2.40) | 50.00–2.10 (2.14–2.10) | 50.00–1.60 (1.63–1.60) | 50.00–1.75 (1.78–1.75) | 50.00–1.80 (1.83–1.80) |
| Total reflections | 2,184,371 | 522,451 | 1,291,267 | 642,592 | 830,557 |
| Unique reflections | 49,159 | 72,225 | 163,884 | 124,297 | 114,626 |
| Completeness (%)a | 98.6 (98.7) | 99.5 (100.0) | 100.0 (100.0) | 98.2 (100.0) | 99.5 (100.0) |
| Redundancya | 23.3 (23.3) | 7.3 (7.2) | 7.9 (7.6) | 5.3 (5.4) | 7.3 (7.4) |
| Mean I/σ(I)a | 38.2 (6.8) | 17.6 (2.6) | 37.3 (2.9) | 20.8 (2.1) | 24.6 (4.1) |
| Rmerge (%)a | 10.9 (50.7) | 11.2 (73.0) | 4.3 (41.1) | 6.4 (46.8) | 9.2 (54.9) |
| Refinement | |||||
| Resolution (Å) | 41.44–2.10 | 31.14–1.60 | 41.66–1.75 | 40.50–1.80 | |
| No. of reflections | 63,592 | 155,047 | 115,743 | 108,205 | |
| R/Rfree (%) | 17.0/21.9 | 15.6/17.7 | 16.9/20.1 | 15.5/18.8 | |
| No. of atoms | 6,750 | 7,118 | 6,964 | 6,932 | |
| No. of solvents | 477 (water), 3 (glycerol), 6 (SO4), 1 (Cl−) | 809 (water), 1 (CbA), 8 (glycerol), 2 (SO4), 1 (Cl−) | 688 (water), 1 (GlcA), 1 (GcL), 2 (glycerol), 2 (SO4), 1 (Cl−) | 629 (water), 1 (Glc-β1,3-GlcUA), 3 (glycerol), 3 (SO4), 1 (Cl−) | |
| r.m.s.d.b from ideal values | |||||
| Bond lengths (Å) | 0.020 | 0.031 | 0.025 | 0.027 | |
| Bond angles (°) | 1.92 | 2.56 | 2.09 | 2.23 | |
| Ramachandran plot (%) | |||||
| Favored | 97.4 | 98.2 | 98.2 | 98.3 | |
| Allowed | 2.3 | 1.7 | 1.5 | 1.4 | |
| Outlier | 0.3 | 0.1 | 0.3 | 0.3 | |
a Values in parentheses correspond to the highest resolution shell.
b Root mean square deviation.
FIGURE 2.

Overall structure of SdCBAP shown as monomer (A) and dimer (B and C). The CbA complex structure is shown and colored by domains: N-terminal β sandwich domain (residues 1–277; blue), helical linker region (278–308; green), a catalytic (α/α)6 barrel domain (319–709; yellow), and a C-terminal β sheet domain (309–318 and 710–785; red). A prominent loop (186–203) in the N-terminal domain is shown in magenta. CbA (slate blue) and sulfate (yellow) are shown as sticks. In B and C, one monomer is shown in gray, and views from two different orientations (90° rotation around a horizontal axis) are shown.
Active Site Structure
Because the crystals grew in high concentrations of ammonium sulfate, all of the crystal structures contained a sulfate ion (SO42−) at the Pi binding site formed by Arg-341, Tyr-624, His-626, Thr-694, and Gly-695 as shown in the ligand-free structure (Fig. 3A). These residues at the Pi binding site, except for Tyr-624, are basically conserved in GH94 phosphorylases (Fig. 4). In the ligand-free structure, a glycerol and an additional sulfate are bound at subsites −1 (the donor glucoside site) and +1 (the acceptor site), respectively. The sulfate at subsite +1 is held by Arg-609, Lys-613, and Gln-190′ (the prime mark indicate that it is a residue from the neighboring molecule). Gln-190′ is located in the prominent 186–203 loop of the N-terminal domain.
FIGURE 3.

Stereoviews of the active site of SdCBAP. A, ligand-free. B, CbA complex (yellow). C, GlcA-GcL complex (cyan). D, Glc-β1,3-GlcUA complex (magenta). The |Fo| − |Fc| omit electron density maps (4.0σ) of ligands are shown as blue mesh.
FIGURE 4.

Partial amino acid sequence alignment of GH94 phosphorylases. Residues at the Pi binding site and subsites −1 and +1 are indicated above (SdCBAP) and below (CgCBP) the sequences. Residues involved in the unique subsite +1 of the three CBAPs are shown in the thick frame. Residues that have similarity across the sequences are shown in the thin frame, and inverted characters indicate residues that have complete conservation. PspLBP, Paenibacillus sp. YM-1 laminaribiose phosphorylase.
In the CbA complex, a clear electron density of the bound CbA was observed (Fig. 3B). The glucose moiety at subsite −1 is in a standard 4C1 conformation with its C6 hydroxyl group in a gauche-gauche conformation. At this site, the side chains of Gln-347, Arg-349, Asn-350, and Glu-619 and the main chain of Trp-470 form hydrogen bonds to the hydroxyl groups of the glucopyranoside sugar. The Asp-472 residue of SdCBAP corresponds to the general acid residue Asp-490 of CgCBP and Asp-492 of VpChBP (20, 23). Asp-472 is located near the glycosidic bond oxygen atom (3.6 Å) so that it can interact with the glycosidic oxygen by a slight movement of the side chain rotamer. These residues at subsite −1 are also highly conserved in GH94 phosphorylases (Fig. 4). The sulfate ion at the Pi binding site is located near the anomeric C1 atom of the glucose moiety (3.2 Å). Thus, the Pi binding site is suitable for the direct nucleophilic attack of the inverting phosphorylase reaction mechanism (23). The gluconate moiety of CbA is bound at subsite +1, and its carboxylate group forms salt bridges with Arg-609 and Lys-613. Gln-190′ is located at a relatively far position (4.5 Å) in this structure. Hydroxyl groups of the gluconate moiety are recognized by hydrogen bonds from the SdCBAP protein. The C2 and C3 hydroxyl groups form hydrogen bonds with Lys-613, Glu-619, and Asp-472, and the C5 hydroxyl group forms hydrogen bonds with Tyr-624 and Asn-672.
In the GlcA complex, the electron density of GlcA (linear form) was observed at subsite +1, whereas a pyranose sugar molecule was found at subsite −1 (Fig. 3C). Because bonds around the C1 anomeric carbon atom appeared flat, we could confidently place a model of d-glucono-1,5-lactone (GcL) in the electron density map without any skewing of the atoms. It is unclear why a GcL molecule was observed in the complex structure with GlcA. Because subsite −1 appears to be highly suitable for binding of a glucopyranose-like molecule, we presume that the high concentration of GlcA in the soaking solution (50 mm) may have caused occupation of its dehydrated compound (GcL) in this site even though the lactone is not dominant in aqueous solution after the equilibrium (33). At subsite +1, GlcA is bound at a similar but slightly different position from the gluconate moiety of CbA. The carboxylate group was recognized by Arg-609, Lys-613, and Gln-190′. The C2, C3, and C5 hydroxyls form hydrogen bonds to Lys-613, Asp-472, and Gln-347, respectively. Fig. 5A shows a superimposition of the CbA (yellow) and GlcA (cyan) complex structures. The glucopyranose at subsite −1 almost completely overlaps. At subsite +1, the carboxylate groups of CbA and GlcA are located in a position near Arg-609, Lys-613, and Gln-190′, indicating that these residues are critical for recognition of the aldonic acid substrates. The remaining part of the GlcA (C2–C6) is located in a significantly displaced position from CbA. Because the C4 hydroxyl group of GlcA is located far from subsite −1, this binding mode does not perfectly represent the acceptor molecule in the synthetic reaction. This is probably because of the steric hindrance with the C1 ketone group of GcL occupying subsite −1. In the synthetic reaction with G1P, the acceptor GlcA molecule is assumed to bind as in the gluconate moiety in the CbA complex structure.
FIGURE 5.

Stereoviews of superimpositions of SdCBAP complex structures. A, CbA (yellow) and GlcA-GcL (cyan). B, CbA (yellow) and Glc-β1,3-GlcUA (magenta). C, CbA (yellow), Glc-β1,3-GlcUA (magenta thick sticks), and a GlcUA molecule modeled at subsite +1 by superimposing the C2–C6 atoms to CbA (magenta thin sticks). Hydrogen bonds present in the CbA, GlcA-GcL, and Glc-β1,3-GlcUA complex structures are shown as yellow, cyan, and magenta dashed lines, respectively.
We also determined the complex structure with Glc-β1,3-GlcUA, which is a product of the synthetic reaction using GlcUA as the acceptor (Fig. 3D). In the electron density map, the glucuronate moiety was observed in a pyranose (ring) form. A superimposition of complex structures with CbA (yellow) and Glc-β1,3-GlcUA (magenta) (Fig. 5B) indicates that the non-reducing end glucose moiety is bound in a similar manner. In the Glc-β1,3-GlcUA complex, however, an additional sulfate is bound at the carboxylate binding pocket in subsite +1, and the glucuronate moiety is significantly displaced from the canonical subsite +1. The carboxylate group forms a water-mediated hydrogen bond with a glycerol molecule, which is derived from the cryoprotectant. Arg-341, Asn-672, and Asn-693 are involved in the recognition of the GlcUA moiety. Therefore, from this complex structure, Arg-609, Lys-613, and Gln-190′ appear not to recognize the carboxylate group of Glc-β1,3-GlcUA.
Mutational Analysis
We constructed site-directed mutants of the three residues at the carboxylate binding site in subsite +1 (Q190A, R609A, and K613A) and measured their synthetic (using either GlcA or GlcUA as the acceptor) and phosphorolytic activities. As shown in Table 1, the activities of the mutants at positively charged residues (R609A and K613A) were either not detectable or very low. For the Q190A mutant, the activities were also impaired. The decrease of synthetic activity of Q190A mutant was due to the significant increase and decrease of the Km and kcat values, respectively. We could not determine the Km value of the synthetic activity of Q190A mutant using GlcUA as the acceptor because we did not observe any activity saturation up to 20 mm GlcUA. The results indicated that all three of these residues were important for substrate recognition and activity against both GlcA and GlcUA acceptors contrary to the observation in the Glc-β1,3-GlcUA complex structure.
Implication for the Glc-β1,3-GlcUA Complex Structure
When a free GlcUA molecule was modeled in subsite +1 of SdCBPA by superimposing the C2–C6 atoms to CbA, the C6 carboxylate and the C3 hydroxyl could be placed in appropriate positions for the carboxylate binding pocket and the glycosidic bond oxygen without any steric clash (Fig. 5C). Therefore, the modeled GlcUA appears to be in a position suitable for the synthetic reaction that forms a β1,3-bond. According to the results of mutational analysis, which emphasize the importance of the three key residues for the reaction using GlcUA, we concluded that the conformation of the Glc-β1,3-GlcUA complex structure was an artifact. Because the concentration of ammonium sulfate in the buffer (600 mm) was higher than that of Glc-β1,3-GlcUA (20 mm), the sulfate anion may have expelled the GlcUA moiety from the canonical position surrounded by Arg-609, Lys-613, and Gln-190′.
Reaction Mechanism
Fig. 6 shows a proposed reaction and substrate recognition mechanism of SdCBAP from both directions. In the phosphorolysis reaction, an oxygen atom of Pi attacks the anomeric C1 atom of the subsite +1 glucoside, and Asp-472 functions as a general acid by donating a proton to the glycosidic oxygen atom. Arg-609 and Lys-613 play a crucial role in the recognition of the C1 carboxylate group of the aldonic acid. In the CbA complex structure, Gln-190 from the neighboring protomer was observed in a relatively distant position. However, this residue contributed to the phosphorolysis reaction because mutation at this site (Q190A) significantly impaired the activity. Glu-619, Asn-672, and Tyr-624 also contributed to the recognition of the gluconate group by forming hydrogen bonds to C2, C3, and C5 hydroxyls. In the synthetic reaction, Asp-472 works as a general base by accepting a proton from the C4 hydroxyl of GlcA to facilitate the nucleophilic attack to the anomeric C1 atom of G1P. In the GlcA-GcL complex structure (Fig. 5A), the C4 hydroxyl oxygen of GlcA is located far from the C1 atom of GcL (4.7 Å) by the significant displacement of the C4-C5-C6 group compared with CbA. This is probably due to the steric hindrance with the GcL molecule that occupied subsite +1, but G1P would not hinder the correct positioning of GlcA. Residues recognizing the hydroxyls at subsite +1 (Glu-619, Asn-672, and Tyr-624) did not move at all (Fig. 5A). Therefore, we assume that GlcA in the synthetic reaction binds to the protein similarly to the gluconate moiety of CbA. As confirmed by the mutational analysis, Arg-609, Lys-613, and Gln-190′ critically contributed to the GlcA recognition in the synthetic reaction.
FIGURE 6.

Proposed reaction and substrate recognition mechanism of SdCBAP from both the phosphorolytic and synthetic directions.
Comparison with GH94 Phosphorylases
Partial amino acid sequence alignment of GH94 phosphorylases is shown in Fig. 4. Because CABP, CBP, ChBP, and laminaribiose phosphorylase act on CbA (Glc-β1,4-gluconic acid), cellobiose (Glc-β1,4-Glc), N,N′-diacetylchitobiose (GlcNAc-β1,4-GlcNAc), and laminaribiose (Glc-β1,3-Glc), respectively, these enzymes are expected to have a discrete subsite +1. For comparison, structures of SdCBAP (CbA complex) and CgCBP were superimposed at the active site (Fig. 7). The CgCBP structure complexed with Pi, glycerol (subsite −1), and β-d-glucose (subsite +1) was used for comparison (Protein Data Bank code 2CQT) (20). As expected, residues at the Pi binding site (Fig. 7A) and subsite −1 (Fig. 7B) are basically conserved except for several residues (Asn-672, Gln-347, and Asn350 in SdCBAP). In CgCBP, Gln-712 forms a hydrogen bond with Pi, whereas the corresponding residue in SdCBAP (Asn-672) is involved in subsite +1 recognition (Fig. 7, A and C). Gln-347 in SdCBAP forms a hydrogen bond with the C2 hydroxyl of the glucose moiety at subsite −1, but the corresponding residue is Gly in CBP and other enzymes (Figs. 7B and 4). Asn-350 in SdCBAP forms hydrogen bonds with C2 and C3 hydroxyls at subsite −1 but is replaced with Asp in other GH94 phosphorylases (Figs. 7B and 4).
FIGURE 7.

Stereoviews of a comparison with CgCBP at the Pi binding site (A), subsite −1 (B), and subsite +1 (C). A superimposition of SdCBAP (green) complexed with CbA (yellow) and sulfate (orange) and CgCBP (cyan) complexed with Pi (yellow), glycerol (subsite −1; gray), and β-d-glucose (subsite +1; gray) are shown. Hydrogen bonds present in the SdCBAP and CgCBP structures are shown as yellow and gray dashed lines, respectively.
Interestingly, the residues involved in subsite +1 recognition of SdCBAP are completely conserved in CBAPs (XcCBAP and NcCBAP), but most of them are not conserved in CBP, ChBP, and laminaribiose phosphorylase (Fig. 4). The region around Gln-190 (186–203 loop in SdCBAP) shows low sequence conservation among GH94 phosphorylases. In CgCBP, Gln-165 in this region comes from the neighboring protomer and participated in the formation of subsite +1 (Fig. 7C). However, Gln-165 in CgCBP and Gln-190 in SdCBAP come from different sides. The other two key residues for the carboxylate recognition (Arg-609 and Lys-613) are replaced by Glu-649 and Tyr-653 in CgCBP, respectively. Moreover, Tyr-624 and Asn-672 are also not conserved. At subsite +1, Glu-619 is solely conserved because this residue is also involved in subsite −1 recognition (Fig. 7B). In summary, CBAPs have a unique subsite +1 compared with other GH94 phosphorylases.
Sulfate ion and GcL were weak competitive inhibitors of CgCBP against phosphate and cellobiose with Ki values of 79 and 1.1 mm, respectively (34). Those reagents also showed weak inhibition to SdCBAP with apparent Ki values of >30 and ∼1 mm, respectively (data not shown).
A Possible Role of CBAP in Fungal Cellulase Systems
In this study, we demonstrated that CBAPs from cellulolytic bacteria and fungi shared a conserved active site specific for the binding of aldonic acids. N. crassa has a complete oxidative cellulose degradation system, including endoglucanase, cellobiohydrolase, cellobiose dehydrogenase, and C1-oxidizing auxiliary activity (AA) family 9 lytic polysaccharide mono-oxygenases that accept electrons from cellobiose dehydrogenase (35, 36). It has been shown that cellobiose dehydrogenase enhances cellobiohydrolase activity by relieving product inhibition (37), indicating that the enzymes in the oxidative cellulose degradation system synergistically work to produce CbL. CbL is rapidly and spontaneously hydrolyzed into CbA with a time constant of 61 min at 26 °C (11) and likely transported into the cytoplasm thereafter. The intracellular enzyme CBAP can efficiently degrade CbA, and the products (G1P and GlcA) are further catabolized by the glycolysis and pentose phosphate pathways. The involvement of the glycoside phosphorylase has an energetic advantage in microbial catabolism because CBAP produces the phosphorylated sugar (G1P) without consuming ATP. In addition to N. crassa, various fungi of the phylum Ascomycota possess CBAP homologs in their genomes (16), suggesting that the phosphorolytic pathway is also present in these organisms.
Bacterial Enzymes in Oxidative Cellulose Degradation
Bacteria do not possess cellobiose dehydrogenase (AA3_1 subfamily in the CAZy database) at all and must have different systems from fungi. Interestingly, the bacterium Fibrobacter succinogenes S85, which does not have cellobiose dehydrogenase, produced CbA and CbL in the culture fluid when grown on cellulosic substrates (38). It has been suggested that pyrroloquinoline quinone-dependent soluble aldose sugar dehydrogenase is responsible for bacterial CbL formation. Aldose sugar dehydrogenase from E. coli has wide substrate specificity and can efficiently catalyze the C1 oxidation of various mono-, di-, and trisaccharides, including cellobiose (39). For lytic polysaccharide mono-oxygenases, cellulose-oxidizing AA9 enzymes were exclusively found from eukaryotes. However, several AA10 (CBM33) enzymes from bacteria (Streptomyces coelicolor and Thermobifida fusca) also oxidize and cleave cellulose (40, 41). Close homologs (amino acid identities >60%) of SdCBAP and XcCBAP are found in the genomes of cellulose-degrading and plant-pathogenic bacteria, including Cellvibrio, Bacillus, Paenibacillus, and Xanthomonas species, which are generally aerobic and possess aldose sugar dehydrogenase and/or AA10 lytic polysaccharide mono-oxygenase genes. This result suggests that bacterial CBAPs also play key roles in oxidative cellulose degradation and infection by decomposing the plant cell wall.
Role of SdCBAP in the Oxidative Cellulose Degradation System of S. degradans
The marine bacterium S. degradans strain 2-40 can decompose at least 10 distinct complex polysaccharides from algae, plants, and invertebrates such as cellulose, hemicellulose, and agarose (24). This organism has a novel and complete multienzyme system for cellulose degradation as its enzymes exhibit unusual architecture via combinations of catalytic and substrate-binding modules (25, 42). Although S. degradans apparently lacks a cellobiohydrolase in its genome, three GH5 endoglucanases were shown to be processive and mainly release cellobiose (43). Interestingly, in the genome of S. degradans, there are two oxidative enzymes that possibly convert cellobiose into CbL. A putative aldose sugar dehydrogenase gene (sde_1897) shows a certain homology to the cellobiose-active aldose sugar dehydrogenase from E. coli (32% amino acid identity). In addition, there is a putative AA10 gene (sde_0633) attached to the carbohydrate-binding module family 2 domain at its C terminus. The AA10 domain of sde_0633 gene exhibits a high homology (54% identity) to cellulose-active and C1-specific lytic polysaccharide mono-oxygenase from Cellvibrio japonicus (44). These facts indicate that the cellulolytic system of S. degradans also produces CbL and CbA. A study on possible cellobiose-active enzymes revealed that genes of an extracellular GH3 β-glucosidase (bgl3C), an intracellular GH1 β-glucosidase (bgl1A), an intracellular GH94 CBP (cep94A), and SdCBAP (cep94B) were induced by Avicel (32), suggesting the presence of multiple metabolic pathways for cellobiose and CbA by hydrolysis and phosphorolysis. The gene architecture of S. degradans near the SdCBAP (sde_0906) gene locus is interesting as there are genes for a putative major facilitator superfamily for sugar (sde_0907) and a putative gluconate kinase (sde_0904). Therefore, these genes possibly function as an operon to import and metabolize CbA and subsequently send the products into the pentose phosphate and glycolysis pathways. Because the unique and novel cellulolytic system of S. degradans has the potential for application to biomass processing (45), more detailed studies are required for a better understanding of its molecular system.
Concluding Remarks
This report has provided the first structural basis of CBAP, revealing the key residues for aldonic acid recognition. The discovery of CBAP and its structural basis have expanded our knowledge on microbial cellulose degradation systems. The conservation pattern of the key residues for the CbA recognition is a good indicator to find putative GH94 CBAP genes from genomic and metagenomic information. Consistent with previous studies, this study provided concrete evidence that CBAP participates in the oxidative cellulose degradation systems of cellulolytic bacteria and fungi. In addition to understanding the microbial cellulosic biomass degradation mechanisms, the three-dimensional structure of CBAP will contribute to the future design and engineering of glycoside phosphorylases, which have the potential for application in large scale production of functional oligosaccharides (46).
Author Contributions
S. F., H. N., and M. K. conceived and designed the study. S. F., T. A., H. N., and M. K. coordinated the study. S. F., Y.-W. N., and T. N. wrote the paper. Y.-W. N. purified and crystallized the protein and determined the crystal structure. Y.-W. N., T. A., and S. F. collected the x-ray data. Y. S. and T. N. designed and constructed expression vectors. Y.-W. N. constructed mutant expression vectors and purified mutant proteins. T. N. and Y.-W. N. purified and performed enzymatic assay of the wild-type and mutant enzymes. T. N., H. N., and M. K. analyzed kinetic data. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Drs. Kiyohiko Igarashi and Takayoshi Wakagi for helpful discussion and the staff of the Photon Factory and SPring-8 for the x-ray data collection. We thank the staffs of Instrumental Analysis Center for Food Chemistry of National Food Research Institute, National Agriculture and Food Research Organization for recording NMR spectra.
This work was supported by the Science and Technology Research Promotion Program for Agriculture, Forestry, Fisheries and Food Industry (Grant 25010A); in part by the Platform for Drug Discovery, Informatics, and Structural Life Science funded by the Ministry of Education, Culture, Sports, Science and Technology, Japan, by Japan Society for the Promotion of Science KAKENHI (Grants 2380053 and 15H02443 to S. F.); and by the Promotion of Environmental Improvement for the Independence of Young Researchers under the Special Coordination Funds for Promoting Science and Technology. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 4ZLE, 4ZLF, 4ZLG, and 4ZLI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CbL
- cellobiono-1,5-lactone
- AA
- auxiliary activity
- CbA
- cellobionic acid
- CBAP
- cellobionic acid phosphorylase
- CBP
- cellobiose phosphorylase
- CgCBP
- C. gilvus CBP
- ChBP
- N,N′-diacetylchitobiose phosphorylase
- G1P
- α-d-glucose 1-phosphate
- GcL
- d-glucono-1,5-lactone
- GH
- glycoside hydrolase
- GlcA
- d-gluconic acid
- GlcUA
- d-glucuronic acid
- Glc-β1,3-GlcUA
- 3-O-β-d-glucopyranosyl-d-glucuronic acid
- NcCBAP
- N. crassa CBAP or NCU09425
- SdCBAP
- S. degradans CBAP or Sde_0906
- VpChBP
- V. proteolyticus ChBP
- XcCBAP
- X. campestris CBAP or XCC4077
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 2. Bornscheuer U., Buchholz K., Seibel J. (2014) Enzymatic degradation of (ligno)cellulose. Angew. Chem. Int. Ed. Engl. 53, 10876–10893 [DOI] [PubMed] [Google Scholar]
- 3. Harris P. V., Welner D., McFarland K. C., Re E., Navarro Poulsen J. C., Brown K., Salbo R., Ding H., Vlasenko E., Merino S., Xu F., Cherry J., Larsen S., Lo Leggio L. (2010) Stimulation of lignocellulosic biomass hydrolysis by proteins of glycoside hydrolase family 61: structure and function of a large, enigmatic family. Biochemistry 49, 3305–3316 [DOI] [PubMed] [Google Scholar]
- 4. Vaaje-Kolstad G., Westereng B., Horn S. J., Liu Z., Zhai H., Sørlie M., Eijsink V. G. (2010) An oxidative enzyme boosting the enzymatic conversion of recalcitrant polysaccharides. Science 330, 219–222 [DOI] [PubMed] [Google Scholar]
- 5. Quinlan R. J., Sweeney M. D., Lo Leggio L., Otten H., Poulsen J. C., Johansen K. S., Krogh K. B., Jørgensen C. I., Tovborg M., Anthonsen A., Tryfona T., Walter C. P., Dupree P., Xu F., Davies G. J., Walton P. H. (2011) Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U.S.A. 108, 15079–15084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horn S. J., Vaaje-Kolstad G., Westereng B., Eijsink V. G. (2012) Novel enzymes for the degradation of cellulose. Biotechnol. Biofuels 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Leggio L. L., Welner D., De Maria L. (2012) A structural overview of GH61 proteins—fungal cellulose degrading polysaccharide monooxygenases. Comput. Struct. Biotechnol. J. 2, e201209019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hemsworth G. R., Davies G. J., Walton P. H. (2013) Recent insights into copper-containing lytic polysaccharide mono-oxygenases. Curr. Opin. Struct. Biol. 23, 660–668 [DOI] [PubMed] [Google Scholar]
- 9. Fushinobu S. (2014) Metalloproteins: a new face for biomass breakdown. Nat. Chem. Biol. 10, 88–89 [DOI] [PubMed] [Google Scholar]
- 10. Eriksson K. E., Habu N., Samejima M. (1993) Recent advances in fungal cellobiose oxidoreductases. Enzyme Microb. Technol. 15, 1002–1008 [Google Scholar]
- 11. Higham C. W., Gordon-Smith D., Dempsey C. E., Wood P. M. (1994) Direct 1H NMR evidence for conversion of β-D-cellobiose to cellobionolactone by cellobiose dehydrogenase from Phanerochaete chrysosporium. FEBS Lett. 351, 128–132 [DOI] [PubMed] [Google Scholar]
- 12. Henriksson G., Johansson G., Pettersson G. (2000) A critical review of cellobiose dehydrogenases. J. Biotechnol. 78, 93–113 [DOI] [PubMed] [Google Scholar]
- 13. Cannella D., Hsieh C. W., Felby C., Jørgensen H. (2012) Production and effect of aldonic acids during enzymatic hydrolysis of lignocellulose at high dry matter content. Biotechnol. Biofuels 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dale M. P., Ensley H. E., Kern K., Sastry K. A., Byers L. D. (1985) Reversible inhibitors of β-glucosidase. Biochemistry 24, 3530–3539 [DOI] [PubMed] [Google Scholar]
- 15. Langston J. A., Shaghasi T., Abbate E., Xu F., Vlasenko E., Sweeney M. D. (2011) Oxidoreductive cellulose depolymerization by the enzymes cellobiose dehydrogenase and glycoside hydrolase 61. Appl. Environ. Microbiol. 77, 7007–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nihira T., Saito Y., Nishimoto M., Kitaoka M., Igarashi K., Ohtsubo K., Nakai H. (2013) Discovery of cellobionic acid phosphorylase in cellulolytic bacteria and fungi. FEBS Lett. 587, 3556–3561 [DOI] [PubMed] [Google Scholar]
- 17. Hildebrand A., Szewczyk E., Lin H., Kasuga T., Fan Z. (2015) Engineering Neurospora crassa for improved cellobiose and cellobionate production. Appl. Environ. Microbiol. 81, 597–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Desai S. H., Rabinovitch-Deere C. A., Fan Z., Atsumi S. (2015) Isobutanol production from cellobionic acid in Escherichia coli. Microb. Cell Fact. 14, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lombard V., Golaconda Ramulu H., Drula E., Coutinho P. M., Henrissat B. (2014) The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hidaka M., Kitaoka M., Hayashi K., Wakagi T., Shoun H., Fushinobu S. (2006) Structural dissection of the reaction mechanism of cellobiose phosphorylase. Biochem. J. 398, 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Hoorebeke A., Stout J., Kyndt J., De Groeve M., Dix I., Desmet T., Soetaert W., Van Beeumen J., Savvides S. N. (2010) Crystallization and x-ray diffraction studies of cellobiose phosphorylase from Cellulomonas uda. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 346–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bianchetti C. M., Elsen N. L., Fox B. G., Phillips G. N., Jr. (2011) Structure of cellobiose phosphorylase from Clostridium thermocellum in complex with phosphate. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 67, 1345–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hidaka M., Honda Y., Kitaoka M., Nirasawa S., Hayashi K., Wakagi T., Shoun H., Fushinobu S. (2004) Chitobiose phosphorylase from Vibrio proteolyticus, a member of glycosyl transferase family 36, has a clan GH-L-like (α/α)6 barrel fold. Structure 12, 937–947 [DOI] [PubMed] [Google Scholar]
- 24. Ekborg N. A., Gonzalez J. M., Howard M. B., Taylor L. E., Hutcheson S. W., Weiner R. M. (2005) Saccharophagus degradans gen. nov., sp. nov., a versatile marine degrader of complex polysaccharides. Int. J. Syst. Evol. Microbiol. 55, 1545–1549 [DOI] [PubMed] [Google Scholar]
- 25. Weiner R. M., Taylor L. E., 2nd, Henrissat B., Hauser L., Land M., Coutinho P. M., Rancurel C., Saunders E. H., Longmire A. G., Zhang H., Bayer E. A., Gilbert H. J., Larimer F., Zhulin I. B., Ekborg N. A., Lamed R., Richardson P. M., Borovok I., Hutcheson S. (2008) Complete genome sequence of the complex carbohydrate-degrading marine bacterium, Saccharophagus degradans strain 2-40 T. PLoS Genet. 4, e1000087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 27. Terwilliger T. C., Berendzen J. (1999) Automated MAD and MIR structure solution. Acta Crystallogr. D Biol. Crystallogr. 55, 849–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 30. Lebedev A. A., Young P., Isupov M. N., Moroz O. V., Vagin A. A., Murshudov G. N. (2012) JLigand: a graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 68, 431–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 32. Zhang H., Moon Y. H., Watson B. J., Suvorov M., Santos E., Sinnott C. A., Hutcheson S. W. (2011) Hydrolytic and phosphorolytic metabolism of cellobiose by the marine aerobic bacterium Saccharophagus degradans 2-40T. J. Ind. Microbiol. Biotechnol. 38, 1117–1125 [DOI] [PubMed] [Google Scholar]
- 33. Pocker Y., Green E. (1973) Hydrolysis of D-glucono-δ-lactone. I. General acid-base catalysis, solvent deuterium isotope effects, and transition state characterization. J. Am. Chem. Soc. 95, 113–119 [DOI] [PubMed] [Google Scholar]
- 34. Fushinobu S., Hidaka M., Hayashi A. M., Wakagi T., Shoun H., Kitaoka M. (2011) Interactions between glycoside hydrolase family 94 cellobiose phosphorylase and glucosidase inhibitors. J. Appl. Glycosci. 58, 91–97 [Google Scholar]
- 35. Kittl R., Kracher D., Burgstaller D., Haltrich D., Ludwig R. (2012) Production of four Neurospora crassa lytic polysaccharide monooxygenases in Pichia pastoris monitored by a fluorimetric assay. Biotechnol. Biofuels 5, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vu V. V., Beeson W. T., Phillips C. M., Cate J. H., Marletta M. A. (2014) Determinants of regioselective hydroxylation in the fungal polysaccharide monooxygenases. J. Am. Chem. Soc. 136, 562–565 [DOI] [PubMed] [Google Scholar]
- 37. Igarashi K., Samejima M., Eriksson K. E. (1998) Cellobiose dehydrogenase enhances Phanerochaete chrysosporium cellobiohydrolase I activity by relieving product inhibition. Eur. J. Biochem. 253, 101–106 [DOI] [PubMed] [Google Scholar]
- 38. Nouaille R., Matulova M., Pätoprstý V., Delort A. M., Forano E. (2009) Production of oligosaccharides and cellobionic acid by Fibrobacter succinogenes S85 growing on sugars, cellulose and wheat straw. Appl. Microbiol. Biotechnol. 83, 425–433 [DOI] [PubMed] [Google Scholar]
- 39. Southall S. M., Doel J. J., Richardson D. J., Oubrie A. (2006) Soluble aldose sugar dehydrogenase from Escherichia coli: a highly exposed active site conferring broad substrate specificity. J. Biol. Chem. 281, 30650–30659 [DOI] [PubMed] [Google Scholar]
- 40. Forsberg Z., Vaaje-Kolstad G., Westereng B., Bunæs A. C., Stenstrøm Y., MacKenzie A., Sørlie M., Horn S. J., Eijsink V. G. (2011) Cleavage of cellulose by a CBM33 protein. Protein Sci. 20, 1479–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forsberg Z., Mackenzie A. K., Sørlie M., Røhr Å. K., Helland R., Arvai A. S., Vaaje-Kolstad G., Eijsink V. G. (2014) Structural and functional characterization of a conserved pair of bacterial cellulose-oxidizing lytic polysaccharide monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 111, 8446–8451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor L. E., 2nd, Henrissat B., Coutinho P. M., Ekborg N. A., Hutcheson S. W., Weiner R. M. (2006) Complete cellulase system in the marine bacterium Saccharophagus degradans strain 2-40T. J. Bacteriol. 188, 3849–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Watson B. J., Zhang H., Longmire A. G., Moon Y. H., Hutcheson S. W. (2009) Processive endoglucanases mediate degradation of cellulose by Saccharophagus degradans. J. Bacteriol. 191, 5697–5705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gardner J. G., Crouch L., Labourel A., Forsberg Z., Bukhman Y. V., Vaaje-Kolstad G., Gilbert H. J., Keating D. H. (2014) Systems biology defines the biological significance of redox-active proteins during cellulose degradation in an aerobic bacterium. Mol. Microbiol. 94, 1121–1133 [DOI] [PubMed] [Google Scholar]
- 45. Suvorov M., Kumar R., Zhang H., Hutcheson S. (2011) Novelties of the cellulolytic system of a marine bacterium applicable to cellulosic sugar production. Biofuels 2, 59–70 [Google Scholar]
- 46. Nakai H., Kitaoka M., Svensson B., Ohtsubo K. (2013) Recent development of phosphorylases possessing large potential for oligosaccharide synthesis. Curr. Opin. Chem. Biol. 17, 301–309 [DOI] [PubMed] [Google Scholar]