

Background: Regulation of NMDA receptors is critical for excitatory neurotransmission.
Results: N-glycans are essential for NMDA receptor release from the endoplasmic reticulum and for receptor affinity for the agonist.
Conclusion: N-glycosylation regulates the trafficking and function of NMDA receptors.
Significance: We identified a novel mechanism that could ensure that postsynaptic membranes contain sufficient numbers of NMDA receptors.
Keywords: endoplasmic reticulum (ER), glutamate receptor, glycosylation, neuron, synapse, trafficking
Abstract
NMDA receptors (NMDARs) comprise a subclass of neurotransmitter receptors whose surface expression is regulated at multiple levels, including processing in the endoplasmic reticulum (ER), intracellular trafficking via the Golgi apparatus, internalization, recycling, and degradation. With respect to early processing, NMDARs are regulated by the availability of GluN subunits within the ER, the presence of ER retention and export signals, and posttranslational modifications, including phosphorylation and palmitoylation. However, the role of N-glycosylation, one of the most common posttranslational modifications, in regulating NMDAR processing has not been studied in detail. Using biochemistry, confocal and electron microscopy, and electrophysiology in conjunction with a lentivirus-based molecular replacement strategy, we found that NMDARs are released from the ER only when two asparagine residues in the GluN1 subunit (Asn-203 and Asn-368) are N-glycosylated. Although the GluN2A and GluN2B subunits are also N-glycosylated, their N-glycosylation sites do not appear to be essential for surface delivery of NMDARs. Furthermore, we found that removing N-glycans from native NMDARs altered the receptor affinity for glutamate. Our results suggest a novel mechanism by which neurons ensure that postsynaptic membranes contain sufficient numbers of functional NMDARs.
Introduction
The main ionotropic glutamate receptors are AMPA receptors and NMDA receptors (NMDARs),5 both of which are present, usually together, at the postsynaptic membrane of most excitatory synapses in the mammalian brain (1–3). Recent studies suggest that abnormal regulation of NMDARs plays a fundamental role in the development of many neurological and psychiatric disorders, including Alzheimer disease, Parkinson disease, epilepsy, and schizophrenia (1, 4). Therefore, understanding the mechanisms that regulate both the trafficking and function of NMDARs is essential for developing novel treatments for these disorders. NMDARs contain a combination of seven principal subunits (GluN1, GluN2A through GluN2D, and GluN3A and GluN3B), all of which share a common membrane topology that includes four membrane domains (called M1 through M4), an extracellular N terminus, an extracellular loop connecting the M3 and M4 domains, and an intracellular C terminus (3). Both the number and type of NMDARs at the cell surface are regulated at multiple levels, including protein synthesis, subunit assembly, processing in the endoplasmic reticulum (ER), intracellular trafficking via the Golgi apparatus (GA), internalization, recycling, and degradation (1–4).
Previous studies have shown that both endogenous and recombinant GluN subunits are extensively N-glycosylated (5–9) and that the presence of N-glycans regulates the trafficking and function of NMDARs in heterologous expression systems (10, 11). However, the precise role of specific N-glycosylation sites on GluN subunits is not clearly understood with respect to regulating ER processing, intracellular trafficking, and/or gating of functional NMDARs. Given the wide diversity of glycan structures in mammalian cells (more than 7000 structures have been identified to date), N-glycosylation likely provides an additional level of regulation with respect to the molecular mechanisms that regulate NMDARs (12).
In this study, we examined the role of conventional N-glycosylation sites in the trafficking and function of NMDARs. We identified two N-glycosylation sites in the GluN1 subunit (Asn-203 and Asn-368) that are essential for releasing functional NMDARs from the ER. Interestingly, other N-glycosylation sites in the GluN1, GluN2A, and GluN2B subunits likely do not play a major role in the surface delivery of NMDARs. Finally, we found that removing N-glycans alters the affinity of native NMDARs for agonist NMDA, thereby altering the functional properties of these receptors. Therefore, we identified a novel molecular mechanism that controls the ER processing and functioning of NMDARs in mammalian neurons.
Experimental Procedures
Mammalian Expression Vectors, Antibodies, and Lentivirus Production
The following cDNAs encoding the respective full-length NMDAR subunits were used: extracellular tagged yellow fluorescent protein GluN1-1a (YFP-GluN1-1a); extracellular tagged green fluorescent protein GluN2A (GFP-GluN2A); GFP-GluN2B; and untagged versions of GluN1, GluN2A, and GluN2B (13–15). Point mutations were introduced using the QuikChange site-directed mutagenesis kit (Agilent Technologies) in accordance with the instructions of the manufacturer. All mutations in this study were introduced in the YFP/GFP-tagged GluN subunits, and the entire GluN-coding region of each cDNA construct was sequenced to verify the DNA sequences.
The following primary antibodies were used in this study: mouse anti-protein disulfide isomerase (PDI) (1:200, Abcam), rabbit anti-GM130 (1:200, Sigma), rabbit anti-GFP (1:1000, Merck Millipore), mouse anti-GFP (1:1000, Abcam), mouse anti-GluN1 (1:2000, Affinity BioReagents), and mouse anti-PSD-95 (1:1000, Merck Millipore). The following secondary antibodies were used: Alexa Fluor 488/647-conjugated goat anti-mouse/rabbit (1:1000, Life Technologies) and horseradish peroxidase-conjugated donkey anti-rabbit IgG (1:1000, Amersham Biosciences).
The FHUGW lentivirus vector construct was used to generate lentiviruses (16). To knock down endogenous GluN1, 20 sense nucleotides in the GluN1 target sequence (gac cgg aag ttt gcc aac ta; with a short hairpin (AAGCTT) and 20 antisense nucleotides were cloned downstream of the H1 promoter (17). For GluN1 rescue studies, silent mutations (gac cgC aaA ttC gcG aac ta, the mutated nucleotides are indicated in capital letters) that resist the target shRNA knockdown were introduced using PCR-based site-directed mutagenesis in the YFP-GluN1-1a or non-glycosylated mutants. For the non-glycosylated GluN1-10N→10Q subunit, an oligonucleotide primer sequence (gac cgC aaA ttC gcG CAA ta) was used to retain the N350Q mutation. YFP-GluN1-1a (or non-glycosylated mutant cDNAs containing silent mutations) were then cloned into the same FHUGW lentiviral vector (pH1-GluN1 shRNA-pUb-YFP-GluN1) under the control of the Ub promoter (18). The following lentiviruses were used in this study: control::GFP (GFP), GluN1-KD::GFP (GluN1-KD), GluN1-KD::YFP-rGluN1-1a (rGluN1), GluN1-KD::YFP-rGluN1-1a-N203Q-N368Q (rGluN1-N203Q-N368Q), and GluN1-KD::YFP-rGluN1-1a-10N→10Q (rGluN1-10N→10Q) (KD, knockdown; r, shRNA-resistant).
To produce lentivirus particles, HEK293T cells were cotransfected with vesicular stomatitis virus glycoprotein, Δ8.9, and lentiviral vectors using X-tremeGENE HD (Roche) in accordance with the instructions of the manufacturer. After 12 h of incubation, the medium was replaced with plain Neurobasal medium (Life Technologies) containing 2 mm l-glutamine and 1× ITS (5 μg/ml insulin, 5 μg/ml transferrin, and 5 ng/ml sodium selenite; Sigma). The culture supernatant containing the lentivirus particles was harvested after 36 h of incubation and then centrifuged at 1800 × g for 15 min at 4 °C to remove cellular debris. The supernatant was then aliquoted or concentrated by centrifugation at 82,700 × g for 90 min and frozen at −80 °C.
Mammalian Cell Culture and Transfection
Heterologous African green monkey kidney fibroblast (COS-7) and HEK293 cells were maintained in Opti-MEM I medium containing 5% FBS (v/v) and transfected with the cDNA constructs using Lipofectamine 2000 (Life Technologies) as described previously (14). To obtain individual HEK293 cells suitable for electrophysiology, cells were trypsinized after transfection and resuspended in Opti-MEM I containing 1% FBS, 20 mm MgCl2, 1 mm d,l-2-amino-5-phosphonopentanoic acid, and 3 mm kynurenic acid (to prevent cell death caused by activation of NMDARs). The cells were then plated on poly-l-lysine-coated glass coverslips. The cells used for microscopy and biochemistry were cultured in medium containing the NMDAR antagonists but were not trypsinized. The experiments were performed within 24–48 h of transfection.
Preparation of Primary Hippocampal Neurons and Cerebellar Granule Cells, Including DNA Transfection/Infection
All animal experiments were performed in accordance with relevant institutional ethical guidelines and regulations protecting animal welfare. Primary cultures of hippocampal neurons were prepared from embryonic day 18 Sprague-Dawley rats. In brief, fetal rat hippocampi were isolated in cold dissection solution consisting of Hanks' balanced salt solution supplemented with 10 mm HEPES (pH 7.4) and penicillin-streptomycin (Life Technologies). The tissue was then incubated for 12 min at 37 °C in chopping solution, which consisted of dissection solution supplemented with 0.1 mg/ml deoxyribonuclease I and 0.05% trypsin (Sigma). Neurons were washed three times with chopping solution and dissociated by triturating 10–15 times through a fire-polished glass pipette. The dissociated neurons were pelleted by centrifugation at 900 × g for 3 min at 4 °C and resuspended in plating medium, which consisted of serum-free Neurobasal medium with B-27 supplement and l-glutamine (Life Technologies). Neurons were plated on poly-d-lysine (Sigma)-coated dishes at a density of ∼2 × 104 cells/cm2. Neurons were fed every 2–3 days with plating medium. The neurons were infected on days in vitro 5–7 and used for experiments 10 days after infection.
Primary cultures of rat cerebellar granule cells (CGCs) were prepared from postnatal day 6–8 rat cerebella as described previously (19, 20). In brief, cells were cultured for the entire period in basal Eagle's medium (Life Technologies) supplemented with 10% FBS (v/v), 2 mm glutamine, and 25 mm KCl. Where appropriate, the neurons were transfected on day in vitro 5 using a modified calcium phosphate method (19, 20). Microscopy experiments were performed within 48–72 h of transfection. Electrophysiology experiments were performed on cultures on days in vitro 6–8.
Quantitative Measurements of Surface and Total Expression
Transfected COS-7 cells grown in 12-well plates were washed with PBS, fixed for 15 min in 4% paraformaldehyde (PFA) dissolved in PBS, and incubated for 1 h in PBS containing 10% normal goat serum either without (for surface labeling) or with (for total labeling) 0.1% Triton X-100, followed by 1 h of incubation in the primary rabbit anti-GFP antibody (in PBS containing 3% normal goat serum (21)). The cells were then washed with PBS, incubated with horseradish peroxidase-conjugated donkey anti-rabbit IgG for 1 h, washed with PBS, and incubated for 30 min in ortho-phenylenediamine dissolved in phosphate citrate buffer containing sodium phosphate (Sigma). The reaction was terminated with 3 m HCl, and optical density was measured at 492 nm using a Personal Densitometer SI (Molecular Dynamics). In each experiment, we calculated the average background surface as well as the total signal from three separate wells containing cells transfected with an empty pcDNA3 vector. These values were then subtracted from the data obtained from the cells that were transfected with NMDAR subunits. We performed each experiment in triplicate and repeated each experiment three times, resulting in nine separate values for each combination of GluN subunits. The data were calculated as the ratio of surface to total expression and then normalized to average data obtained from cells expressing a control NMDAR subunit combination.
Electrophysiology
Whole-cell patch clamp recordings from transfected HEK293 cells and cultured CGCs were performed using an Axopatch 200B patch clamp amplifier (Molecular Devices). The extracellular solution contained 160 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 10 mm glucose, 0.2 mm EDTA, and 0.7 mm CaCl2 (pH adjusted to 7.3 with NaOH). The intracellular solution contained 125 mm gluconic acid, 15 mm CsCl, 5 mm BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N,N-tetraacetic acid), 10 mm HEPES, 3 mm MgCl2, 0.5 mm CaCl2, and 2 mm ATP-magnesium salt (pH adjusted to 7.2 with CsOH). Patch pipettes (3–5 MΩ tip resistance) were prepared using a model P-97 horizontal micropipette puller (Sutter Instrument Co.). A microprocessor-controlled multibarrel rapid perfusion system (with a time constant for solution exchange around the cell of 10–20 ms) was used to apply extracellular solutions containing various agonists and antagonists (20). Stock solutions of NMDA, glutamate, and glycine (Sigma) were prepared in distilled water. MK-801 maleate (dizocilpine, Tocris) was dissolved in dimethyl sulfoxide. The final concentration of dimethyl sulfoxide in the working solution was <0.1%. The peptide-N-glycosidase F (PNGase F) treatment of live CGCs was performed in PBS containing 1 mm MgCl2 and 0.1 mm CaCl2 (PBS++) solution at 37 °C. All electrophysiology experiments were performed at room temperature.
Immunofluorescence Microscopy
Surface NMDARs were labeled as described previously (20, 22). In brief, the cells were washed in PBS and then incubated on ice for 10 min in blocking solution containing PBS and 10% normal goat serum (v/v). Then the cells were incubated in the primary antibody (in blocking solution) for 30 min. After the cells were washed in PBS, they were incubated in the secondary antibody (conjugated with fluorescent dye and diluted in blocking solution) for 30 min. The cells were washed twice in PBS, fixed in 4% PFA in PBS (w/v) for 20 min, and mounted using ProLong Antifade reagent (Life Technologies). Intracellular NMDAR subunits and the various cellular compartments were labeled at room temperature (22). First, cells were fixed with 4% PFA for 30 min and then permeabilized in 0.1% Triton X-100 in PBS for 5 min. The cells were then blocked with blocking solution containing 0.1% Triton X-100 for 1 h, incubated in primary antibody for 1 h, and then incubated in secondary antibody for 1 h. Images were acquired at room temperature using a confocal scanning microscope (Leica TCS SPE) fitted with solid-state lasers and a ×63/1.30 oil immersion apochromat objective. The images were analyzed using ImageJ software (National Institutes of Health). For hippocampal neurons, the intensity of the surface and total YFP signals was analyzed in an area of 10 μm2 in segments of secondary and tertiary dendrites. The numbers of PSD-95 and YFP puncta were counted in 10-μm-long segments of secondary and tertiary dendrites, as indicated in the figures. The NMDAR subunits expressed in the CGCs were quantified as described previously (20).
Surface Biotinylation Assays in Primary Hippocampal Neurons and HEK293 Cells
The surface biotinylation assay was performed as described previously (18). In brief, primary hippocampal neurons or HEK293 cells were washed three times with ice-cold PBS++ and then incubated in 1 mg/ml membrane-impermeable EZ-Link Sulfo-NHS-SS-biotin (Thermo Scientific) in PBS for 20 min at 4 °C with gentle rocking. The biotinylation reaction was terminated by adding biotin quenching solution (ice-cold PBS++ containing 50 mm glycine). The biotinylated cells were washed three additional times in biotin-quenching solution for 4 min with gentle rocking and then harvested in TNE buffer (50 mm Tris-HCl (pH 8.0), 150 mm NaCl, and 2 mm EDTA) containing complete protease inhibitors. The cells were sonicated for 10 s and then centrifuged at 120,000 × g for 20 min at 4 °C. The resulting pellet was resuspended in TNE buffer plus complete protease inhibitor and then solubilized in 1% SDS plus 1% Triton X-100. Insoluble materials were removed by centrifugation at 20,000 × g for 15 min at 4 °C. The biotinylated lysate was incubated with streptavidin-agarose beads (Thermo Scientific) for 3 h at 4 °C, followed by four washes. The isolated surface fractions were analyzed by Western blot using the appropriate antibodies.
Pre-embedding Electron Microscopy Immunoperoxidase
Cell cultures were fixed and labeled as reported previously (23, 24). In brief, cultures were fixed in 4% PFA and 0.1% glutaraldehyde. The cells were then incubated sequentially with the primary and secondary antibodies, followed by 3,3′-diaminobenzidine. The cells were again fixed in 4% PFA and 1% glutaraldehyde, followed by 1% osmium tetroxide, 2% glutaraldehyde, and dehydration in a series of alcohol solutions (including en bloc staining with uranyl acetate). After embedding in epon resin, the glass coverslips were dissolved in hydrofluoric acid, and the epon-embedded cultures were sectioned and examined using a JEOL 2100 transmission electron microscope at 200 kV. Images were captured using a Gatan digital camera and analyzed using Digital Micrograph software. Diaminobenzidine produces an intense black reaction product, visible by transmission electron microscopy, that is centered on the epitope site of the antibody. This signal can occasionally spread slightly to adjacent structures.
Statistical Analysis
Group differences were analyzed using unpaired Student's t test or one-way ANOVA followed by the Student-Newman-Keuls test.
Results
Specific N-Glycosylation Sites within GluN1 but Not GluN2A or GluN2B Regulate the Surface Delivery of NMDARs
The GluN1, GluN2A, and GluN2B subunits contain twelve, seven, and seven N-glycosylation consensus sites (N-X-S/T, Fig. 1A), respectively. We hypothesized that specific N-glycosylation sites in GluN1 and/or GluN2 subunits are essential for early trafficking of NMDARs. We first tested this hypothesis in COS-7 cells expressing GluN1/GluN2 receptors in which single N-glycosylation consensus sites were mutated (by replacing the Asn residue with a Gln residue, which is not glycosylated) using a quantitative colorimetric surface expression assay (14). We found that there are two essential N-glycosylation sites in GluN1 (N203Q and N368Q, Fig. 1B). However, mutating any one N-glycosylation site in GluN2A or GluN2B did not reduce surface delivery of the GluN1/GluN2 receptors (Fig. 1, C and D). To confirm that the observed reduction in the surface delivery of GluN1-N203Q/GluN2A and GluN1-N368Q/GluN2A receptors is specifically due to a lack of N-glycosylation at their respective sites, we replaced critical adjacent residues with an alanine (e.g. Thr → Ala) or a proline residue to disrupt N-glycosylation, as reported previously (25). Our quantitative assay revealed that replacing Val-204 with an Ala (V204A), which should not prevent N-glycosylation at this site, did not affect the surface delivery of NMDARs. However, the single V204P and T205A mutations and the double V204A/T205A mutation significantly reduced surface delivery of NMDARs, supporting our hypothesis that glycosylation of Asn-203 is required for surface delivery of the NMDAR (Fig. 1E). Similarly, we examined the effect of mutating residues adjacent to Asn-368 and found that the single G369A and T370A mutations and the double G369A/T370A mutation significantly reduced surface delivery of the NMDAR (Fig. 1F). We suggest that the essential role of the Gly-369 residue in the trafficking of NMDARs may be explained by the presence of a non-conventional N-glycosylation site (NG), which, in some cases, can be functionally similar to a conventional N-X-S/T consensus site (12).
FIGURE 1.
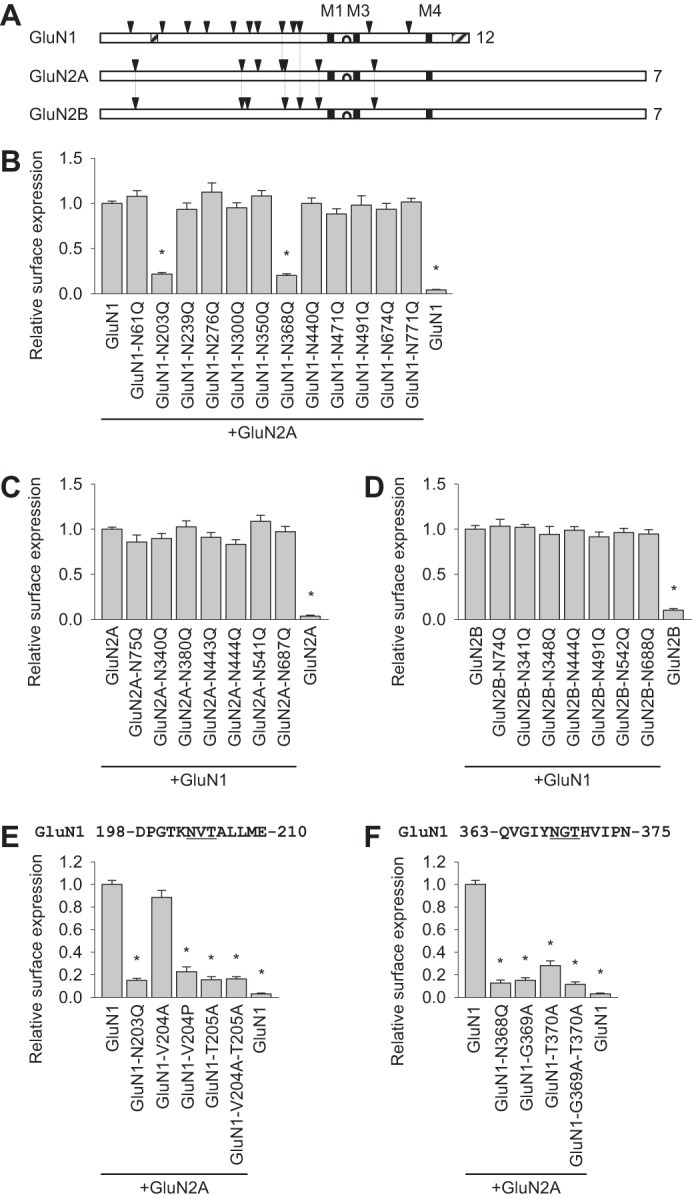
Specific N-glycosylation sites within the GluN1 subunit regulate the surface expression of NMDARs in mammalian cell lines. A, schematic of the GluN1, GluN2A, and GluN2B subunits. Arrowheads indicate the location of extracellular N-glycosylation consensus sites (N-X-S/T), and the total number of sites is shown at the right. The vertical lines indicate the sites that are conserved at homologous positions among the subunits. The black squares indicate the transmembrane domains (M1, M3, and M4), and the hairpin loop that forms the channel pore is indicated between M1 and M3. The hatched rectangles indicate alternatively spliced domains in the GluN1 subunit. Adapted from Ref. 10. B–F, COS-7 cells expressing the indicated wild-type or mutant YFP-GluN1-1a subunits (GluN1; B, E, and F) with or without the GluN2A subunit or wild-type or mutant GFP-GluN2A (GluN2A, C) and GFP-GluN2B subunits (GluN2B, D) with or without the GluN1-1a subunit were stained using anti-GFP antibodies under non-permeabilizing (surface labeling) or permeabilizing (to measure total receptor levels) conditions and measured using a quantitative colorimetric assay. The bar graphs show the relative surface expression (surface/total expression signals) of the indicated NMDAR subunit combinations measured in triplicate from three independent experiments (n = 9). *, p < 0.05 versus control (wild-type GluN1/GluN2A or GluN1/GluN2B), analyzed by one-way ANOVA. In E and F, the sequences of the GluN1 subunits surrounding the Asn-203 (E) and Asn-368 (F) residues are shown. The conventional N-glycosylation consensus sites are underlined. In this and subsequent figures, the summary data are presented as mean ± S.E.
Next we further analyzed NMDARs containing GluN1 subunits that lack specific N-glycosylation sites. Then we generated a GluN1-1a subunit lacking both critical N-glycosylation sites (GluN1-N203Q-N368Q) and, to complement this mutant, a GluN1-1a subunit lacking the other ten N-glycosylation sites (but retaining the Asn-203 and Asn-368 residues, GluN1-10N→10Q) or lacking all 12 N-glycosylation sites (GluN1-12N→12Q). These subunits were then expressed together with GluN2A in HEK293 cells and analyzed using a gel shift assay. We observed altered gel mobility between the GluN1 subunits containing one (GluN1-N203Q and GluN1-N368Q), two (GluN1-N203Q-N368Q), ten (GluN1-10N→10Q), and 12 (GluN1-12N→12Q) asparagine replacements, indicating that at least two of these asparagines (Asn-203 and Asn-368) are glycosylated in our cell expression system (Fig. 2A). This conclusion is consistent with a recent structural model of the NMDAR, which shows glycans present at Asn-203 and Asn-368 in the GluN1 subunit (PDB code 4PE5 26) and with a recent study of glycosylation at Asn-368 (25). Next we incubated recombinant NMDARs with PNGase F, an enzyme that removes all forms of N-glycans from glycoproteins. We observed a large shift in the mobility of control GluN1/GluN2A receptors as well as GluN1-N203Q-N368Q/GluN2A receptors (Fig. 2B). Interestingly, however, the GluN1-12N→12Q subunit had no shift in gel mobility after treatment with PNGase F (Fig. 2B). Together, our data indicate that GluN1/GluN2A receptors are heavily glycosylated at multiple N-glycosylation sites, including the Asn-203 and Asn-368 residues in the GluN1 subunit.
FIGURE 2.

Biochemical characterization of NMDARs containing GluN1 subunits that lack specific N-glycosylation sites. A, HEK293 cells were cotransfected with either wild-type or mutant YFP-GluN1-1a subunits (GluN1) together with the GluN2A subunit. 24 h after transfection, the GluN1 subunit was examined by Western blot analysis. B, HEK293 cells were cotransfected with either wild-type or mutant YFP-GluN1-1a subunits (GluN1) together with the GluN2A subunit. 24 h after transfection, cell homogenates were incubated in the presence or absence of PNGase F and analyzed by Western blot using either the anti-GFP antibody (to detect GluN1) or the anti-GluN2A antibody. C, cell surface biotinylation assay for the indicated YFP-GluN1-1a (GluN1) subunits coexpressed with the GluN2A subunit. HEK293 cells were transfected with the indicated YFP-GluN1-1a and GluN2A subunits. The surface receptors were biotinylated and pulled down using streptavidin-agarose resin. Total input (5% of the lysate) and the surface GluN1 receptor were detected using anti-GluN1 antibody. Anti-tubulin antibody was used to confirm the integrity of the assay. D, summary of the experiments in C. The band intensities of the surface and total NMDAR pools were quantified as described under “Experimental Procedures” (n = 5). *, p < 0.05 versus wild-type GluN1/GluN2A, analyzed by one-way ANOVA. E and F, COS-7 cells expressing the indicated wild-type or mutant YFP-GluN1-1a subunit (GluN1) with or without the GluN2A (E) or GluN2B (F) subunits were labeled using anti-GFP under non-permeabilizing and permeabilizing conditions. Expression was measured using a quantitative colorimetric assay. The bar graphs show the relative surface expression of the indicated GluN subunit combinations from three independent experiments (n = 9). *, p < 0.05 versus wild-type GluN1/GluN2A or GluN1/GluN2B receptors, analyzed by one-way ANOVA.
Next we investigated further whether glycosylation of the Asn-203 and Asn-368 residues in GluN1 is essential for surface delivery of GluN1/GluN2A receptors by performing a surface biotinylation assay in HEK293 cells (Fig. 2, C and D). We also performed a quantitative assay using COS-7 cells (Fig. 2E). In all cases, receptors containing the GluN1-N203Q-N368Q mutant subunits had markedly reduced numbers of receptors at the cell surface compared with both wild-type GluN1/GluN2A and GluN1-10N→10Q/GluN2A receptors. Similar results were observed when the mutant GluN1 subunits were expressed with GluN2B (Fig. 2F). Together, these results support the hypothesis that glycosylation at Asn-203 and Asn-368 in the GluN1 subunit plays an essential role in intracellular processing of NMDARs in mammalian cells.
The Asn-203 and Asn-368 Residues in the GluN1 Subunit Are Essential for Releasing the NMDAR from the ER
Next we examined whether Asn-203 and Asn-368 in GluN1 are essential for the delivery of NMDARs in neurons, including the cell surface and excitatory synapses. Because the GluN1 subunit is expressed in excess levels in neurons (27), we used a lentivirus-based molecular replacement system in which the endogenous GluN1 subunit was reduced by specific shRNA knockdown (GluN1-KD) and replaced with an shRNA-resistant, YFP-tagged wild-type or mutant recombinant GluN1-1a subunit (rGluN1, rGluN1-N203Q-N368Q, rGluN1-10N→10Q) (18). Using biochemistry in lentivirus-infected primary hippocampal neurons, we confirmed that the GluN1-KD lentivirus effectively knocks down expression of the endogenous GluN1 subunit. Moreover, the rGluN1 subunit restored both total and surface expression of the GluN1 subunit (Fig. 3, A and B). Similar to our experiments performed in HEK293 and COS-7 cells, replacing Asn-203 and Asn-368 with glutamines significantly reduced surface expression compared with wild-type rGluN1. Mutating the other ten N-glycosylation sites (rGluN1-10N→10Q) also significantly reduced surface expression, although not to the same extent as rGluN1-N203Q-N368Q (Fig. 3, A and B). Therefore, these results show that, in hippocampal neurons, the Asn-203 and Asn-368 residues in the GluN1 subunit are essential for delivering NMDARs to the cell surface. In the GluN1 blot, the exogenously expressed proteins run as double bands, which may be due to the posttranslational modification of overexpressed proteins such as O-glycosylation. We suggest that the lower band is not likely a truncated form of overexpressed proteins, but, rather, it is a regular form because N-terminal tagged anti-YFP antibody appears to detect a lower band. In addition, the surface-expressed GluN1s in the upper position were detected less efficiently by anti-GluN1 antibody. The posttranslational modification may reduce the affinity of GluN1 antibody, which recognizes the extracellular domain of GluN1.
FIGURE 3.
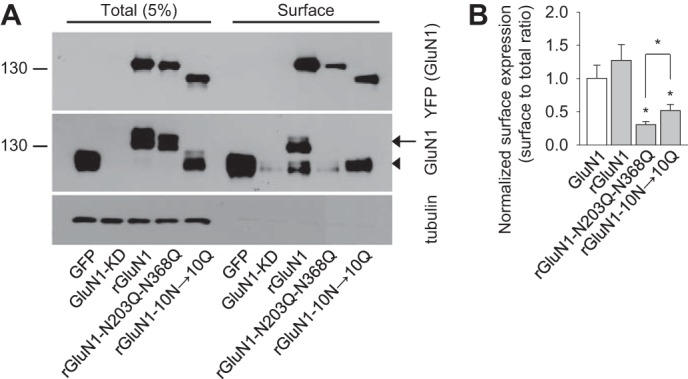
GluN1-N203Q-N368Q subunits have reduced surface expression in hippocampal neurons. A, example of a cell-surface biotinylation assay from dense cultures of hippocampal neurons infected with the following lentiviruses: control::GFP (GFP), GluN1-KD::GFP (GluN1-KD), GluN1-KD::YFP-rGluN1-1a (rGluN1), GluN1-KD::YFP-rGluN1-1a-N203Q-N368Q (rGluN1-N203Q-N368Q), or GluN1-KD::YFP-rGluN1-1a-10N→10Q (rGluN1-10N→10Q). The surface NMDARs were biotinylated and pulled down using streptavidin-agarose resin. Total input (5% of the lysate) and surface NMDARs were detected using anti-GluN1 and anti-GFP antibodies. Anti-tubulin antibody was used to confirm the integrity of the assay. In the GluN1 blot, the arrow indicates either rGluN1 or rGluN1-N203Q-N368Q. The arrowhead indicates either endogenous GluN1 or YFP-rGluN1-10N→10Q. B, summary of the band intensities of the surface and total NMDAR pools from the blots in A (n = 6). *, p < 0.05 versus wild-type rGluN1, analyzed by one-way ANOVA. The surface to total ratio of endogenous GluN1 level (GluN1), which was quantified from the anti-GluN1 blot, is included in the graph, together with the data obtained with the recombinant rGluN1 subunits (rGluN1, rGluN1-N203Q-N368Q, rGluN1-10N→10Q), quantified from the anti-YFP blot.
Next we used confocal microscopy to determine in which subcellular compartment the mutant GluN1 subunits are retained. First we labeled the surface pool of recombinant GluN1 subunits expressed in hippocampal neurons (Fig. 4, A and B). These experiments yielded similar results as our biotinylation assays. Specifically, wild-type rGluN1 and rGluN1-10N→10Q subunits were present at the cell surface, whereas rGluN1-N203Q-N368Q subunits were not. We found similar results using cultured CGCs transfected with GluN1 subunits (Fig. 4, C and D). Interestingly, when expressed in hippocampal neurons, the rGluN1-N203Q-N368Q subunit showed a different pattern in terms of total expression compared with wild-type rGluN1 and rGluN1-10N→10Q subunits. Specifically, no clusters were observed (Fig. 4A). This finding suggests that the rGluN1-N203Q-N368Q subunit may not reach excitatory synapses. To test this possibility, we stained infected hippocampal neurons using anti-PSD-95 antibody to label the postsynaptic compartment (Fig. 4, E and F). We found that wild-type rGluN1 and rGluN1-10N→10Q subunits were present in PSD-95-positive clusters, whereas the rGluN1-N203Q-N368Q subunit was clearly retained in other somatic compartments and dendritic structures.
FIGURE 4.

Confocal analysis of fixed neurons expressing recombinant GluN1 subunits. A, dense cultures of hippocampal neurons were immunostained for surface and total YFP under non-permeabilizing and permeabilizing conditions, respectively. Representative images of total (left column) and surface (right column) immunoreactivity are shown. Below each image is a higher magnification image of the indicated region indicated. B, summary of the fluorescence intensity per unit area of surface and total YFP expression for the indicated rGluN1 subunits expressed in hippocampal neurons obtained from a 10-μm2 region of interest in segments of secondary and tertiary dendrites (n ≥ 25 segments from ≥ 5 different cells). *, p < 0.05, analyzed by one-way ANOVA. C, dense cultures of CGCs were immunostained for surface and total YFP under non-permeabilizing and permeabilizing conditions, respectively. Representative images of total (left column) and surface (right column) immunoreactivity are shown. Below each image is a higher magnification image of the region indicated. D, summary of the fluorescence intensity per unit area of surface and total YFP expression for the indicated rGluN1 subunits expressed in CGCs obtained from a 10-μm2 region of interest in segments of primary dendrites (n ≥ 10 different cells). *, p < 0.05, analyzed by one-way ANOVA (20). E, hippocampal neurons were immunostained for YFP (GluN1) and PSD-95 under permeabilizing conditions. Representative images of the YFP channel (left column), the PSD-95 channel (center column), and merged channels (right column) are shown. Below each image is a higher magnification image of the region indicated. F, summary of the fraction of PSD-95 puncta that colocalized with the indicated rGluN1 subunits measured in 10-μm segments of secondary and tertiary dendrites (n ≥ 30 segments from ≥ 20 different hippocampal neurons). *, p < 0.05, analyzed by one-way ANOVA. G and H, the distribution of the indicated rGluN1 subunits in hippocampal neurons colocalizes with the ER marker PDI (G) but not with the GA marker GM130 (H). In each panel, YFP is shown in the left column, PDI or GM130 is shown in the center column, and the merge is shown in the right column. Below each image is a higher magnification image of the region indicated. The primary antibodies used in this figure were as follows: rabbit anti-GFP for surface rGluN1 labeling in A–D and total rGluN1 labeling in E–G, mouse anti-GFP for total rGluN1 labeling in A–D and H, mouse anti-PSD-95 in E and F, mouse anti-PDI in G, and rabbit anti-GM130 in H. The following corresponding secondary antibodies were used: Alexa Fluor 647-conjugated goat anti-rabbit and Alexa Fluor 488-conjugated goat anti-mouse in A–D and H and Alexa Fluor 647-conjugated goat anti-mouse and Alexa Fluor 488-conjugated goat anti-rabbit in E–G.
To determine the subcellular compartment in which the rGluN1-N203Q-N368Q subunit was retained, we stained cells with antibodies that selectively label the ER and GA. We found that the rGluN1-N203Q-N368Q subunit colocalizes with the anti-PDI antibody, which labels the ER (Fig. 4G). The wild-type rGluN1 subunit also colocalized with PDI, and this result may be explained by the fact that the majority of GluN1 subunits expressed in neurons accumulate in the ER because of the limiting number of GluN2 subunits in neurons (27). Our confocal microscopy experiments also revealed that neither wild-type rGluN1 nor the rGluN1-N203Q-N368Q subunit accumulated in the GA, which was labeled using the anti-GM130 antibody (Fig. 4H). Because the rGluN1-N203Q-N368Q subunit is absent from both the cell surface and the GA, we conclude that this subunit is likely retained in the ER.
Despite our best efforts, we were unable to detect either the dendritic ER subcompartments using the anti-PDI antibody or the Golgi outposts using the anti-GM130 antibody (data not shown). Therefore, we used pre-embedding EM immunoperoxidase labeling in hippocampal neurons infected with wild-type rGluN1 or rGluN1-N203Q-N368Q (Fig. 5, two experiments for each). Uninfected controls showed only a low background reaction (data not shown). Neurons infected with wild-type rGluN1 (Fig. 5) had abundant immunolabeling at the synapse, in early/sorting endosomes, and in the rough endoplasmic reticulum. In contrast, neurons infected with rGluN1-N203Q-N368Q (Fig. 5) had abundant immunolabeling in the rough endoplasmic reticulum but virtually no labeling at synapses or in early/sorting endosomes. Together, these data suggest that the Asn-203 and Asn-368 N-glycosylation sites in the GluN1 subunit are essential for releasing the NMDAR from the ER.
FIGURE 5.

Transmission electron microscopy of immunoperoxidase-labeled wild-type rGluN1 and rGluN1-N203Q-N368Q in infected cultured hippocampal neurons. A–G, rGluN1 immunolabeling is prevalent in synapses (arrows), early/sorting endosomes (arrowheads), and rough endoplasmic reticulum (asterisk). A–F show dendrites, and G shows a soma. A and E show spine synapses, and F shows a dendrite shaft synapse. Note that the intense labeling in the postsynaptic membrane by the diaminobenzidine reaction product appears to have spread slightly into the presynaptic membrane. The synaptic structure in G may be an early contact but is not distinct. D shows a higher magnification image of the fourth marked endosome in C. H–J, in contrast to rGluN1, the rGluN1-N203Q-N368Q subunit (H–J) is absent from synapses (the double arrow shows a spine synapse in H with little or no labeling) and early/sorting endosomes (not shown). In contrast, the rough endoplasmic reticulum (asterisks) shows clear labeling of rGluN1-N203Q-N368Q subunits (H and I are dendrites, and J is a higher magnification of H). Scale bars = 100 nm except for C (200 nm).
The Effect of N-Glycosylation on the Functional Properties of NMDARs
Next we performed electrophysiology experiments in HEK293 cells (Fig. 6, A–D). In agreement with our previous data, receptors containing the GluN1-N203Q-N368Q mutant subunits had markedly reduced peak current amplitudes compared with both wild-type GluN1/GluN2A and GluN1-10N→10Q/GluN2A receptors (Fig. 6, A and B). Then we compared the ratio of the steady-state current amplitudes induced by 10 μm and 1 mm glutamate between control and mutant GluN1/GluN2A receptors. For these experiments, the coagonist glycine was present at a saturating concentration (50 μm, Fig. 6C). Similarly, we measured the ratio of the steady-state current amplitudes mediated by 1 and 50 μm glycine in the presence of glutamate at a saturating concentration (1 mm) among NMDARs containing mutations in the GluN1 subunit (Fig. 6D). These experiments revealed no significant difference in the glutamate and glycine ratios among three studied subunit combinations, even though the GluN1 subunit contains a glycine-binding site (see “Discussion”) (3).
FIGURE 6.

N-glycosylation regulates the functional properties of NMDARs. A and B, whole-cell patch clamp recordings were performed in HEK293 cells expressing the indicated wild-type or mutant YFP-GluN1-1a subunits (GluN1) together with the GluN2A subunit. Currents were elicited in the continuous presence of 50 μm glycine with a 5-s pulse of 1 mm glutamate (horizontal bar). The cells were held at a membrane potential of −60 mV. The graph in B summarizes the peak current amplitude measured for each combination of NMDARs (n ≥ 12 cells). *, p < 0.05 versus wild-type GluN1/GluN2A, analyzed by one-way ANOVA. C and D, whole-cell patch clamp recordings of HEK293 cells transfected with the indicated NMDAR subunits. The bar graph summarizes the ratio of steady-state current amplitudes induced by 10 μm and 1 mm glutamate (C) or 0.5 μm and 100 μm glycine (D) at the indicated NMDAR combinations. n ≥ 8 cells; p > 0.05 versus wild-type GluN1/GluN2A receptors, analyzed by one-way ANOVA. E, H, and J, CGCs were incubated with PNGase F for 60 min (or control-treated) and then subjected to electrophysiological recordings. Shown are representative currents elicited by 1 mm and 50 μm NMDA (black and white horizontal bars, respectively) in the continued presence of 10 μm glycine (E), elicited by 100 μm and 0.5 μm glycine (black and white horizontal bars, respectively) in the presence of 1 mm NMDA (H), and elicited by 1 mm NMDA (black bar) and 1 μm MK-801 (white bar, J). F, G, I, and K, bar graphs summarizing the peak current amplitude induced by 1 mm NMDA (F), the ratio of steady-state currents induced by 50 μm and 1 mm NMDA (G), the ratio of steady-state currents induced by 100 μm and 0.5 μm glycine (I), and the time constant of MK-801-induced inhibition (K). In each graph, the values obtained from PNGase F-treated CGCs were normalized to control responses obtained on the same recording day culture (n ≥ 8 cells). *, p < 0.05 versus control-treated CGCs, analyzed by Student's t test. Note that receptor desensitization was not significantly affected by incubation with PNGase F (data not shown; p > 0.05 versus control, analyzed by Student's t test). L--N, cultured CGCs were incubated for 48 h in the presence of 1 μg/ml tunicamycin and then subjected to electrophysiological recordings as described above. M and N, bar graphs summarizing the peak current amplitude induced by 1 mm NMDA (M) and the ratio of steady-state currents induced by 50 μm and 1 mm NMDA (N). The values obtained from the tunicamycin-treated CGCs were normalized to the control responses obtained on the same recording day. n ≥ 7 cells. *, p < 0.05 versus control-treated CGCs, analyzed by Student's t test. Note that receptor desensitization was not significantly affected by incubation with tunicamycin (data not shown). O, COS-7 cells expressing GluN1-1a/GFP-GluN2A subunits (GluN1/GluN2A) were incubated for 36 h in the presence of 1 μg/ml tunicamycin and labeled with anti-GFP and the appropriate secondary antibody under non-permeabilizing (to measure surface receptors) and permeabilizing conditions, followed by analysis using a quantitative colorimetric assay. The bar graph shows the relative surface expression (surface/total expression signals) of NMDARs normalized to control-treated cells. Each bar represents data obtained from three independent experiments (n = 9). *, p < 0.05 versus control-treated cells, analyzed by Student's t test. Note that the total expression level of the GFP-GluN2A subunit was significantly reduced by treatment with tunicamycin (data not shown; p < 0.05 versus control, analyzed by Student's t test).
Finally we recorded cultured CGCs to test whether the presence of N-glycans on native NMDARs affects their functional properties. Cultured neurons were incubated with a PNGase F, which targets accessible N-glycans on the NMDAR. We observed that the ratios of the steady-state current amplitudes induced by 50 μm and 1 mm NMDA were altered but not the steady-state current amplitudes induced by 0.5 μm and 100 μm glycine and the time constant of MK-801-mediated inhibition (which is used to study the open probability of the NMDARs; Fig. 6, E–K) (28). Indeed, tunicamycin, an inhibitor of N-glycosylation in the ER, significantly reduced the NMDAR peak current amplitudes recorded from the CGCs and the ratios of the steady-state current amplitudes induced by 50 μm and 1 mm NMDA (Fig. 6, L–N) but not the steady-state current amplitudes induced by 0.5 μm and 100 μm glycine (data not shown) (29). In addition, COS-7 cells treated with tunicamycin also exhibited a reduced number of surface NMDARs (Fig. 6O). Together, our data show that the presence of N-glycans affects the trafficking and functional properties of native NMDARs.
Discussion
Here we examined the role of conventional N-glycosylation sites in the GluN1, GluN2A, and GluN2B NMDAR subunits with respect to receptor trafficking and function using a combination of biochemistry, microscopy, and electrophysiology in heterologous expression systems and neurons. Our findings show that N-glycosylation plays an essential role in the release of NMDARs from the ER. Moreover, we found that N-glycosylation regulates the functional properties of native NMDARs. These results could drive future research regarding the diverse roles glycosylation plays in the central nervous system.
With respect to NMDARs, some GluN1 splice variants, and all GluN2 subunits, are retained in the ER until they are assembled into a complete receptor (30–32). Moreover, specific regions within GluN subunits have been suggested to regulate the assembly and/or ER retention of NMDARs. These regions include the C terminus of some GluN1 splice variants (21, 33, 34), the extracellular glycine-binding site in the GluN1 subunit (8), the glutamate-binding site in the GluN2B subunit (35), the membrane domains of both the GluN1 and GluN2 subunits (14, 20, 22), and the N-terminal A2 segments of GluN2A and GluN2C (9, 20). Here we identified a novel molecular mechanism that employs two N-glycosylation sites in the GluN1 subunit, but not in GluN2A or GluN2B, that are essential for the release of the receptor from the ER. Because we used a variety of cell types, including COS-7 cells, HEK293 cells, and cultured hippocampal neurons and CGCs, our findings support the view that ER quality control mechanisms are generally present in mammalian cells and monitor the structure and/or presence of N-glycans on the two critical asparagine residues in the GluN1 subunit.
Did this mechanism develop early in the evolution of NMDARs, or is it a newly emerged mechanism in mammalian cells? A bioinformatics analysis revealed that the N-glycosylation sites at Asn-203 and Asn-368 have been conserved within GluN1 subunits throughout evolution (Fig. 7A), therefore underscoring the high importance of these two sites. Previous studies revealed that some GluN1 splice variants can traffic to the cell surface, even without the GluN2 subunit, because of the lack of an ER retention signal in the C1 cassette and/or the presence of a PDZ-binding motif within the C2′ cassette (21, 33). We found that introducing a single Asn → Gln mutation in GluN1-4a, the main GluN1 subtype that reaches the cell surface without the presence of the GluN2 subunit, reduced the surface delivery of GluN1-4a-N203Q and GluN1-4a-N368Q subunits in mammalian cell lines (Fig. 7B). Therefore, given that the GluN1 subunit is present in the ER in large excess relative to GluN2 subunits, N-glycosylation may provide another dimension in terms of regulating the ER processing of NMDARs (6, 36). In addition, we found that eliminating N-glycosylation at the Asn-203 and Asn-368 residues did not significantly alter the functional properties of the receptors. Therefore, the quality control system in the ER does not allow these receptors to be released from the ER, even if their gating properties may be checked by this system prior to release, as is the case with some AMPA receptors (37).
FIGURE 7.

The N-glycosylation sites at Asn-203 and Asn-368 have been conserved within GluN1 subunits throughout evolution. A, sequences of GluN1 homologs from the indicated species. The top and bottom sets of sequences contain the Asn-203 and Asn-368 residues, respectively, that were identified in rat GluN1 as essential for trafficking of NMDARs. The conventional N-glycosylation consensus sites are underlined. B, COS-7 cells expressing the indicated wild-type or mutant YFP-GluN1-4a (GluN1-4a) subunits were labeled with anti-GFP and the appropriate secondary antibody under non-permeabilizing or permeabilizing conditions and then analyzed using a quantitative colorimetric assay. The bar graphs summarize the relative surface expression (surface/total expression signal) of the indicated subunit combinations normalized to wild-type GluN1-4a. The data were obtained from three independent experiments (n = 9). *, p < 0.05 versus wild-type GluN1-4a, analyzed by one-way ANOVA. Note that the total expression levels of GluN1 were similar among all studied GluN1 subunit combinations (data not shown).
Ensuring the precise timing of NMDAR gating is essential to the fidelity of excitatory synaptic transmission (3). NMDARs have specific conformations that are associated with the closed, open, and desensitized states of the channel. The functional properties of NMDARs are regulated by several domains in GluN subunits, including the extracellular N terminus (38–40). Several lines of evidence support the hypothesis that N-glycosylation regulates the gating of NMDARs. First, multiple N-glycosylation sites are present in critical regions that control receptor function, including the ligand-binding domains (Fig. 1). Second, electrophysiology studies using Xenopus oocytes revealed that treating GluN1/GluN2B receptors with the N-glycosylation inhibitor tunicamycin decreases the EC50 for glutamate of the receptor (10). In our experiments, we found that removing N-glycans from native NMDARs (using PNGase F or tunicamycin) alters the affinity for NMDA of the receptor, which is consistent with the aforementioned study in Xenopus oocytes (10). In addition, removing all accessible N-glycans from the NMDARs had no effect on the open probability or desensitization properties of the receptor (Fig. 6 and data not shown). At the moment, it is unknown whether any specific glycosylation sites within the GluN1 and/or GluN2 subunit contribute to this phenomena.
N-glycans serve a wide range of functions and can be divided into two main categories on the basis of processing and maturation. High-mannose forms of N-glycans affect the folding and sorting of proteins within the ER, and, after glycan remodeling in the GA, the hybrid and complex forms of N-glycans mediate intracellular glycoprotein sorting and interactions between the cell and the extracellular environment (12, 29). Previous biochemical data have shown that NMDARs are heavily N-glycosylated (5–8). However, we currently do not know which asparagine residues, aside from Asn-203 and Asn-368 in the GluN1 subunit, are N-glycosylated in native subunits because only two-thirds of the conventional N-glycosylation consensus sites are occupied by glycans (12, 29). Furthermore, we cannot exclude the possibility that the presence of N-glycans at non-conventional site(s) and/or the presence of other glycan structures (including O-glycans) may be essential for the trafficking and/or function of NMDARs. This is an important point given that a non-conventional N-glycosylation site in the GluN2B subunit is required for trafficking NMDARs to synapses in an activity-independent manner (41). In our experiments, we observed that mutating the other ten conventional N-glycosylation sites within the GluN1 subunit (instead of the Asn-203 and Asn-368 residues), diminishes the surface delivery of the NMDARs slightly differently in the tested cell types. We suggest that the differences in the expression of the native GluN subunits and/or specific protein complexes regulating the assembly, ER retention, and/or intracellular trafficking of NMDARs among the various cell types could account for this phenomenon.
Interestingly, recent studies reported that the N-glycosylation pattern of AMPA receptors in the cortex is altered in schizophrenia (42), the ionotropic glutamate receptor function is modulated by vertebrate galectins (43), and O-GlcNAcylation of the GluA2 subunit is associated with long-term depression in hippocampal synapses (44). Therefore, it is likely that future studies will reveal more information regarding the precise roles that specific glycosylation of NMDARs play in regulating neuronal function. Finally, because patients with congenital disorders of glycosylation often exhibit neuropsychiatric symptoms, including mental retardation, seizures, and epilepsy (12, 45), understanding the role that N-glycosylation plays in the mammalian brain is highly relevant from a clinical perspective.
Author Contributions
R. S. P., Y. H. S., and M. H. designed the study. K. L., M. K., S. P. P., K. S., Y. X. W., and R. S. P. performed the research. K. L., M. K., S. P. P., K. S., Y. X. V., and R. S. P. analyzed the data. R. S. P., Y. H. S., and M. H. wrote the paper.
This work was supported by project 14-02219S from the Grant Agency of the Czech Republic (to M. H., K. L., M. K., and K. S.), Marie Curie International Reintegration Grant PIRG-GA-2010-276827 (to M. H.), Grant Agency of Charles University Grant 1520-243-253483 (to K. L.), and the Research Project of the AS CR RVO:67985823 and BIOCEV/Biotechnology and Biomedicine Center of Academy of Sciences and Charles University in Vestec (project supported by the European Regional Development Fund (to M. H., K. L., and K. S.). The authors declare that they have no conflicts of interest with the contents of this article.
- NMDAR
- NMDA receptor
- ER
- endoplasmic reticulum
- GA
- Golgi apparatus
- CGC
- cerebellar granule cell
- PFA
- paraformaldehyde
- PNGase F
- peptide-N-glycosidase F
- ANOVA
- analysis of variance.
References
- 1. Lau C. G., Zukin R. S. (2007) NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 8, 413–426 [DOI] [PubMed] [Google Scholar]
- 2. Sanz-Clemente A., Nicoll R. A., Roche K. W. (2013) Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Traynelis S. F., Wollmuth L. P., McBain C. J., Menniti F. S., Vance K. M., Ogden K. K., Hansen K. B., Yuan H., Myers S. J., Dingledine R. (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Petralia R. S., Al-Hallaq R. A., Wenthold R. J. (2009) Trafficking and Targeting of NMDA Receptors. CRC Press, Boca Raton, FL, p. 149–227 [PubMed] [Google Scholar]
- 5. Clark R. A., Gurd J. W., Bissoon N., Tricaud N., Molnar E., Zamze S. E., Dwek R. A., McIlhinney R. A., Wing D. R. (1998) Identification of lectin-purified neural glycoproteins, GPs 180, 116, and 110, with NMDA and AMPA receptor subunits: conservation of glycosylation at the synapse. J. Neurochem. 70, 2594–2605 [DOI] [PubMed] [Google Scholar]
- 6. Huh K. H., Wenthold R. J. (1999) Turnover analysis of glutamate receptors identifies a rapidly degraded pool of the N-methyl-d-aspartate receptor subunit, NR1, in cultured cerebellar granule cells. J. Biol. Chem. 274, 151–157 [DOI] [PubMed] [Google Scholar]
- 7. Kaniakova M., Lichnerova K., Vyklicky L., Horak M. (2012) Single amino acid residue in the M4 domain of GluN1 subunit regulates the surface delivery of NMDA receptors. J. Neurochem. 123, 385–395 [DOI] [PubMed] [Google Scholar]
- 8. Kenny A. V., Cousins S. L., Pinho L., Stephenson F. A. (2009) The integrity of the glycine co-agonist binding site of N-methyl-d-aspartate receptors is a functional quality control checkpoint for cell surface delivery. J. Biol. Chem. 284, 324–333 [DOI] [PubMed] [Google Scholar]
- 9. Qiu S., Zhang X. M., Cao J. Y., Yang W., Yan Y. G., Shan L., Zheng J., Luo J. H. (2009) An endoplasmic reticulum retention signal located in the extracellular amino-terminal domain of the NR2A subunit of N-Methyl-d-aspartate receptors. J. Biol. Chem. 284, 20285–20298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Everts I., Villmann C., Hollmann M. (1997) N-Glycosylation is not a prerequisite for glutamate receptor function but is essential for lectin modulation. Mol. Pharmacol. 52, 861–873 [DOI] [PubMed] [Google Scholar]
- 11. Chazot P. L., Cik M., Stephenson F. A. (1995) An investigation into the role of N-glycosylation in the functional expression of a recombinant heteromeric NMDA receptor. Mol. Membr. Biol. 12, 331–337 [DOI] [PubMed] [Google Scholar]
- 12. Moremen K. W., Tiemeyer M., Nairn A. V. (2012) Vertebrate protein glycosylation: diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horak M., Vlcek K., Chodounska H., Vyklicky L., Jr. (2006) Subtype-dependence of N-methyl-d-aspartate receptor modulation by pregnenolone sulfate. Neuroscience 137, 93–102 [DOI] [PubMed] [Google Scholar]
- 14. Kaniakova M., Krausova B., Vyklicky V., Korinek M., Lichnerova K., Vyklicky L., Horak M. (2012) Key amino acid residues within the third membrane domains of NR1 and NR2 subunits contribute to the regulation of the surface delivery of N-methyl-d-aspartate receptors. J. Biol. Chem. 287, 26423–26434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Luo J. H., Fu Z. Y., Losi G., Kim B. G., Prybylowski K., Vissel B., Vicini S. (2002) Functional expression of distinct NMDA channel subunits tagged with green fluorescent protein in hippocampal neurons in culture. Neuropharmacology 42, 306–318 [DOI] [PubMed] [Google Scholar]
- 16. Schlüter O. M., Xu W., Malenka R. C. (2006) Alternative N-terminal domains of PSD-95 and SAP97 govern activity-dependent regulation of synaptic AMPA receptor function. Neuron 51, 99–111 [DOI] [PubMed] [Google Scholar]
- 17. Alvarez V. A., Ridenour D. A., Sabatini B. L. (2007) Distinct structural and ionotropic roles of NMDA receptors in controlling spine and synapse stability. J. Neurosci. 27, 7365–7376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng C. Y., Chang K., Suh Y. H., Roche K. W. (2015) TARP γ-8 glycosylation regulates the surface expression of AMPA receptors. Biochem. J. 465, 471–477 [DOI] [PubMed] [Google Scholar]
- 19. Prybylowski K., Chang K., Sans N., Kan L., Vicini S., Wenthold R. J. (2005) The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron 47, 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lichnerova K., Kaniakova M., Skrenkova K., Vyklicky L., Horak M. (2014) Distinct regions within the GluN2C subunit regulate the surface delivery of NMDA receptors. Front Cell Neurosci. 8, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horak M., Wenthold R. J. (2009) Different roles of C-terminal cassettes in the trafficking of full-length NR1 subunits to the cell surface. J. Biol. Chem. 284, 9683–9691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Horak M., Chang K., Wenthold R. J. (2008) Masking of the endoplasmic reticulum retention signals during assembly of the NMDA receptor. J. Neurosci. 28, 3500–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petralia R. S., Wang Y. X., Hua F., Yi Z., Zhou A., Ge L., Stephenson F. A., Wenthold R. J. (2010) Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167, 68–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petralia R. S., Wenthold R. J. (1999) Immunocytochemistry of NMDA receptors. Methods Mol. Biol. 128, 73–92 [DOI] [PubMed] [Google Scholar]
- 25. Gleichman A. J., Spruce L. A., Dalmau J., Seeholzer S. H., Lynch D. R. (2012) Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J. Neurosci. 32, 11082–11094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Karakas E., Furukawa H. (2014) Crystal structure of a heterotetrameric NMDA receptor ion channel. Science 344, 992–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Prybylowski K., Fu Z., Losi G., Hawkins L. M., Luo J., Chang K., Wenthold R. J., Vicini S. (2002) Relationship between availability of NMDA receptor subunits and their expression at the synapse. J. Neurosci. 22, 8902–8910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen N., Luo T., Raymond L. A. (1999) Subtype-dependence of NMDA receptor channel open probability. J. Neurosci. 19, 6844–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vagin O., Kraut J. A., Sachs G. (2009) Role of N-glycosylation in trafficking of apical membrane proteins in epithelia. Am. J. Physiol. Renal Physiol. 296, F459–F469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McIlhinney R. A., Le Bourdellès B., Molnár E., Tricaud N., Streit P., Whiting P. J. (1998) Assembly intracellular targeting and cell surface expression of the human N-methyl-d-aspartate receptor subunits NR1a and NR2A in transfected cells. Neuropharmacology 37, 1355–1367 [DOI] [PubMed] [Google Scholar]
- 31. Fukaya M., Kato A., Lovett C., Tonegawa S., Watanabe M. (2003) Retention of NMDA receptor NR2 subunits in the lumen of endoplasmic reticulum in targeted NR1 knockout mice. Proc. Natl. Acad. Sci. U.S.A. 100, 4855–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Okabe S., Miwa A., Okado H. (1999) Alternative splicing of the C-terminal domain regulates cell surface expression of the NMDA receptor NR1 subunit. J. Neurosci. 19, 7781–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Standley S., Roche K. W., McCallum J., Sans N., Wenthold R. J. (2000) PDZ domain suppression of an ER retention signal in NMDA receptor NR1 splice variants. Neuron 28, 887–898 [DOI] [PubMed] [Google Scholar]
- 34. Scott D. B., Blanpied T. A., Swanson G. T., Zhang C., Ehlers M. D. (2001) An NMDA receptor ER retention signal regulated by phosphorylation and alternative splicing. J. Neurosci. 21, 3063–3072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. She K., Ferreira J. S., Carvalho A. L., Craig A. M. (2012) Glutamate binding to the GluN2B subunit controls surface trafficking of N-methyl-d-aspartate (NMDA) receptors. J. Biol. Chem. 287, 27432–27445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chazot P. L., Stephenson F. A. (1997) Biochemical evidence for the existence of a pool of unassembled C2 exon-containing NR1 subunits of the mammalian forebrain NMDA receptor. J. Neurochem. 68, 507–516 [DOI] [PubMed] [Google Scholar]
- 37. Penn A. C., Williams S. R., Greger I. H. (2008) Gating motions underlie AMPA receptor secretion from the endoplasmic reticulum. EMBO J. 27, 3056–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yuan H., Hansen K. B., Vance K. M., Ogden K. K., Traynelis S. F. (2009) Control of NMDA receptor function by the NR2 subunit amino-terminal domain. J. Neurosci. 29, 12045–12058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhu S., Stroebel D., Yao C. A., Taly A., Paoletti P. (2013) Allosteric signaling and dynamics of the clamshell-like NMDA receptor GluN1 N-terminal domain. Nat. Struct. Mol. Biol. 20, 477–485 [DOI] [PubMed] [Google Scholar]
- 40. Borschel W. F., Murthy S. E., Kasperek E. M., Popescu G. K. (2011) NMDA receptor activation requires remodelling of intersubunit contacts within ligand-binding heterodimers. Nat. Commun. 2, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Storey G. P., Opitz-Araya X., Barria A. (2011) Molecular determinants controlling NMDA receptor synaptic incorporation. J. Neurosci. 31, 6311–6316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tucholski J., Simmons M. S., Pinner A. L., Haroutunian V., McCullumsmith R. E., Meador-Woodruff J. H. (2013) Abnormal N-linked glycosylation of cortical AMPA receptor subunits in schizophrenia. Schizophr. Res. 146, 177–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Copits B. A., Vernon C. G., Sakai R., Swanson G. T. (2014) Modulation of ionotropic glutamate receptor function by vertebrate galectins. J. Physiol. 592, 2079–2096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor E. W., Wang K., Nelson A. R., Bredemann T. M., Fraser K. B., Clinton S. M., Puckett R., Marchase R. B., Chatham J. C., McMahon L. L. (2014) O-GlcNAcylation of AMPA receptor GluA2 is associated with a novel form of long-term depression at hippocampal synapses. J. Neurosci. 34, 10–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Freeze H. H. (2006) Genetic defects in the human glycome. Nat. Rev. Genet. 7, 537–551 [DOI] [PubMed] [Google Scholar]