

Background: Translationally stalled proteins (including those aberrantly translated beyond their stop codons) pose dangers for eukaryotic cells.
Results: The ubiquitin ligase Rkr1/Ltn1 targets translationally stalled ER-associated proteins for degradation.
Conclusion: Cytosolic and ER-associated translationally stalled proteins are targeted for destruction by related mechanisms.
Significance: Mechanisms regulating the degradation of translationally stalled ER-associated proteins may represent therapeutic targets for human disease.
Keywords: E3 ubiquitin ligase, endoplasmic reticulum-associated protein degradation (ERAD), ER quality control, protein translocation, ribosome, Saccharomyces cerevisiae, translation, yeast genetics, ribosome-associated degradation
Abstract
Aberrant nonstop proteins arise from translation of mRNA molecules beyond the coding sequence into the 3′-untranslated region. If a stop codon is not encountered, translation continues into the poly(A) tail, resulting in C-terminal appendage of a polylysine tract and a terminally stalled ribosome. In Saccharomyces cerevisiae, the ubiquitin ligase Rkr1/Ltn1 has been implicated in the proteasomal degradation of soluble cytosolic nonstop and translationally stalled proteins. Rkr1 is essential for cellular fitness under conditions associated with increased prevalence of nonstop proteins. Mutation of the mammalian homolog causes significant neurological pathology, suggesting broad physiological significance of ribosome-associated quality control. It is not known whether and how soluble or transmembrane nonstop and translationally stalled proteins targeted to the endoplasmic reticulum (ER) are detected and degraded. We generated and characterized model soluble and transmembrane ER-targeted nonstop and translationally stalled proteins. We found that these proteins are indeed subject to proteasomal degradation. We tested three candidate ubiquitin ligases (Rkr1 and ER-associated Doa10 and Hrd1) for roles in regulating abundance of these proteins. Our results indicate that Rkr1 plays the primary role in targeting the tested model ER-targeted nonstop and translationally stalled proteins for degradation. These data expand the catalog of Rkr1 substrates and highlight a previously unappreciated role for this ubiquitin ligase at the ER membrane.
Introduction
Life depends on accurate protein synthesis. Faulty protein synthesis can lead to a variety of pathological states, including neurodegenerative conditions and cancer. Not surprisingly, cells have evolved a host of co- and post-translational quality control mechanisms to reduce the abundance of aberrant proteins. One type of aberration in protein synthesis is the translation of an mRNA molecule beyond its open reading frame into the 3′-untranslated region (UTR), resulting in the synthesis of so-called nonstop (NS)3 proteins. 3′ UTR translation may arise from stop codon mutations, mutations in genes encoding factors involved in translational termination, errors in transcription, or errors in ribosomal decoding of stop codons. In the absence of a fortuitously positioned stop codon in the 3′ UTR, translation continues into the poly(A) tail. This results in an abnormal, nonstop protein with a C-terminal polylysine extension. Eukaryotic cells have developed at least two complementary strategies for reducing the abundance of potentially harmful NS proteins: nonstop decay (NSD) of NS mRNA molecules and ribosome-associated degradation of NS proteins (1).
The consequences of faulty NS protein degradation on health are severe. Mice lacking the ribosome-associated quality control ubiquitin ligase (E3) Listerin exhibit severe neurodegeneration and impaired motor function (2). Under genetic or pharmacological conditions of excessive NS protein production, Saccharomyces cerevisiae lacking the yeast homolog (Rkr1/Ltn1) display a significant growth defect (3). Furthermore, yeast lacking Rkr1 exhibit transcriptional defects (4) and sensitivity to the yeast prion [PSI+], particularly in the absence of enzymes that regulate histone modifications (5).
Stop codon mutations that result in 3′ UTR and poly(A) translation in individual genes have been implicated in several pathologies, including 2,8-dihydroxyadenine urolithiasis (6), congenital adrenal hyperplasia (7), idiopathic hypogonadotropic hypogonadism (8), and muscular dystrophy (9). In such cases, disease phenotypes may arise from the loss of protein function due to translation of a C-terminal extension, decreased protein abundance due to functional NS quality control mechanisms, or both. Reduced expression of disease-associated NS variants suggests that NS quality control mechanisms may represent viable therapeutic targets for such conditions (6, 7, 9).
A current model of ribosome-associated protein degradation of NS proteins is based largely on genetic and biochemical studies of protein and mRNA quality control in S. cerevisiae. Translation of the poly(A) tail of an NS mRNA molecule is likely to result in a terminally stalled complex containing a ribosome, an mRNA molecule, and a tRNA-linked protein with a C-terminal polylysine extension (10). In addition to the absence of a stop codon to trigger termination, electrostatic attraction between the translated basic polylysine tract and the acidic ribosome exit tunnel is believed to prolong ribosome occupancy (3, 11, 12). Indeed, translational pausing and ribosome-associated degradation can be induced by artificially inserting a sequence encoding several consecutive basic amino acids upstream of a wild-type (WT) stop codon (3, 11, 13). The heterodimeric complex of Hbs1 (a GTPase with homology to translational release factors) and Dom34 (which adopts a conformation that mimics tRNA structure) stimulates ribosome dissociation into large and small subunits (10, 14–16), potentially with assistance from the ATPase Rli1 (1). The stalled polypeptide remains associated with the large subunit (17). A ribosome quality control (RQC) complex composed of the E3 Rkr1 and scaffolding proteins Rqc1 and Rqc2/Tae2 ubiquitylates translationally paused ribosome-associated proteins (3, 17, 18). The AAA-ATPase (ATPase associated with various cellular activities) Cdc48 (with co-factors Npl4 and Ufd1) recognizes and extracts stalled ubiquitylated proteins from large ribosome subunits and facilitates their proteasomal degradation (18, 19). Recent in vitro analyses with mammalian counterparts confirm that these mechanisms have been highly conserved (20, 21).
Characterization of the genetic requirements for degradation of translationally stalled proteins has chiefly relied upon variants of model soluble cytosolic proteins (e.g. His3, protein A, and green fluorescent protein (GFP)) engineered to be translated beyond their stop codons or to possess internal polybasic sequences that mimic a translated poly(A) tail (3, 11, 13, 19, 22). Following translational pausing and ribosome dissociation, translationally stalled cytosolic proteins are expected to have their N-terminal portions exposed to the cytosol where the RQC complex would have access (17, 23). By contrast, it is unclear how or whether cells regulate the abundance of translationally stalled proteins targeted to the endoplasmic reticulum (ER). Many ER-targeted proteins are co-translationally translocated, during which the nascent polypeptide moves directly from the ribosome exit tunnel into the protein-conducting translocon. The ribosome and translocon shield many ER-targeted proteins from cytosolic exposure (24, 25). If a ribosome translates a pause-inducing sequence in a soluble ER-targeted protein and Hbs1-Dom34 trigger ribosome dissociation, very little (or none) of the nascent polypeptide would be expected to be exposed to the cytosol. It is therefore not a priori evident how or whether Rkr1 could access such a stalled polypeptide. It is equally unapparent how or whether translationally stalled integral membrane proteins are recognized by the ribosome-associated quality control machinery.
Two other E3s, Doa10 and Hrd1/Der3, represent candidate mediators of ribosome-associated quality control at the ER membrane. These transmembrane E3s catalyze the quality control degradation of aberrant ER-localized proteins via multiple mechanisms of ER-associated degradation (ERAD) (26–31). Doa10 and Hrd1 ubiquitylate distinct substrate classes in a manner that depends, in general, on degradation signal (degron) localization with respect to the ER membrane (32). Doa10 typically targets proteins with cytosolic degrons (ERAD-C substrates), whereas Hrd1 targets proteins with degrons in the ER lumen (ERAD-L substrates) or within membrane-spanning segments (ERAD-M substrates) (33–38). However, Doa10 has also recently been shown to recognize an intramembrane (ERAD-M) degron (39). Additionally, Hrd1 may target for degradation proteins that persistently or aberrantly engage the ER-localized translocon (ERAD-T substrates) (40–42). Given that translationally stalled ER-targeted proteins may be expected to remain translocon-engaged, it may be hypothesized that Hrd1 targets such proteins for degradation. An alternative hypothesis is that Doa10 recognizes the abnormal, persistent presence of an intact or dissociated ribosome tethered to the ER membrane by a translationally stalled ER-targeted polypeptide as an ERAD-C degron.
In this study, we investigated whether Rkr1, Doa10, or Hrd1 regulate the abundance of translationally stalled ER-targeted proteins. We found that model NS and polylysine-containing proteins targeted to the ER are proteasomally degraded. Although Doa10 and Hrd1 are required for cells to cope with conditions associated with increased frequency of stop codon read-through, degradation of the tested model translationally stalled ER-targeted proteins depends principally on Rkr1. Our data indicate that ER-targeted proteins, like soluble proteins, are subject to ribosome-associated quality control and reveal a previously unappreciated role for Rkr1 at the ER membrane, where it targets translationally paused ER-targeted proteins for degradation. Furthermore, the mode of translocation (i.e. co- versus post-translational) influences the efficiency of translational pausing and Rkr1-dependent degradation of aberrant ER-targeted proteins.
Experimental Procedures
Yeast and Bacterial Methods
Yeast cells were cultured in rich yeast extract/peptone/dextrose (YPD) or synthetic defined (SD) medium as described previously (43). Yeast cells were transformed with DNA molecules (plasmids or PCR products) using standard techniques (43). To delete genes by homologous recombination, antibiotic selection markers were amplified from donor yeast strains or plasmids with flanking sequences that possess homology to sequence immediately upstream and downstream of target gene start and stop codons. Gene deletions were confirmed by PCR. Plasmids were manipulated using standard restriction enzyme-based cloning, PCR-based mutagenesis, and gap repair. Detailed cloning and gene knock-out strategies, plasmid sequences, and primer sequences are available upon request. Yeast growth assays were performed by spotting 4 μl of 6-fold serial dilutions of yeast cultures (beginning with cells at an A600 of 0.2) onto plates containing yeast growth medium. Plates were incubated at the indicated temperatures. Yeast strains used in this study are presented in Table 1. Plasmids used in this study are presented in Table 2.
TABLE 1.
Yeast strains used in this study
All yeast strains are congenic with BY4741 (69).
| Name | Genotype | Source |
|---|---|---|
| VJY22 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hrd1Δ::kanMX4 | 68 |
| VJY33 (Alias BY4741) | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 | 68, 69 |
| VJY102 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 | 68 |
| VJY245 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ski7Δ::kanMX4 | 68 |
| VJY246 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rkr1Δ::kanMX4 | 68 |
| VJY314 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 hrd1Δ::kanMX4 rkr1Δ::hphMX4 ski7Δ::natMX4 | This study |
| VJY317 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hrd1Δ::kanMX4 ski7Δ::natMX4 | This study |
| VJY318 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 ski7Δ::natMX4 | This study |
| VJY320 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rkr1Δ::hphMX4 ski7Δ::natMX4 | This study |
| VJY328 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 hrd1Δ::kanMX4 rkr1Δ::natMX4 | This study |
| VJY331 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 rkr1Δ::natMX4 | This study |
| VJY334 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 hrd1Δ::kanMX4 rkr1Δ::natMX4 | This study |
| SKY122 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 pdr5Δ::kanMX4 | 68 |
| SKY252 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 hrd1Δ::kanMX4 | 39 |
| SKY342 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 rkr1Δ::hphMX4 | This study |
| SKY347 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 doa10Δ::kanMX4 hrd1Δ::kanMX4 rkr1Δ::hphMX4 | This study |
| SKY415 | MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0 ski7Δ::kanMX4 dom34Δ::hphMX4 | This study |
TABLE 2.
Plasmids used in this study
All plasmids used in this study are yeast CEN plasmids and harbor the AmpR gene for selection of ampicillin-resistant Escherichia coli. All genes encoding model proteins are driven by glyceraldehyde-3-phosphate dehydrogenase (GPD) promoter (72). Furthermore, all genes are followed by the HIS3 3′ UTR, as described previously (73) (except for FLAG-Vma12-ProtA-K12–13myc and FLAG-Vma12(glyc)-ProtA-K12–13myc; STK 07.4.3 and pVJ485) and the CYC1 transcriptional terminator sequence. In all cases, K12 was encoded by 5′-(AAGAAA)6-3′. See Fig. 2 for schematic depictions of constructs used in this study.
| Name | Alias | Yeast selection marker | Description | Source |
|---|---|---|---|---|
| pVJ26 | pRS313 | HIS3 | Empty vector | 70 |
| pVJ48 | YCplac33 | URA3 | Empty vector | 71 |
| pVJ380 | pKK7 | URA3 | GFP-2A-FLAG-HIS3-K12 followed by stop codon in YCplac33 vector backbone | 13 |
| pVJ382 | pSA158 | URA3 | GFP-2A-FLAG-HIS3 followed by stop codon in YCplac33 vector backbone | 13 |
| pVJ383 | pSA159 | URA3 | GFP-2A-FLAG-HIS3 with mutated stop codon in YCplac33 vector backbone | 13 |
| pVJ485 | p413-GPD-FLAG-Vma12 (glyc)-ProtA-K12–13myc | HIS3 | Vma12-ProtA-K12–13myc followed by stop codon in pRS413 vector backbone. Luminal loop of Vma12 replaced with 3 × HA and two N-glycosylation sites from invertase (Suc2) | This study |
| pVJ492 | p413GPD-OPY-ProtA-STOP | HIS3 | OPY-ProtA followed by stop codon in pRS413 vector backbone | This study |
| pVJ494 | p413GPD-OPY-ProtA-NS | HIS3 | OPY-ProtA with mutated stop codon in pRS413 vector backbone | This study |
| pVJ496 | p413GPD-OPY-ProtA-K12–13myc | HIS3 | OPY-ProtA-K12–13myc followed by a stop codon in pRS413 vector backbone | This study |
| STK 07.2.8 | p413GPD-FLAG-Vma12-ProtA | HIS3 | FLAG-Vma12-ProtA followed by stop codon in pRS413 vector backbone | This study |
| STK 07.4.3 | p413GPD-FLAG-Vma12-ProtA-K12–13myc | HIS3 | FLAG-Vma12-ProtA-K12–13myc followed by stop codon in pRS413 vector backbone | This study |
| STK 07.4.4 | p413GPD-FLAG-Vma12-ProtA-NS | HIS3 | FLAG-Vma12-ProtA with mutated stop codon in pRS413 vector backbone | This study |
| STK 07.4.5 | p413GPD-FLAG-Vma12-ProtA-STOP | HIS3 | FLAG-Vma12-ProtA followed by stop codon in pRS413 vector backbone | This study |
| STK 07.4.9 | p413GPD-CPY-ProtA-STOP | HIS3 | CPY-ProtA followed by stop codon in pRS413 vector backbone | This study |
| STK 07.5.1 | p413GPD-CPY-ProtA-NS | HIS3 | CPY-ProtA with mutated stop codon in pRS413 vector backbone | This study |
| STK 07.5.2 | p413GPD-dSP-CPY-ProtA-NS | HIS3 | CPY-ProtA lacking ER-targeting signal peptide with mutated stop codon in pRS413 vector backbone | This study |
| STK 07.6.9 | p413GPD-CPY-ProtA-K12–13myc | HIS3 | CPY-ProtA-K12–13myc followed by stop codon in pRS413 vector backbone | This study |
Protein Extraction for Steady State Analysis and Endoglycosidase H Treatment
Protein extracts were prepared using a post-alkaline lysis method (40, 44). Briefly, 2.5 A600 eq of mid-log phase yeast cells were suspended in 200 μl of 0.1 m NaOH and incubated for 5 min at room temperature. Cells were pelleted by centrifugation. Pelleted cells were resuspended and lysed in 1× Laemmli sample buffer. Lysates were boiled for 5 min and cleared by centrifugation prior to separation by SDS-PAGE. For analysis of protein N-glycosylation, protein extracts (0.375 A600 eq) were supplemented with 0.83 m potassium acetate, pH 5.6, to a final concentration of 80 mm and incubated at 37 °C for 3 h in the presence or absence of 0.005 units of endoglycosidase H (Endo H; Roche Applied Science).
Proteasome Inhibition
Six A600 eq of mid-log phase yeast cells were pelleted by centrifugation and resuspended in 2 ml of pre-warmed (30 °C) medium. Cells were split into 2 aliquots and incubated with the proteasome inhibitor MG132 (Sigma) dissolved in DMSO (final concentration 50 μm) or an equivalent amount of DMSO. Cells were incubated for 3 h and pelleted by centrifugation. Cells were lysed as described previously (45). Briefly, yeast cells were suspended and vortexed in 1.15 ml of 0.25 m β-mercaptoethanol and 0.125 m NaOH. Proteins were precipitated in 5% trichloroacetic acid. The pellet was resuspended in SDS gel loading buffer.
Western Blotting
Proteins were transferred from SDS-PAGE to polyvinylidene fluoride membranes (Millipore). Membranes were blocked in 5% skim milk in Tris-buffered saline (TBS; 50 mm Tris, 150 mm NaCl, pH 7.5) for 60 min at room temperature or overnight at 4 °C. All antibody incubations were performed for 60–90 min at room temperature in 1% skim milk in TBS supplemented with 0.1% Tween 20 (TBS/T) followed by three 5-min washes in TBS/T. The following antibody dilutions were used for experiments presented in Figs. 3 and 5–7: mouse anti-FLAG (Sigma catalog no. F3165) at 1:10,000; mouse anti-phosphoglycerate kinase 1 (Pgk1; clone 22C5D8; Life Technologies, Inc., catalog no. 459250) at 1:80,000; and rabbit anti-glucose-6-phosphate dehydrogenase (G6PDH; Sigma catalog no. A9521) at 1:10,000. Mouse primary antibodies were followed by incubation with AlexaFluor-680-conjugated rabbit anti-mouse secondary antibody (Life Technologies, Inc., catalog no. A-21065) at 1:40,000. Rabbit primary antibodies were followed by incubation with IRDye-680RD-conjugated goat anti-rabbit secondary antibody (Li-Cor catalog no. 925-68071) at 1:40,000. The AlexaFluor-680-conjugated rabbit anti-mouse antibody (1:40,000 dilution) was also used to directly detect the Staphylococcus aureus protein A epitope (which binds to mammalian immunoglobulins (46)). The following antibody dilutions were used for experiments presented in Fig. 4: peroxidase-anti-peroxidase-soluble complex (PAP; antibody produced in rabbit; Sigma catalog no. P1291) at 1:20,000 to directly detect the S. aureus protein A epitope; mouse monoclonal anti-phosphoglycerate kinase 1 (Pgk1; clone 22C5; Molecular Probes catalog no. A-6457) at 1:20,000, and rabbit anti-glucose-6-phosphate dehydrogenase (G6PDH; Sigma catalog no. A9521) at 1:10,000. Anti-Pgk1 mouse primary antibody was followed by incubation with peroxidase-conjugated goat anti-mouse antibody (IgG1-specific; Jackson ImmunoResearch catalog no. 115-035-205) at 1:10,000. Anti-G6PDH rabbit primary antibody was followed by incubation with peroxidase-conjugated goat anti-rabbit (Dianova catalog no. 111-035-003) at 1:10,000. No secondary antibody was used for detection of the peroxidase-anti-peroxidase-soluble complex. Membranes were imaged using an Odyssey CLx Infrared Imaging System and Image Studio Software (Li-Cor) (Figs. 3 and 5–7) or an LAS-3000 imaging system (Fuji) and Aida Image Analyzer software (Bio Imaging) (Fig. 4).
FIGURE 3.

Rkr1 regulates GFP-NS and GFP-K12 abundance. A, C, and D, GFP, GFP-NS, and GFP-K12 proteins were detected in yeast strains of the indicated genotypes by anti-FLAG Western blotting. Pgk1 served as a loading control and was detected with an anti-Pgk1 antibody. B, number of samples from multiple experiments that exhibited increased abundance of GFP-NS relative to the appropriate control (SKI7 DOA10 HRD1 RKR1 or ski7Δ DOA10 HRD1 RKR1 yeast strains) is aggregated and presented as an absolute number and a percentage of trials. See Fig. 2 for schematic depiction of constructs in this figure.
FIGURE 5.
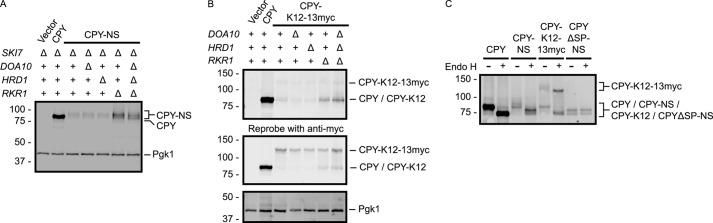
Rkr1 regulates CPY-NS and CPY-K12 abundance. A and B, CPY, CPY-NS, and CPY-K12–13myc proteins were detected in yeast strains of the indicated genotypes by anti-ProtA Western blotting. The membrane in B was reprobed with an anti-Myc antibody. Pgk1 served as a loading control and was detected with an anti-Pgk1 antibody. C, lysates prepared from cells expressing the indicated constructs were incubated in the presence or absence of Endo H. See Fig. 2 for schematic depiction of constructs in this figure.
FIGURE 6.
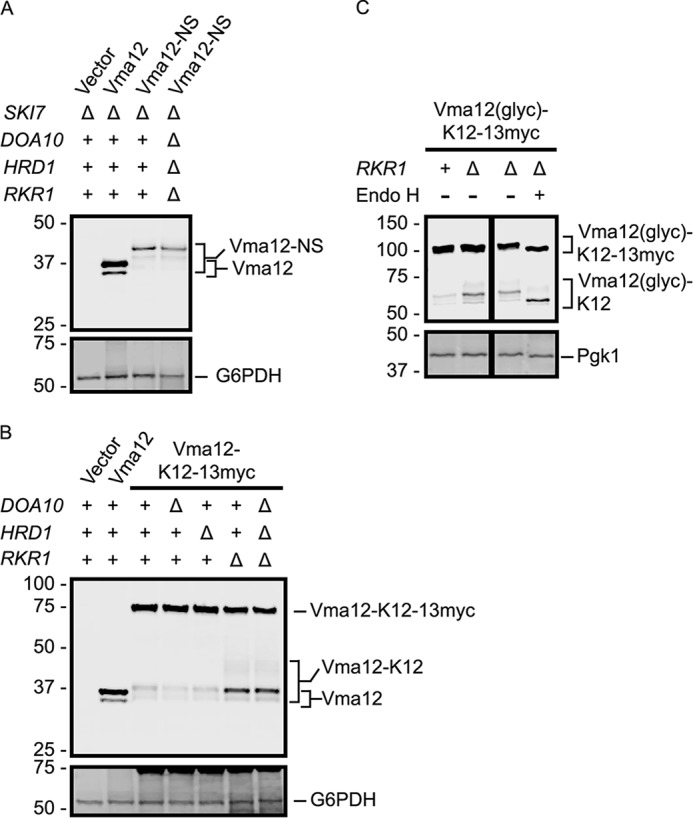
Rkr1 regulates Vma12-K12 abundance. A and B, Vma12, Vma12-NS, and Vma12-K12–13myc proteins were detected in yeast strains of the indicated genotypes by anti-ProtA Western blotting. G6PDH served as a loading control and was detected with an anti-G6PDH antibody. C, N-glycosylation sequences were artificially inserted into the ER luminal loop of Vma12-K12–13myc to generate Vma12(glyc)-K12–13myc. Lysates from cells of the indicated genotypes expressing Vma12(glyc)-K12–13myc were incubated in the presence or absence of Endo H. The black lines indicate that intervening lanes have been spliced out. See Fig. 2 for schematic depiction of constructs in this figure.
FIGURE 7.

Rkr1 regulates OPY-K12 abundance. A, lysates prepared from cells expressing the indicated constructs were incubated in the presence or absence of Endo H. B and C, OPY, OPY-NS, and OPY-K12–13myc proteins were detected in yeast strains of the indicated genotypes by anti-ProtA Western blotting. Pgk1 served as a loading control and was detected with an anti-Pgk1 antibody. D, lysates from cells expressing CPY-K12–13myc, Vma12-K12–13myc, or OPY-K12–13myc were analyzed by anti-ProtA Western blotting. 20% volume of lysate harboring Vma12-K12–13myc was loaded to facilitate clearer comparison of relative abundance of truncated (K12) and full-length (K12–13myc) species. See Fig. 2 for schematic depiction of constructs in this figure.
FIGURE 4.
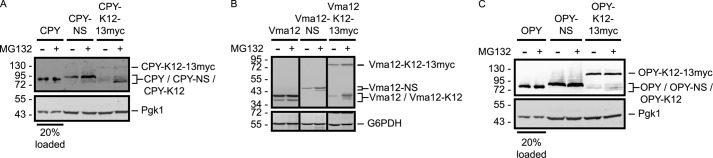
ER-targeted NS and K12 proteins are proteasome substrates. pdr5Δ yeast cells expressing the indicated variants of CPY (A), Vma12 (B), or OPY (C) were incubated in the presence or absence of MG132 for 3 h followed by anti-ProtA Western blot analysis. G6PDH and Pgk1 served as loading controls, as indicated, and were detected with anti-G6PDH and anti-Pgk1 antibodies, respectively. Where noted, 20% of lysate volumes were loaded for clearer comparison of protein abundance in the presence or absence of MG132. The black lines in B indicate that intervening lanes have been spliced out. See Fig. 2 for schematic depiction of constructs in this figure.
Results
Rkr1, Doa10, and Hrd1 Confer Resistance to Hygromycin B
Mutations in genes required for co-translational degradation of NS and translationally stalled cytosolic proteins render cells sensitive to conditions associated with excess NS protein production (3, 19). We hypothesized that the ERAD E3s contribute to co-translational degradation of NS and translationally stalled proteins targeted to the ER membrane. We predicted that growth of yeast cells lacking Doa10 or Hrd1 would be impaired under conditions associated with elevated NS protein production. Yeast cells expressing or lacking all combinations of Doa10, Hrd1, and Rkr1 were cultured on medium containing the aminoglycoside hygromycin B (Fig. 1). Hygromycin B induces ribosome A site distortion and errors in mRNA decoding, likely resulting in frequent stop codon read-through (47, 48). Consistent with previous studies (3), we observed a growth defect in yeast cells lacking RKR1 in the presence of hygromycin B. Cells lacking either DOA10 or HRD1 also exhibited a growth defect, although less severe, under these conditions. Combined deletion of DOA10 and HRD1 further impaired growth. Yeast with deletions of genes encoding all three E3s (DOA10, HRD1, and RKR1) exhibited a very strong growth defect.
FIGURE 1.

Rkr1 and ERAD E3s confer resistance to hygromycin B. 6-Fold serial dilutions of yeast of the indicated genotypes were spotted onto rich medium or rich medium containing the indicated compounds and imaged after 1 day (no drug or cycloheximide) or 2 days (hygromycin B or anisomycin) of incubation at 30 °C. Note: data in all figures are representative of at least three experiments.
To confirm the specificity of the growth defect in the presence of hygromycin B, we also analyzed growth in the presence of compounds that inhibit the ribosome by other mechanisms. Anisomycin and cycloheximide inhibit translational elongation by preventing peptidyltransferase activity and interfering with ribosome translocation, respectively (49, 50). Individual or simultaneous deletion of DOA10 and HRD1 did not specifically sensitize cells to either compound. Loss of RKR1 (but not of DOA10 or HRD1) mildly retarded growth in the presence of anisomycin. We note that cycloheximide slowed growth of cells of each genotype to a similar extent, confirming compound bioactivity. The specific sensitivity of cells lacking the ERAD E3s to hygromycin B is consistent with a role for Doa10 and Hrd1 in NS protein degradation.
Rkr1 Regulates Abundance of a Model Translationally Stalled Cytosolic Soluble Protein
Given their subcellular localization, we hypothesized that Rkr1 and the ERAD E3s catalyze ribosome-associated degradation of cytosolic and ER-targeted proteins, respectively. Previous analyses have indicated a role for Rkr1 in the degradation of the cytosolic protein GFP lacking a stop codon or possessing a C-terminal polylysine tract preceding a stop codon (3). To confirm a role for Rkr1 in degradation of a model NS cytosolic protein, we compared the abundance of an NS GFP construct (GFP-NS; see Fig. 2) in WT cells and cells lacking any one or all three of Doa10, Hrd1, and Rkr1.
FIGURE 2.

Constructs used in this study. A, schematic representation of proteins analyzed in this study, drawn to scale. For simplicity, throughout this report, each protein is referred to by the name at left. The following abbreviations are used: N, N terminus; C, C terminus; 2A, sequence from foot-and-mouth disease virus; F, FLAG epitope tag; TM, transmembrane segment; ProtA, protein A epitope from S. aureus; N-term, N-terminal cytosolic tail; C-term, C-terminal cytosolic tail; HA, human influenza hemagglutinin epitope; Invertase glyc sites, portion of the invertase protein possessing 2 N-glycosylation consensus sequences; CPY, vacuolar carboxypeptidase Y; SP, ER-targeting signal peptide. B, schematic representation of alterations made to the constructs depicted in A. mRNA molecules are depicted. WT, possessing normal stop codon. Non-Stop, stop codon mutated (no stop codon is present in the 3′-untranslated region). K12, hard-coded polylysine tract inserted upstream of a normal stop codon. K12–13myc, hard-coded polylysine tract and 13 Myc epitope tags inserted upstream of a normal stop codon. C, predicted localization of wild-type versions of proteins analyzed in this study shortly after synthesis. GFP is a soluble cytosolic protein. Vma12 is a two-transmembrane ER-localized protein. CPY is a soluble protein translocated into the ER (before it is trafficked to the vacuole). D, translation of classes of proteins analyzed in this study. Case 1, translation of soluble, cytosolic, or post-translationally translocated ER-targeted protein on free ribosome. Case 2, translation of soluble co-translationally translocated ER-targeted protein. After translation has stalled, NS variants of soluble post-translationally translocated proteins may also reach the translocon while still ribosome-engaged (57). Case 3, translation of ER-targeted membrane protein with cytosolic C-terminal tail. Also pictured are the three ubiquitin ligases analyzed in this study (Rkr1, Doa10, and Hrd1). T, translocon. Note: the GFP-based constructs have been used in previous studies (13). The other constructs were generated specifically for this study.
Unexpectedly, we observed variable changes in GFP-NS protein abundance in the absence of each individual E3 (Fig. 3A). The variability was observed between independent transformants in single experiments (e.g. the experiment depicted in Fig. 3A) and across experiments. Relative to WT cells, deletion of RKR1 increased GFP-NS abundance in 7 out of 12 transformants (Fig. 3B). Mutation of DOA10 resulted in a similar increase of GFP-NS abundance in 6 out of 11 transformants. GFP-NS was found in increased abundance in hrd1Δ cells in 3 out of 11 transformants. Simultaneous deletion of DOA10, HRD1, and RKR1 increased the abundance of GFP-NS in 7 out of 12 transformants. To our knowledge, variability in the effect of E3 gene deletion on NS protein abundance has not been reported. The fact that deletion of DOA10 increased the abundance of GFP-NS at the approximate frequency, and to the approximate extent, as deletion of RKR1 suggested a potential previously uncharacterized role for Doa10 in NS protein quality control of cytosolic proteins.
The abundance of proteins arising from mRNA molecules lacking stop codons is influenced by quality control at two levels, NSD and NS protein degradation. Conceivably, fluctuations in abundance of NS proteins could be attributed to variations in either pathway. To focus exclusively on NS protein quality control, we analyzed GFP-NS abundance in the context of a defective NSD pathway (deletion of SKI7, which mediates NSD (51)). Elimination of Ski7 rendered E3-dependent alterations in abundance highly reproducible (Fig. 3C). When Ski7 was absent, deletion of RKR1 increased GFP-NS abundance in all (11/11) independent transformants tested (Fig. 3B). Neither DOA10 nor HRD1 affected GFP-NS abundance in any yeast transformant lacking Ski7. Simultaneous elimination of all three E3s did not increase GFP-NS abundance compared with individual deletion of RKR1. Thus, in the absence of a functional NSD pathway, Rkr1, but not Doa10 or Hrd1, regulates the abundance of a model NS cytosolic protein.
A second strategy to focus exclusively on protein, rather than mRNA, quality control mechanisms is to analyze the abundance of proteins possessing a hard-coded polylysine (K12) tract (mimicking a translated poly(A) tail) upstream of a functional stop codon and 3′ UTR (13). The NSD pathway is not expected to recognize the polylysine-encoding mRNA, due to the presence of a functional stop codon. Abundance of K12-possessing constructs may therefore be analyzed in cells expressing Ski7. Like GFP-NS in ski7Δ cells, GFP-K12 abundance was consistently increased in cells lacking RKR1 (Fig. 3D). Deletion of neither DOA10 nor HRD1 detectably impacted GFP-K12 levels.
Together, these data confirm that Rkr1 regulates the abundance of model NS and K12 cytosolic proteins. When the NSD pathway is not active, ER-localized E3s play little to no role in regulating protein abundance. However, when NSD is intact, loss of Rkr1 and Doa10 results in similar, inconsistent increases of GFP-NS abundance. Subsequent analyses of model NS proteins were therefore conducted in the absence of Ski7, and studies of variants possessing hard-coded polylysine tracts and stop codons were performed in the presence of Ski7.
Proteasome Inhibition Stabilizes Model Translationally StalledER-targeted Proteins
We sought to determine whether NS versions of ER-targeted proteins are subject to proteasome-dependent degradation. We generated epitope-tagged variants of model soluble (vacuolar carboxypeptidase Y (CPY)) and transmembrane (Vma12) ER-targeted proteins that lack a stop codon (see Fig. 2) (52, 53). Cells lacking the Pdr5 drug efflux pump and expressing CPY-NS or Vma12-NS were treated with the proteasome inhibitor MG132. NS variants of both CPY and Vma12 increased in abundance following proteasome inhibition (to a greater extent than stop-codon possessing counterparts), confirming that both proteins are subject to proteasomal degradation (Fig. 4, A and B).
To determine whether a polylysine tract is sufficient to destabilize these ER-targeted proteins in a proteasome-dependent manner, a second set of tagged constructs was engineered to possess the coding sequence for the ER-targeted protein, a hard-coded polylysine tract, a 13myc extension, and a stop codon (see Fig. 2). The 13myc extension allows visualization of proteins that have been translated beyond the polylysine tract. Thus, the frequency of translational pausing relative to read-through for each construct may be compared, as translationally paused polypeptides migrate faster than their full-length counterparts on SDS-PAGE. When pdr5Δ cells expressing either CPY-K12–13myc or Vma12-K12–13myc were treated with MG132, a truncated polypeptide fragment (consistent with translational stalling at the internal polylysine tract) increased in abundance (Fig. 4, A and B). A markedly smaller increase in abundance of the full-length (13myc-possessing) proteins was observed following MG132 treatment, indicating that proteins translated beyond the polylysine tract are not subject to the same quality control mechanism. Thus, as for soluble cytosolic proteins, a polylysine tract is sufficient to trigger translational stalling and proteasomal degradation of nascent ER-targeted polypeptides.
Intriguingly, the frequency of translational read-through beyond the polylysine sequence differs for CPY and Vma12. Following proteasome inhibition, the fraction of protein translationally paused at the polylysine tract is markedly greater for CPY than for Vma12 (cf. Fig. 4, A and B). This suggests some feature(s) upstream of the polylysine tract regulates the efficiency of translational pausing and protein degradation. We note that Vma12 and variants thereof migrate as two bands. The faster migrating species is present in the wild-type, stop-codon possessing version and likely represents a subpopulation of Vma12 for which translation has begun at an internal start codon.
Rkr1 Regulates Abundance of a Model Translationally Stalled ER-targeted Soluble Protein
Elimination of Doa10 or Hrd1 sensitizes cells to conditions associated with increased frequency of stop codon misreading (Fig. 1). Neither contributes to quality control regulation of the NS and K12 variants of the cytosolic protein GFP when NSD is not functional (Fig. 3). Given their ER localization, we hypothesized that Doa10 and Hrd1 contribute to ribosome-associated quality control of ER-targeted proteins. Similar to GFP, stop codon mutation reduced CPY abundance (Fig. 5A). However, in contrast to our hypothesis, deletion of RKR1, and not DOA10 or HRD1, partially restored CPY-NS abundance. CPY-NS abundance was not further increased in doa10Δ hrd1Δ rkr1Δ cells.
The C-terminal addition of a polylysine tract and 13myc extension to CPY resulted in two species detectable by Western analysis (translationally stalled CPY-K12 and full-length CPY-K12–13myc). In WT cells, the combined abundance of both species (CPY-K12 and CPY-K12–13myc) was markedly reduced compared with WT CPY (Fig. 5B). RKR1 deletion increased abundance of the translationally stalled CPY-K12 without a comparable increase in the full-length CPY-K12–13myc. Deletion of DOA10 or HRD1 did not increase CPY-K12 levels. Reactivity with an anti-Myc antibody confirmed that the more slowly migrating species possesses Myc epitopes.
We confirmed ER localization of these CPY variants, each of which contains four N-glycosylation sites, by incubating lysates with Endo H, which removes ER luminally added N-linked glycans in yeast cells. Consistent with ER localization, WT CPY, CPY-NS, and translationally paused and full-length species of CPY-K12–13myc exhibited increased migration in the presence of Endo H (Fig. 5C). A control version of CPY-NS lacking an ER-targeting signal sequence was not sensitive to Endo H. Together, these data indicate that the cytosolic E3 Rkr1, but not the canonical ERAD E3s Doa10 or Hrd1, regulates the abundance of a model soluble ER luminal protein lacking a stop codon or possessing a polylysine tract.
Rkr1 Regulates Abundance of a Model Translationally Stalled ER-targeted Transmembrane Protein
An NS variant of the two-transmembrane protein Vma12, Vma12-NS, was reduced in abundance relative to Vma12 possessing a wild-type stop codon (albeit not to the extent observed for GFP-NS or CPY-NS) (Fig. 6A). Vma12-NS increased in abundance when the proteasome was inhibited (Fig. 4B). However, simultaneous deletion of DOA10, HRD1, and RKR1 did not detectably increase Vma12-NS abundance. The population of Vma12-NS detectable by Western blot likely represents a mixture of translationally stalled polypeptide at the poly(A) tail and NS protein that has been released from the ribosome. A preponderance of the latter would visually obscure changes in the former.
A hard-coded polylysine tract and 13myc extension (K12–13myc) allowed us to electrophoretically distinguish translationally stalled protein (at the polylysine tract) from translational read-through products. In WT cells, although translationally stalled Vma12-K12 was clearly detectable, the majority of the protein migrated at a rate consistent with full-length protein (Vma12-K12–13myc), demonstrating considerable translational read-through of the polylysine tract (Fig. 6B). Direct comparison of Vma12-K12–13myc and CPY-K12–13myc indicates a stronger propensity for pausing at the internal polylysine tract in the soluble ER luminal protein CPY than in the integral ER membrane protein Vma12 (cf. Figs. 5B and 6B). Deletion of RKR1, but not DOA10 or HRD1, increased the abundance of the translationally stalled Vma12-K12. The abundance of the full-length translational read-through protein was not increased in the absence of any of the E3s. Simultaneous deletion of all three genes did not increase the abundance of Vma12-K12 more than deletion of RKR1.
To confirm ER localization of Vma12-K12–13myc, we inserted a segment containing two N-glycosylation sites from the secreted enzyme invertase into the ER luminal loop of Vma12 to generate Vma12(glyc)-K12–13myc (see Fig. 2) (41, 54). Importantly, deletion of RKR1 increased the abundance of the faster migrating (translationally paused) species of this protein (Vma12(glyc)-K12) (Fig. 6C). Treatment with Endo H increased the mobility of truncated Vma12(glyc)-K12 and full-length Vma12(glyc)-K12–13myc, confirming ER localization. Together, these data indicate that Rkr1 is capable of mediating ribosome-associated degradation of transmembrane proteins targeted to the ER.
Switching the ER-targeting Mechanism of an ER-targeted Protein Influences Efficiency of Translational Stalling and Ribosome-associated Degradation
The soluble ER luminal CPY is post-translationally imported into the ER (55). By contrast, the transmembrane Vma12 is predicted to be co-translationally translocated into the ER membrane. Translational pausing at an internal polylysine tract and Rkr1-dependent degradation occurred more readily in the context of CPY-derived constructs than in those derived from Vma12 (cf. Figs. 5 and 6). We hypothesized that differences in frequency of translational read-through and efficiency of degradation correlate with mechanism of ER targeting (i.e. co- versus post-translational translocation). To test this hypothesis, we generated versions of CPY, CPY-NS, and CPY-K12–13myc that are co-translationally translocated. The post-translational ER-targeting signal sequence of CPY was replaced with the co-translational ER-targeting signal sequence of Ost1 (a subunit of the ER luminal oligosaccharyltransferase complex) to generate OPY (Ost1-CPY; see Fig. 2) (56). Endo H sensitivity confirmed ER localization of these OPY-derived proteins (Fig. 7A).
Consistent with our hypothesis, the co-translationally translocated OPY-NS and OPY-K12–13myc behave more similarly to Vma12- than CPY-derived constructs. Stop codon mutation or insertion of a polylysine tract destabilized OPY (Fig. 7, B and C), and proteasome inhibition increased abundance of OPY-K12 (while mildly, if at all, influencing OPY-NS abundance) (Fig. 4C). Similar to Vma12-NS, simultaneous deletion of DOA10, HRD1, and RKR1 did not markedly increase OPY-NS abundance (Fig. 7B). OPY-K12–13myc migrated as two species (Fig. 7C), consistent with translational pausing at the polylysine tract (OPY-K12) and translational read-through to the C-terminal 13myc extension (OPY-K12–13myc). Deletion of RKR1 preferentially increased the abundance of the translationally paused OPY-K12 relative to the full-length read-through product OPY-K12–13myc. Finally, in a manner more closely resembling Vma12-K12–13myc than CPY-K12–13myc, OPY-K12–13mycexhibited a significant propensity for translational read-through of the polylysine tract to the 13myc extension (Fig. 7D). In all of these respects, the co-translationally translocated ER-soluble protein OPY more closely resembled the co-translationally translocated transmembrane Vma12 protein than the post-translationally translocated soluble CPY protein. These data support the hypothesis that the mode of translocation influences efficiency of translational pausing and Rkr1-mediated ribosome-associated degradation.
Deletion of RKR1 Impairs Cell Growth in the Presence of a Model Nonstop ER-targeted Soluble Protein
A previous study indicated that a translationally stalled, tRNA-linked NS version of CPY persistently engages the ER translocon (57). In the absence of a functional NSD pathway (e.g. in the context of mutations in SKI7 or DOM34), this persistent engagement prevented efficient translocation of other ER-resident proteins and impaired growth, particularly at lower temperatures (57). We also observed a growth defect in ski7Δ dom34Δ cells when CPY-NS was expressed at elevated levels (Fig. 8A). Eliminating Dom34 is expected to disable both NS mRNA and NS protein quality control mechanisms. If the growth defect observed in these cells is due in part to deficiencies in ribosome-associated degradation, we predicted that ski7Δ rkr1Δ cells would phenocopy ski7Δ dom34Δ cells in the presence of CPY-NS. Indeed, high-level expression of CPY-NS (but not CPY with a wild-type stop codon) mildly, but reproducibly, impaired growth in ski7Δ rkr1Δ cells (Fig. 8B). This effect was specific to Rkr1, as ski7Δ doa10Δ and ski7Δ hrd1Δ cells expressing CPY-NS did not exhibit a growth defect.
FIGURE 8.

Loss of Rkr1 sensitizes cells to expression of CPY-NS. 6-Fold serial dilutions of yeast cells of the indicated genotypes harboring a plasmid encoding CPY-NS driven by the glyceraldehyde-3-phosphate dehydrogenase promoter were spotted onto minimal selective medium and incubated at 30 or 23 °C. See Fig. 2 for schematic depiction of constructs in this figure.
Discussion
In this report, we have shown that the ribosome-associated E3 Rkr1 regulates the abundance of NS and translationally stalled proteins that are targeted to the ER membrane in a novel quality control mechanism we term ERAD-RA (ERAD of ribosome-associated proteins). Stalled nascent chains of soluble ER proteins and transmembrane proteins are subject to Rkr1-dependent degradation, with an efficiency that depends on the ER-targeting mechanism. Although the canonical ERAD E3s Doa10 and Hrd1 are required for optimal growth under conditions associated with elevated NS protein production, neither regulates the abundance of the model NS proteins analyzed in this study. The physiological significance of ERAD-RA is underscored by the toxicity of elevated levels of a model NS protein in rkr1Δ ski7Δ cells. Furthermore, an NS allele of at least one gene encoding an ER-targeted protein (GPR54) has been associated with human pathology (8).
A previous report suggested that ER-targeted NS proteins are not subject to proteasome-dependent quality control (57). However, in our experiments, proteasome inhibition increased the abundance of model ER-targeted NS and K12 proteins (Fig. 4). Rkr1 regulates the abundance of NS and K12 versions of the post-translationally translocated ER-targeted CPY protein (Fig. 5). Although variants of the co-translationally translocated proteins Vma12 (Fig. 6) and OPY (Fig. 7) exhibit frequent translational read-through, translational stalling at the polylysine tract and Rkr1-dependent degradation do occur, albeit less robustly than for cytosolic or post-translationally translocated ER proteins. These data suggest that sequence elements upstream of the polylysine tract or the mechanism of translocon engagement influence the efficiency of translational stalling, presumably the critical factor in triggering Rkr1-dependent proteasomal degradation. It is possible that ER luminal chaperones, such as the HSP70 proteins Kar2 (58) or Lhs1 (59), exert a pulling force on co-translationally translocated proteins in a manner that counteracts translational stalling, thereby increasing translational read-through of polybasic sequences.
Vma12 and OPY are both co-translationally targeted to the ER membrane. In NS and K12 variants of both, translocation is predicted to begin prior to translational pausing at polylysine sequences. CPY, however, is a post-translationally translocated protein. Therefore, it may be expected that NS and K12 variants of CPY would be translated in the cytosol and detected by Rkr1 in a manner similar to previously characterized soluble cytosolic NS proteins (such as GFP). However, previous studies indicate that CPY-NS interacts with the translocon while covalently bound to a tRNA molecule (57). Furthermore, CPY-NS impairs translocation of other ER-targeted proteins in the presence of defects in ribosome dissociation (57). Together, these data strongly suggest simultaneous ribosome and translocon engagement. We speculate that CPY-NS and CPY-K12 are translated on free cytosolic ribosomes until the polylysine tract enters the exit tunnel and arrests translation (Fig. 9). The stalled ribosome-nascent chain complex may then be targeted to an ER-resident translocon via the post-translational translocation mechanism. Translocation would proceed until the portion of the protein occluded within the ribosome exit tunnel is reached. Consistent with partial translocation into the ER lumen, all variants of CPY analyzed in this study become N-glycosylated, unless the signal peptide has been mutated (Fig. 5). Given a role for Hrd1 in the degradation of other proteins that aberrantly engage the translocon (40, 41), we anticipated that Hrd1 would contribute to the degradation of CPY-NS. This was not the case. Therefore, at least two distinct quality control mechanisms appear to act on proteins that persistently or aberrantly engage the ER translocon.
FIGURE 9.
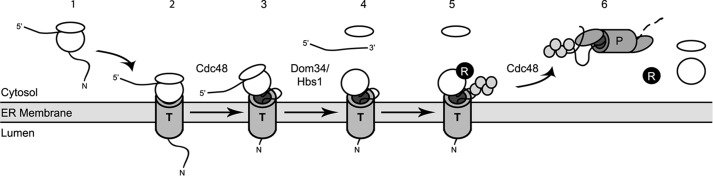
Hypothetical model for translocon engagement and Rkr1-dependent ERAD-RA. Step 1, translation of an NS protein begins in the cytosol on a free ribosome. Step 2, co-translationally translocated NS proteins are delivered to the translocon through interactions with signal recognition particle. Post-translationally translocated NS proteins on free ribosomes stall when the ribosome translates the polylysine tract. Translocation of the protein proceeds, and the nascent polypeptide remains ribosome-engaged. Step 3, upon detection by quality control machinery, the translationally and translocationally stalled protein is partially or completely retrotranslocated, in a manner that may depend on Cdc48. Step 4, as for soluble cytosolic ribosome-associated substrates of Rkr1, Dom34 and Hbs1 trigger dissociation of ribosome small and large subunits and release of mRNA. The translationally stalled protein remains engaged with the large subunit. Step 5, Rkr1 binds to the dissociated ribosome and ubiquitylates the associated aberrant protein. Step 6, ubiquitylated protein is extracted from the ribosome, presumably in a Cdc48-dependent manner, and degraded by the proteasome. N, N terminus of nascent chain; R, Rkr1; T, translocon; P, proteasome. Gray circles, ubiquitin monomers.
Recent structural data suggest that the catalytic RING domain of Rkr1 is poised at the opening of the exit tunnel of the dissociated 60S subunit (23, 60, 61). Nascent polytopic membrane proteins (such as Vma12) are likely to expose amino acids to the cytosol during translocation. Ribosome-associated Rkr1 may be readily envisaged to ubiquitylate such sequences. However, nascent co-translationally translocated soluble ER-targeted polypeptides, which are thought to move directly from the ribosome exit tunnel into the translocon, may never experience cytosolic exposure (24); hence, it is not apparent how nascent soluble NS and K12 proteins that simultaneously engage the ribosome and translocon could be ubiquitylated by Rkr1. We speculate that the translationally and translocationally arrested protein is at least partially retrotranslocated prior to ubiquitylation by Rkr1 and proteasomal degradation (Fig. 9). This may be accomplished by retrograde movement of the nascent polypeptide through the translocon (62). Alternatively, the partially translocated protein may be laterally transferred from the translocon to a distinct retrotranslocating channel.
The sensitivity of cells lacking Doa10 or Hrd1 to hygromycin B (Fig. 1) initially led us to hypothesize a role for these E3s in ERAD-RA. However, neither detectably contributed to the degradation of the ERAD-RA substrates investigated in this study. To what, then, can the sensitivity of cells lacking Doa10 and Hrd1 to hygromycin B be attributed? In this study, we investigated the E3 specificity for a small subset of ER-targeted proteins. It is possible that Rkr1 fortuitously recognizes the NS and K12 forms of these proteins and that Doa10 and Hrd1 recognize other cytosolic or ER-targeted NS proteins (represented in the panoply of NS proteins likely to be generated by hygromycin B). In support of this notion, deletion of DOA10 and HRD1 has been reported to cause small reductions in ubiquitylation of ribosome-associated proteins (63). Another potential explanation for sensitivity of these cells derives from the mechanism of action of hygromycin B, which induces ribosome A site distortion and errors in mRNA decoding (47). This is likely to result in frequent missense mutation, contributing to a heterogeneous collection of aberrant and potentially harmful proteins. Some of these anomalous species may be substrates of characterized Doa10- and Hrd1-dependent ERAD pathways. It is noteworthy, however, that the absence of Doa10 and Hrd1 is not detrimental in the presence of anisomycin or cycloheximide, both of which are also expected to result in a variety of abnormal proteins. Interestingly, the ERAD E3s and Rkr1 appear to play partially overlapping roles in protecting cells against protein aggregate formation (64). The extent to which this relates to ribosome-associated degradation remains to be determined.
In the presence of an intact NSD pathway, the effect of eliminating specific E3s on GFP-NS protein abundance was highly inconsistent (Fig. 3). Variability in abundance in E3 gene deletion strains was observed in experiments conducted by both labs contributing to this study. In the context of an intact NSD pathway, deleting DOA10 increased the abundance of GFP-NS as frequently and to a similar extent as deleting RKR1. However, when NSD was inactivated, no increase in GFP-NS abundance was observed in the absence of Doa10. These data provide tantalizing evidence that Doa10 may yet play a role in ribosome-associated quality control but in a manner that requires a functional NSD pathway. Alternatively, a functional NSD pathway may reduce the frequency of NS protein translation to a level that reveals the effects of minor stochastic events that impact NS protein abundance.
Proteasome inhibition increased the abundance of ER-targeted NS and K12 proteins to a greater degree than RKR1 deletion. This suggests that E3s other than Rkr1 contribute to ERAD-RA. One candidate E3 is Not4, which has been implicated in the degradation of a subset of cytosolic ribosome-associated degradation substrates (11, 65). Ubr1 has also been shown to co-translationally target for degradation proteins with N-terminal degrons (66). Furthermore, Hel2 and Hul5 have been implicated in co-translational ubiquitylation of nascent polypeptides (63). Additional experiments are necessary to determine the contributions of these and other proteins to ERAD-RA.
While this manuscript was in preparation, others reported a physical complex composed of mammalian RQC proteins, the large subunit of the ribosome, and the Sec61 translocon. Furthermore, elegant in vitro studies provided strong evidence for RQC of soluble ER-targeted polypeptides (67). These biochemical and in vitro data are consistent with and complementary to our in vivo characterization of ERAD-RA.
The E3 Rkr1 targets translationally arrested cytosolic proteins for proteasomal degradation. Our results indicate that Rkr1 also promotes proteasomal degradation of translationally stalled ER-targeted proteins. Transmembrane and soluble (both co- and post-translationally translocated) proteins are subject to Rkr1-mediated ERAD-RA. Loss of RKR1 sensitizes cells to conditions associated with globally elevated NS protein production (Fig. 1) and to high level expression of a single specific NS protein (Fig. 8). An improved understanding of the quality control mechanisms governing recognition of translationally stalled ER-targeted proteins is likely to be clinically significant.
Author Contributions
E. M. R. and S. G. K. designed the study and wrote the paper. J. J. C., M. G., R. T. G., E. S. F., B. W. B., N. S., A. S., and S. G. K. performed and analyzed the experiments. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Amy Wu, Katelynn Mannix, Ian Tesch, and Nagjie Aziraj for generating reagents and preliminary data for these studies. We thank Mark Hochstrasser for continued friendship and advice. We thank Mark Hochstrasser, Elke Deuerling, and Steffen Preissler for helpful discussions in the initial phase of the study. S. G. K. thanks Martin Scheffner for stimulating discussions and continuous support. We thank Toshifumi Inada (Tohoku University) for plasmids.
This work was supported, in whole or in part, by National Institutes of Health Grant R15 GM111713 (to E. M. R.). This work was also supported by Ball State University ASPiRE research awards (to J. J. C., E. S. F., and E. M. R.), an Indiana Academy of Science senior research award (to E. M. R.), funds from the Ball State University Provost's Office and Department of Biology (to E. M. R.), and Deutsche Forschungsgemeinschaft Grant SFB969 (to S. G. K.). The authors declare that they have no conflicts of interest with the contents of this article.
- NS
- nonstop
- NSD
- nonstop decay
- ER
- endoplasmic reticulum
- RQC
- ribosome quality control
- ERAD
- ER-associated degradation
- Endo H
- endoglycosidase H
- G6PDH
- glucose-6-phosphate dehydrogenase
- K12
- 12 consecutive lysine amino acids
- CPY
- vacuolar carboxypeptidase Y
- glyc
- portion of invertase protein possessing two consensus N-glycosylation sequences
- OPY
- CPY with ER-targeting signal sequence replaced with that from Ost1.
References
- 1. Lykke-Andersen J., Bennett E. J. (2014) Protecting the proteome: eukaryotic cotranslational quality control pathways. J. Cell Biol. 204, 467–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Chu J., Hong N. A., Masuda C. A., Jenkins B. V., Nelms K. A., Goodnow C. C., Glynne R. J., Wu H., Masliah E., Joazeiro C. A., Kay S. A. (2009) A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. U.S.A. 106, 2097–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bengtson M. H., Joazeiro C. A. (2010) Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Braun M. A., Costa P. J., Crisucci E. M., Arndt K. M. (2007) Identification of Rkr1, a nuclear RING domain protein with functional connections to chromatin modification in Saccharomyces cerevisiae. Mol. Cell. Biol. 27, 2800–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klucevsek K. M., Braun M. A., Arndt K. M. (2012) The Paf1 complex subunit Rtf1 buffers cells against the toxic effects of [PSI+] and defects in Rkr1-dependent protein quality control in Saccharomyces cerevisiae. Genetics 191, 1107–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Taniguchi A., Hakoda M., Yamanaka H., Terai C., Hikiji K., Kawaguchi R., Konishi N., Kashiwazaki S., Kamatani N. (1998) A germline mutation abolishing the original stop codon of the human adenine phosphoribosyltransferase (APRT) gene leads to complete loss of the enzyme protein. Hum. Genet. 102, 197–202 [DOI] [PubMed] [Google Scholar]
- 7. Pang S., Wang W., Rich B., David R., Chang Y. T., Carbunaru G., Myers S. E., Howie A. F., Smillie K. J., Mason J. I. (2002) A novel non-stop mutation in the stop codon and a novel missense mutation in the type II 3β-hydroxysteroid dehydrogenase (3β-HSD) gene causing, respectively, nonclassic and classic 3β-HSD deficiency congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 87, 2556–2563 [DOI] [PubMed] [Google Scholar]
- 8. Seminara S. B., Messager S., Chatzidaki E. E., Thresher R. R., Acierno J. S., Jr., Shagoury J. K., Bo-Abbas Y., Kuohung W., Schwinof K. M., Hendrick A. G., Zahn D., Dixon J., Kaiser U. B., Slaugenhaupt S. A., Gusella J. F., et al. (2003) The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 349, 1614–1627 [DOI] [PubMed] [Google Scholar]
- 9. Cacciottolo M., Numitone G., Aurino S., Caserta I. R., Fanin M., Politano L., Minetti C., Ricci E., Piluso G., Angelini C., Nigro V. (2011) Muscular dystrophy with marked dysferlin deficiency is consistently caused by primary dysferlin gene mutations. Eur. J. Hum. Gen. 19, 974–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tsuboi T., Kuroha K., Kudo K., Makino S., Inoue E., Kashima I., Inada T. (2012) Dom34:Hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell 46, 518–529 [DOI] [PubMed] [Google Scholar]
- 11. Dimitrova L. N., Kuroha K., Tatematsu T., Inada T. (2009) Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J. Biol. Chem. 284, 10343–10352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lu J., Deutsch C. (2008) Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 384, 73–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ito-Harashima S., Kuroha K., Tatematsu T., Inada T. (2007) Translation of the poly(A) tail plays crucial roles in non-stop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 21, 519–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Graille M., Chaillet M., van Tilbeurgh H. (2008) Structure of yeast Dom34: a protein related to translation termination factor Erf1 and involved in No-Go decay. J. Biol. Chem. 283, 7145–7154 [DOI] [PubMed] [Google Scholar]
- 15. Lee H. H., Kim Y. S., Kim K. H., Heo I., Kim S. K., Kim O., Kim H. K., Yoon J. Y., Kim H. S., Kim do J., Lee S. J., Yoon H. J., Kim S. J., Lee B. G., Song H. K., et al. (2007) Structural and functional insights into Dom34, a key component of no-go mRNA decay. Mol. Cell 27, 938–950 [DOI] [PubMed] [Google Scholar]
- 16. Shoemaker C. J., Eyler D. E., Green R. (2010) Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science 330, 369–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Brandman O., Stewart-Ornstein J., Wong D., Larson A., Williams C. C., Li G. W., Zhou S., King D., Shen P. S., Weibezahn J., Dunn J. G., Rouskin S., Inada T., Frost A., Weissman J. S. (2012) A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Defenouillère Q., Yao Y., Mouaikel J., Namane A., Galopier A., Decourty L., Doyen A., Malabat C., Saveanu C., Jacquier A., Fromont-Racine M. (2013) Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc. Natl. Acad. Sci. U.S.A. 110, 5046–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Verma R., Oania R. S., Kolawa N. J., Deshaies R. J. (2013) Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2, e00308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shao S., von der Malsburg K., Hegde R. S. (2013) Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol. Cell 50, 637–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shao S., Hegde R. S. (2014) Reconstitution of a minimal ribosome-associated ubiquitination pathway with purified factors. Mol. Cell 55, 880–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wilson M. A., Meaux S., van Hoof A. (2007) A genomic screen in yeast reveals novel aspects of non-stop mRNA metabolism. Genetics 177, 773–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shao S., Brown A., Santhanam B., Hegde R. S. (2015) Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Becker T., Bhushan S., Jarasch A., Armache J. P., Funes S., Jossinet F., Gumbart J., Mielke T., Berninghausen O., Schulten K., Westhof E., Gilmore R., Mandon E. C., Beckmann R. (2009) Structure of monomeric yeast and mammalian Sec61 complexes interacting with the translating ribosome. Science 326, 1369–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mitra K., Schaffitzel C., Shaikh T., Tama F., Jenni S., Brooks C. L., 3rd, Ban N., Frank J. (2005) Structure of the E. coli protein-conducting channel bound to a translating ribosome. Nature 438, 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wilhovsky S., Gardner R., Hampton R. (2000) HRD gene dependence of endoplasmic reticulum-associated degradation. Mol. Biol. Cell 11, 1697–1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Swanson R., Locher M., Hochstrasser M. (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev. 15, 2660–2674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Plemper R. K., Bordallo J., Deak P. M., Taxis C., Hitt R., Wolf D. H. (1999) Genetic interactions of Hrd3p and Der3p/Hrd1p with Sec61p suggest a retro-translocation complex mediating protein transport for ER degradation. J. Cell Sci. 112, 4123–4134 [DOI] [PubMed] [Google Scholar]
- 29. Hampton R. Y., Gardner R. G., Rine J. (1996) Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 31. Deak P. M., Wolf D. H. (2001) Membrane topology and function of Der3/Hrd1p as a ubiquitin-protein ligase (E3) involved in endoplasmic reticulum degradation. J. Biol. Chem. 276, 10663–10669 [DOI] [PubMed] [Google Scholar]
- 32. Zattas D., Hochstrasser M. (2015) Ubiquitin-dependent protein degradation at the yeast endoplasmic reticulum and nuclear envelope. Crit. Rev. Biochem. Mol. Biol. 50, 1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sato B. K., Schulz D., Do P. H., Hampton R. Y. (2009) Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol. Cell 34, 212–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gauss R., Sommer T., Jarosch E. (2006) The Hrd1p ligase complex forms a linchpin between ER-lumenal substrate selection and Cdc48p recruitment. EMBO J. 25, 1827–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carvalho P., Goder V., Rapoport T. A. (2006) Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373 [DOI] [PubMed] [Google Scholar]
- 36. Ravid T., Kreft S. G., Hochstrasser M. (2006) Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 25, 533–543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Metzger M. B., Maurer M. J., Dancy B. M., Michaelis S. (2008) Degradation of a cytosolic protein requires endoplasmic reticulum-associated degradation machinery. J. Biol. Chem. 283, 32302–32316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huyer G., Piluek W. F., Fansler Z., Kreft S. G., Hochstrasser M., Brodsky J. L., Michaelis S. (2004) Distinct machinery is required in Saccharomyces cerevisiae for the endoplasmic reticulum-associated degradation of a multispanning membrane protein and a soluble luminal protein. J. Biol. Chem. 279, 38369–38378 [DOI] [PubMed] [Google Scholar]
- 39. Habeck G., Ebner F. A., Shimada-Kreft H., Kreft S. G. (2015) The yeast ERAD-C ubiquitin ligase Doa10 recognizes an intramembrane degron. J. Cell Biol. 209, 261–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Watts S. G., Crowder J. J., Coffey S. Z., Rubenstein E. M. (2015) Growth-based determination and biochemical confirmation of genetic requirements for protein degradation in Saccharomyces cerevisiae. J. Vis. Exp. 96, e52428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rubenstein E. M., Kreft S. G., Greenblatt W., Swanson R., Hochstrasser M. (2012) Aberrant substrate engagement of the ER translocon triggers degradation by the Hrd1 ubiquitin ligase. J. Cell Biol. 197, 761–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hrizo S. L., Gusarova V., Habiel D. M., Goeckeler J. L., Fisher E. A., Brodsky J. L. (2007) The Hsp110 molecular chaperone stabilizes apolipoprotein B from endoplasmic reticulum-associated degradation (ERAD). J. Biol. Chem. 282, 32665–32675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Guthrie C., Fink G. R., Abelson J. N., Simon M. I. (eds) (2004) Guide to Yeast Genetics and Molecular and Cell Biology, pp. 1–41. Elsevier, San Diego [Google Scholar]
- 44. Kushnirov V. V. (2000) Rapid and reliable protein extraction from yeast. Yeast 16, 857–860 [DOI] [PubMed] [Google Scholar]
- 45. Loayza D., Tam A., Schmidt W. K., Michaelis S. (1998) Ste6p mutants defective in exit from the endoplasmic reticulum (ER) reveal aspects of an ER quality control pathway in Saccharomyces cerevisiae. Mol. Biol. Cell 9, 2767–2784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hjelm H., Hjelm K., Sjöquist J. (1972) Protein A from Staphylococcus aureus. Its isolation by affinity chromatography and its use as an immunosorbent for isolation of immunoglobulins. FEBS Lett. 28, 73–76 [DOI] [PubMed] [Google Scholar]
- 47. Brodersen D. E., Clemons W. M., Jr., Carter A. P., Morgan-Warren R. J., Wimberly B. T., Ramakrishnan V. (2000) The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell 103, 1143–1154 [DOI] [PubMed] [Google Scholar]
- 48. Ganoza M. C., Kiel M. C. (2001) A ribosomal ATPase is a target for hygromycin B inhibition on Escherichia coli ribosomes. Antimicrob. Agents Chemother. 45, 2813–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grollman A. P. (1967) Inhibitors of protein biosynthesis. II. Mode of action of anisomycin. J. Biol. Chem. 242, 3226–3233 [PubMed] [Google Scholar]
- 50. Grollman A. P. (1966) Structural basis for inhibition of protein synthesis by emetine and cycloheximide based on an analogy between ipecac alkaloids and glutarimide antibiotics. Proc. Natl. Acad. Sci. U.S.A. 56, 1867–1874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van Hoof A., Frischmeyer P. A., Dietz H. C., Parker R. (2002) Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science 295, 2262–2264 [DOI] [PubMed] [Google Scholar]
- 52. Jackson D. D., Stevens T. H. (1997) VMA12 encodes a yeast endoplasmic reticulum protein required for vacuolar H+-ATPase assembly. J. Biol. Chem. 272, 25928–25934 [DOI] [PubMed] [Google Scholar]
- 53. Stevens T., Esmon B., Schekman R. (1982) Early stages in the yeast secretory pathway are required for transport of carboxypeptidase Y to the vacuole. Cell 30, 439–448 [DOI] [PubMed] [Google Scholar]
- 54. Reddy V. A., Johnson R. S., Biemann K., Williams R. S., Ziegler F. D., Trimble R. B., Maley F. (1988) Characterization of the glycosylation sites in yeast external invertase. I. N-Linked oligosaccharide content of the individual sequons. J. Biol. Chem. 263, 6978–6985 [PubMed] [Google Scholar]
- 55. Ng D. T., Brown J. D., Walter P. (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J. Cell Biol. 134, 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Willer M., Forte G. M., Stirling C. J. (2008) Sec61p is required for ERAD-L: genetic dissection of the translocation and ERAD-L functions of Sec61P using novel derivatives of CPY. J. Biol. Chem. 283, 33883–33888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Izawa T., Tsuboi T., Kuroha K., Inada T., Nishikawa S., Endo T. (2012) Roles of dom34:hbs1 in non-stop protein clearance from translocators for normal organelle protein influx. Cell Rep. 2, 447–453 [DOI] [PubMed] [Google Scholar]
- 58. Brodsky J. L., Goeckeler J., Schekman R. (1995) BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 92, 9643–9646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tyson J. R., Stirling C. J. (2000) LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J. 19, 6440–6452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shen P. S., Park J., Qin Y., Li X., Parsawar K., Larson M. H., Cox J., Cheng Y., Lambowitz A. M., Weissman J. S., Brandman O., Frost A. (2015) Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347, 75–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lyumkis D., Oliveira dos Passos D., Tahara E. B., Webb K., Bennett E. J., Vinterbo S., Potter C. S., Carragher B., Joazeiro C. A. (2014) Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc. Natl. Acad. Sci. U.S.A. 111, 15981–15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ooi C. E., Weiss J. (1992) Bidirectional movement of a nascent polypeptide across microsomal membranes reveals requirements for vectorial translocation of proteins. Cell 71, 87–96 [DOI] [PubMed] [Google Scholar]
- 63. Duttler S., Pechmann S., Frydman J. (2013) Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell 50, 379–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Theodoraki M. A., Nillegoda N. B., Saini J., Caplan A. J. (2012) A network of ubiquitin ligases is important for the dynamics of misfolded protein aggregates in yeast. J. Biol. Chem. 287, 23911–23922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Matsuda R., Ikeuchi K., Nomura S., Inada T. (2014) Protein quality control systems associated with no-go and non-stop mRNA surveillance in yeast. Genes Cells 19, 1–12 [DOI] [PubMed] [Google Scholar]
- 66. Turner G. C., Varshavsky A. (2000) Detecting and measuring cotranslational protein degradation in vivo. Science 289, 2117–2120 [DOI] [PubMed] [Google Scholar]
- 67. von der Malsburg K., Shao S., Hegde R. S. (2015) The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol. Biol. Cell 10.1091/mbc.E15-01-0040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tong A. H., Evangelista M., Parsons A. B., Xu H., Bader G. D., Pagé N., Robinson M., Raghibizadeh S., Hogue C. W., Bussey H., Andrews B., Tyers M., Boone C. (2001) Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science 294, 2364–2368 [DOI] [PubMed] [Google Scholar]
- 69. Brachmann C. B., Davies A., Cost G. J., Caputo E., Li J., Hieter P., Boeke J. D. (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14, 115–132 [DOI] [PubMed] [Google Scholar]
- 70. Sikorski R. S., Hieter P. (1989) A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics 122, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gietz R. D., Sugino A. (1988) New yeast-Escherichia coli shuttle vectors constructed with in vitro mutagenized yeast genes lacking six-base pair restriction sites. Gene 74, 527–534 [DOI] [PubMed] [Google Scholar]
- 72. Mumberg D., Müller R., Funk M. (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156, 119–122 [DOI] [PubMed] [Google Scholar]
- 73. Inada T., Aiba H. (2005) Translation of aberrant mRNAs lacking a termination codon or with a shortened 3′-UTR is repressed after initiation in yeast. EMBO J. 24, 1584–1595 [DOI] [PMC free article] [PubMed] [Google Scholar]