

Background: The DNA methyltransferase DNMT1 and tyrosine-protein kinase KIT are crucial for lung tumorigenesis, and the resistance to their inhibitors invariably develops.
Results: Simultaneously reactivated DNMT1 and KIT endow drug-resistant cells with a survival and growth advantage.
Conclusion: Reciprocal inactivation of DNMT1 and KIT eradicates drug-resistant cells.
Significance: DNMT1 and KIT interplay represents a therapeutic target for relapsed or refractory lung cancer patients.
Keywords: DNA methyltransferase, drug resistance, lung cancer, protein kinase, tumor suppressor gene
Abstract
Lung cancer cells are sensitive to 5-aza-2′-deoxycytidine (decitabine) or midostaurin (PKC412), because decitabine restores the expression of methylation-silenced tumor suppressor genes, whereas PKC412 inhibits hyperactive kinase signaling, which is essential for cancer cell growth. Here, we demonstrated that resistance to decitabine (decitabineR) or PKC412 (PKC412R) eventually results from simultaneously remethylated DNA and reactivated kinase cascades. Indeed, both decitabineR and PKC412R displayed the up-regulation of DNA methyltransferase DNMT1 and tyrosine-protein kinase KIT, the enhanced phosphorylation of KIT and its downstream effectors, and the increased global and gene-specific DNA methylation with the down-regulation of tumor suppressor gene epithelial cadherin CDH1. Interestingly, decitabineR and PKC412R had higher capability of colony formation and wound healing than parental cells in vitro, which were attributed to the hyperactive DNMT1 or KIT, because inactivation of KIT or DNMT1 reciprocally blocked decitabineR or PKC412R cell proliferation. Further, DNMT1 knockdown sensitized PKC412R cells to PKC412; conversely, KIT depletion synergized with decitabine in eliminating decitabineR. Importantly, when engrafted into nude mice, decitabineR and PKC412R had faster proliferation with stronger tumorigenicity that was caused by the reactivated KIT kinase signaling and further CDH1 silencing. These findings identify functional cross-talk between KIT and DNMT1 in the development of drug resistance, implying the reciprocal targeting of protein kinases and DNA methyltransferases as an essential strategy for durable responses in lung cancer.
Introduction
Although chemotherapies have initially achieved a moderate response or even complete remission, most patients with non-small cell lung cancer (NSCLC)2 experience periodic relapses (1). It is believed that the resistance of lung cancer cells to anticancer agents is a major cause of chemotherapeutic failure. Although the precise mechanisms underlying drug-resistant phenotype remain elusive, accumulating evidence indicates that the acquisition of drug resistance involves a complex and multifactorial process, including increased DNA repair, altered cell cycle kinetics, up-regulated oncogenes (2, 3), and deregulated epigenetics (4).
DNA methylation is a major epigenetic change and involves the covalent transfer of a methyl group to the fifth position of a cytosine residue within CpG dinucleotides. Promoter DNA hypermethylation, which causes the silencing of tumor suppressor genes involved in lung carcinogenesis (5–7), is attributed to hyperactive DNA methyltransferases (DNMTs). In accordance, DNMTs are found to be highly expressed in different type of tumors (8–13), and DNMT overexpression correlates significantly with the CpG island methylator phenotype (14). Notably, DNMT1 up-regulation serves as an independent prognostic factor in lung cancer (7, 8, 15). As a consequence, DNA hypomethylating agents, such as 5-aza-2′-deoxycytidine (decitabine), have been developed for targeted therapy in different cancers (16, 17). Decitabine is a deoxycytidine analog that induces a covalent entrapment of DNMT1 to decitabine-substituted DNA leading to a significant loss of maintenance DNMT activity followed by a reduction of DNA methylation (18, 19). Another mechanism underlying decitabine-induced DNA hypomethylation likely involves ubiquitin-dependent DNMT1 degradation. Regardless, the therapeutic effects of decitabine are limited in against cancers, like NSCLC, because decitabine displays cell cycle-dependent activity (20); thus quiescent/slow proliferating lung tumor cell subpopulations could escape from decitabine-induced cell killing. Further, it is reported that the genomic DNA becomes remethylated in patients at the end of decitabine therapy, but the underlying mechanisms remain largely unclear.
Receptor tyrosine kinases (RTKs) are transmembrane proteins translating extracellular signals into active intracellular cues. Under physiological conditions, these receptors are activated only by ligand binding. In cancers, such ligand-dependent activation can be bypassed because of RTK overexpression or activating mutations. In particular, RTK overexpression increases the dynamics of homo/heterodimerization without ligand (21, 22), resulting in hyperactive RTK signaling followed by aggressive NSCLC growth and metastasis (23–25). As such, RTK inhibitors, like midostaurin (PKC412), are in various preclinical and clinical testing stages (26–30) with inhibitory activities against class III RTKs, including KIT and FLT3 (Fms-like tyrosine kinase 3) (31, 32). However, PKC412 failed to achieve long term therapeutic efficacy, and resistant mechanisms against PKC412 occurred even from the very beginning of treatment. The molecular processes underlying the onset of chemoresistance are believed to be the acquired mutations, but kinase gene overamplification could be essential in developing drug resistance (33, 34), given that gene up-regulation is a major mechanism of oncogenic activation (35).
It is reported that DNMT1 and KIT are highly expressed in certain chemotherapeutic resistance (34, 36), and because aberrant DNA methylation appears in resistance to imatinib (Gleevec) in leukemia (37), we proposed that KIT and DNMT1 may work in concert during lung tumor progression to a drug-resistant phenotype. In the present study, we have generated drug-resistant cells by chronic exposure of lung cancer cells to decitabine or PKC412 in vitro. Compared with parental cells, both decitabineR and PKC412R exhibited a higher proliferation rate in vitro and stronger tumorigenicity in xenograft models when the inhibitor treatment was discontinued. Mechanistic investigations revealed that the enhanced proliferative potential in both decitabineR and PKC412R was ascribed to the reactivated kinase signaling and a DNA hypermethylation profile. Our findings offer mechanistic insight into decitabine and PKC412 resistance, and they illustrate how reciprocal application of inhibitors for DNMT1 and KIT oncogenic pathways may improve the anticancer responses of decitabine and PKC412, and potentially, other types of DNA methylation and RTK inhibitors in lung cancer therapy.
Experimental Procedures
Cell Lines and Chemicals
H1975 and A549 cell lines were obtained from American Type Culture Collection (Manassas, VA) and grown in RPMI 1640 medium with 10% FBS (Life Technologies) at 37 °C under 5% CO2. For the drug treatment, cells were treated with the following reagents used at concentrations, times, and schedules indicated under “Results.” PKC412 (Midostaurin) was obtained from LC Laboratories (Woburn, MA), and decitabine (5-aza-2′-deoxycytidine or Dacogen) was from Sigma-Aldrich.
Generation of PKC412 or Decitabine-resistant Cells
H1975 and A549 cells were cultured continuously with a stepwise increase of decitabine or PKC412 concentrations for 6 weeks. Parental cells were cultured in parallel without decitabine or PKC412 and served as control. Resistant cells were maintained in medium containing 0.5 μm of decitabine or PKC412.
Transfections
1 × 106 cells were seeded into 6-well plates overnight before transfection. ON-TARGETplus Smart pool siRNAs containing a mixture of four oligonucleotides against KIT, DNMT1, and Sp1, as well as the scramble oligonucleotides were obtained from Dharmacon (Thermo Scientific). The siRNA oligonucleotides with final concentration of 100 nm were introduced into cells using Lipofectamine® RNAiMAX transfection reagent (Life Technologies) as previously described (10, 38).
Cell Proliferation Assays
After various treatments, the cell proliferation was assessed by methylcellulose colony assays in MethoCult® medium (Stem Cell Technologies) and MTS assays as previously described (10, 38). The colonies were scored in 7–14 days.
Wound Healing Assays
Lung cancer cells were treated with decitabine or PKC412 or transfected with siRNA of DNMT1, KIT, or Sp1 as indicated, and the wound healing assays were performed as previously described (38). The migration of the cells toward the wound was photographed under light microscope, and the migration distance was determined by CorelDRAWX5 Software.
Dot Blotting
The genomic DNA was purified using DNA blood/tissue kit (Qiagen), and the dot-blot was performed as previously described (10, 38). Briefly, ∼2 μg of DNA was denatured, spotted on the prewet positively charged nylon membrane, blocked with 5% nonfat milk, and incubated with mouse anti-5-methylcytosine (Active Motif, Carlsbad, CA). The signal was detected by HRP-conjugated secondary antibody and enhanced chemiluminescence.
Immunoprecipitation and Western Blot
After the various treatments, the whole cellular lysates were prepared in 1× cell lysis buffer (10, 33). Approximately 1 mg of total protein lysates was precleared with 70 μl of 50% slurry of Dynabeads® Protein G (Life Technologies), and the immunoprecipitation was performed as described previously (33). The immunoprecipitates or the whole cellular lysates were resolved by SDS-PAGE and transferred onto PVDF membranes (Amersham Biosciences) for Western blot (10, 33). The antibodies are: Sp1 and β-actin (Santa Cruz Biotechnology, Santa Cruz, CA); KIT, phospho-KIT (Tyr-719), AKT, phospho-AKT (Ser-473), STAT3, phospho-STAT3, STAT5, phospho-STAT5, and CDH1 (Cell Signaling Technology, Danvers, MA); DNMT1 (New England Biolabs, Ipswich, MA); and phosphotyrosine 4G10 (anti-4G10) (Millipore, Billerica, MA).
RNA Isolation, cDNA Preparation, and qPCR
RNA was isolated using miRNAeasy Kit (Qiagen), and cDNA was synthesized by SuperScript® III first strand synthesis system (Invitrogen). qPCR was performed by TaqMan technology (Applied Biosystems, Foster City, CA) for the expression of DNMT1 and KIT or by SYBR Green for the expression of CDH1 normalized by 18S levels. Expression of the target genes was measured using the ΔCT approach. The primers are: CDH1 forward, 5′-AGAACGCATTGCCACATACAC-3′; CDH1 reverse, 5′-GAGGATGGTGTAAGCGATGG-3′; 18S forward, 5′-ACAGGATTGACAGATTGA-3′; and 18S reverse, 5′-TATCGGAATTAACCAG ACA-3′.
Gene Microarray
Total RNA isolated using miRNAeasy kit (Qiagen) was subjected to gene expression analysis using Illumina array expression system. Changes in gene expression were considered statistically significant (p < 0.05) when up- or down-regulated by at least 1.5-fold. Pathway analysis was performed using the DAVID bioinformatics resources 6.7.
Bisulfite Sequencing Analysis
The bisulfite sequencing was performed as previously described (10, 38). Briefly, ∼2 μg of genomic DNA was modified with sodium bisulfite using an EpiTect bisulfite kit (Qiagen). The human CDH1 promoter region (−251 to +139) was amplified by PCR using the bisulfite-treated DNA as template. Ten clones were sequenced with M13R primer in Genewiz. The primers are: forward, 5′-TTTTTTTTGATTTTAGGTTTTAGTGAG-3′; and reverse, 5′-ACTCCAAAAACCCATAACTAACC-3′.
In Vivo Tumorigenesis Assays
Athymic nude mice (4–6 weeks old) were purchased from Harlan Laboratories (Madison, WI). Approximately 1 × 106 decitabineR, PKC412R, or parental cells were subcutaneously injected into the flanks of nude mice. Tumor diameters were measured after 2 days from injection and then every 2 days. Tumor sizes were analyzed using the formula π/6 × A × B × C, where A is the length, B is the width, and C is the height, expressed in cubic millimeters (10). Tumor-bearing mice were sacrificed at the indicated time points after cell injection, and the excised lungs were fixed with 10% formaldehyde for 24 h at 4 °C. The partial tumor tissue was frozen immediately in liquid nitrogen and then transferred to −80 °C freezer for subsequent molecular characterization. The leftover of tumor was fixed in 10% neutral buffered formalin for 24 h at 4 °C and then transferred to phosphate-buffered saline for paraffin-embedded blocks.
Hematoxylin and Eosin (H&E) and Immunohistochemistry Staining
Tumors and tissues collected from the animal studies were fixed in 10% neutral buffered formalin, and the paraffin-embedded samples were cut to 5-μm thickness. H&E and immunohistochemistry staining were performed using our previous protocol (10, 38). Briefly, slides were heated at 60 °C, deparaffinized in Histo-Clear, rehydrated through graded ethanol, and microwaved to unmask antigens. After incubating with 10% goat serum to block the nonspecific binding, the primary anti-Ki-67 (Abcam, Cambridge, MA) was applied, and the slides were visualized with donkey anti-mouse secondary antibody.
Statistical Analysis
The quantification for target changes from Western blot, qPCR, dot-blot, and in vivo tumorigenic assays was performed using the Student's t test. All statistical analysis was conducted by GraphPad Prism 5.0. Differences were considered statistically significant at p < 0.05. All p values are two-tailed.
Results
Long Term Exposure of H1975 and A549 Cells to PKC412 or Decitabine Results in Resistance to Drug-induced Growth Inhibition
Although both KIT and DNMT1 are broadly targeted for cancer therapies, the clinical outcomes are disappointing, because of a subpopulation of cancer cells lacking response to drug-mediated regulation of cell proliferation, apoptosis, and differentiation. To disassemble the underlying molecular rules, we initially established drug resistance in NSCLC H1975 and A549 cells to PKC412 (PKC412R) or decitabine (decitabineR) by the stepwise increase of drug dosages and continuously culture in drug-containing medium for 6 weeks. The final concentrations were 0.5 μm for both PKC412 and decitabine, which exerted sufficient inhibitory action and were in the range of clinically achievable levels (39, 40). Initially, lung cancer cell migration was significantly inhibited when the drug concentrations approached 0.5 μm for both PKC412 (H1975, 973 ± 56 versus 436 ± 58 μm; A549, 923 ± 36 versus 336 ± 68 μm) and decitabine (H1975, 988 ± 35 versus 352 ± 65 μm; A549, 974 ± 56 versus 443 ± 51) (Fig. 1, A and B). However, the resistant cells regained the capability to proliferate and were able to be passaged in the presence of PKC412 or decitabine. When the drugs were temporally removed, H1975 resistant cells migrated and grew at a faster pace than the parental cells, indicated by the enhanced wound healing capability (387 ± 76 μm in parental versus 935 ± 54 μm in PKC412R; 498 ± 87 μm in parental versus 954 ± 33 μm in decitabineR) (Fig. 1C) and the increased colony number (86 ± 8 in parental, 124 ± 12 in decitabineR, 164 ± 16 in PKC412R) (Fig. 1D), suggesting that chronic treatment with PKC412 or decitabine led to a higher cell proliferation rate, which was further confirmed in A549 cells resistant to PKC412 and decitabine, as indicated by the enhanced wound healing capability (417 ± 66 μm in parental versus 962 ± 67 μm in PKC412R; 468 ± 33 μm in parental versus 939 ± 88 μm in decitabineR) (Fig. 1E). To further verify the acquisition of resistance phenotype, we treated PKC412R or decitabineR with the indicated concentrations of PKC412 or decitabine, respectively, for 48 h, and the parental cells were used as a negative control. These experiments revealed that the proliferation of parental cells was significantly inhibited in a dose-dependent manner, whereas there was no obvious change in both H1975 and A549 resistant cells, in the presence of the corresponding inhibitors (Fig. 1, F and G). These results support the notion that therapy-related acquisition of resistance poses significant limitations on therapeutic efficiency.
FIGURE 1.

Characterization of H1975 or A549 drug-resistant cells. A and B, wound healing assays in H1975 or A549 treated with 0.5 μm PKC412 (A) or decitabine (B). Quantitative measurement of cell migration was performed at 72 h post-treatment. C and D, wound healing (C) and colony forming (D) assays in H1975 PKC412R, decitabineR, and parental cells growing in drug-free medium. Quantitative measurement of cell migration was performed at 24 h. D, the graphs show the mean numbers of colonies, and pictured is a representative image (magnification, ×25). E, wound healing assays in A549 PKC412R and decitabineR cells. Quantitative measurement of cell migration was performed at 48 h. F and G, MTS assays in PKC412R (F), decitabineR (G), or parental H1975 and A549 cells treated with the indicated doses of PKC412 or decitabine, respectively, for 48 h. Note: In wound healing assays, the left panels show representative images of wound healing, and the right panels show graphs that indicate the quantification of “gap closure.” The data are representative of three independent experiments. *, p < 0.05.
Resistant Cells Possess Higher Tumorigenic Potential in Vivo
To examine whether decitabineR and PKC412R exhibit higher proliferative activity than parental cells in vivo, ∼1 × 106 of parental, decitabineR, or PKC412R H1975 cells were recovered in drug-free medium for 6 h and subcutaneously injected into the flanks of nude mice. Although the tumor incidence between the parental and decitabineR or PKC412R cells was similar (6 of 6, 100%) (Fig. 2, A and B), the tumor formation rate in parental was much slower than that in decitabineR or PKC412R cells (Fig. 2C), consistent with the higher expansion rate of decitabineR or PKC412R in vitro. Tumor volume increased proportionally between both groups over 16 days, but tumors from decitabineR or PKC412R grew faster with larger final tumor volume and weight than those from parental cells (Fig. 2, C and D). At the termination of the experiment, the mean tumor volume or weight was 252 ± 56 mm3 or 245 ± 52 mg in PKC412R and 244 ± 55 mm3 or 219 ± 43 mg in decitabineR, compared with 51 ± 16 mm3 or 53 ± 11 mg in parental control. Such a highly proliferative feature of resistant-derived tumors was further confirmed by the stronger staining of H&E and Ki-67 in decitabineR or PKC412R than in parental cells (Fig. 2E). Because decitabineR or PKC412R displayed more robust mobility in vitro, we analyzed the pathology of lung. Consistently, both decitabineR and PKC412R had higher metastatic growth in lung than parental control, indicated by the stronger H&E staining (Fig. 2F), the larger lesion areas (Fig. 2G, 5% in parental, 59% in decitabineR, 42% in PKC412R), and more metastatic foci (Fig. 2H; 2.3 ± 1.5 in parental, 11.3 ± 2.5 in decitabineR, 10.7 ± 2.6 in PKC412R). Collectively, these findings suggest that decitabineR and PKC412R cells develop a more aggressive phenotype.
FIGURE 2.

Drug-resistant cells exhibit stronger oncogenic potential in vivo. A and B, ∼1 × 106 PKC412R, decitabineR, or parental cells recovered in drug-free medium for 6 h were subcutaneously injected into the flanks of nude mice. The photographs of tumor-bearing mice (A) (n = 3 mice/group) and tumors (B) (n = 6 tumors/group) derived from parental or resistant cells and dissected at day 16 after implantation. Scale bar, 5 mm. C and D, the curves of tumor growth (C) and the graphs of tumor weight (D) from dissected nude mice injected with parental or resistant cells (n = 6 tumors/group). E, representative images of immunohistochemistry (anti-Ki-67) or H&E-stained tumor sections from nude mice injected with resistant or parental cells (magnification, ×200). F, representative images of lung and H&E-stained lung section for boxed lesion from nude mice injected with parental or resistant cells (magnification, ×200). G and H, the quantification of lesion area (G) and tumor nodules (H) from lung (n = 3 mice/group). Horizontal bars indicate the medians. *, p < 0.05.
Sp1 Links Aberrant DNMT1 Expression to the Hyperactive KIT Kinase Signaling in Lung Cancer Cells
Our previous demonstrations, which showed that Sp1 functions as a transcriptional activator controlling the expression of both DNMT1 and KIT (33, 41), give us a clue that DNA methylation program and KIT signaling could be cooperative in the development of drug resistance. For this, we first answered the question of whether DNMT1 and KIT pathways have regulatory interactions in lung cancer cells. To investigate the role of DNMT1 in KIT signaling, we employed a gain and loss of function strategy to specifically manipulate DNMT1 expression in H1975 and A549. Western blot revealed that siRNA-triggered DNMT1 depletion resulted in down-regulation of Sp1 and KIT, leading to the reduction of KIT autophosphorylation and the dephosphorylation of STAT3 and STAT5, the best defined KIT downstream mediators (Fig. 3A), indicative of inactivated KIT kinase signaling; in contrast, DNMT1 overexpression resulted in the up-regulation of Sp1 and KIT followed by an increase of KIT autophosphorylation and subsequent STAT3 and STAT5 hyperphosphorylation (Fig. 3B), suggesting an activated KIT kinase signaling. To address whether KIT activity modulates DNA methylation machinery, we specifically monitored KIT activity in H1975 and A549 cells. As expected, specific KIT knockdown led to a decrease of Sp1 and DNMT1 expression followed by DNA demethylation (Fig. 3C); conversely, enforced KIT expression caused the up-regulation of DNMT1 and Sp1, leading to an increase of DNA methylation (Fig. 3D). These findings suggest that DNMT1 and KIT pathways have a reciprocal regulation in lung cancer cells.
FIGURE 3.
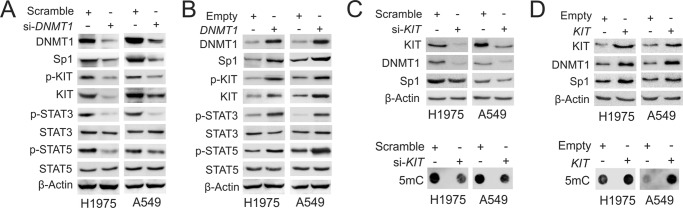
KIT and DNMT1 have reciprocal regulation in lung cancer cells. A, H1975 and A549 cells were transfected with DNMT1 siRNA or scramble for 48 h, and the whole cell lysates were subjected to Western blot. B, H1975 and A549 cells were transfected with DNMT1 expression or empty vectors for 48 h, and the whole cell lysates were subjected to Western blot. C and D, H1975 and A549 cells were transfected with KIT siRNA (C), expression vectors (D), or the corresponding controls for 48 h. The whole cell lysates were subjected to Western blot (upper panels), and the genomic DNA was used for dot-blot (lower panels).
Genomic DNA Becomes Rehypermethylated in Cells Resistant to Decitabine
To elucidate the mechanisms behind the faster growth of decitabineR, we initially assessed the change of DNMT1 protein levels, because decitabine is a typical DNMT1 inhibitor. Unexpectedly, in contrast to the effects of short term exposure, including the fact that decitabine abolishes DNMT1 protein expression (42), chronic treatment with decitabine led to a DNMT1 up-regulation at both protein and RNA levels in decitabineR compared with parental cells (Fig. 4A), which was further confirmed in tumors (4.8-fold increase) from decitabineR cells (Fig. 4B). To address whether DNA methylation levels are changed accordingly, the genomic DNA was subjected to dot-blot using anti-5-methylcytosine that specifically recognizes the methylated DNA at the fifth position of cytosine (10). Consistent with DNMT1 up-regulation in decitabineR, the representative image uncovered a significant increase of DNA methylation, which was verified by densitometric quantification of dot intensities showing that global DNA methylation was increased by 4.5 ± 0.4-fold in decitabineR (Fig. 4C). Equal DNA loading was verified by staining the DNA-spotted membrane with 0.2% methylene blue (not shown). The increased levels of DNMT1 and DNA methylation were confirmed in A549 decitabineR (Fig. 4D). Importantly, ∼6.2-fold increase of global DNA methylation was observed in tumors derived from decitabineR (Fig. 4E). Because CDH1, a tumor suppressor gene, is frequently silenced via promoter DNA hypermethylation and plays a critical role in the oncogenesis and invasiveness of lung cancer cells (38, 43), we reasoned that the enhanced DNA methylation in decitabineR could cause further CDH1 suppression, thus leading to faster lung cancer cell growth. Indeed, qPCR revealed a 67% reduction of CDH1 mRNA expression, and Western blot disclosed a significant decrease of CDH1 protein expression in decitabineR compared with parental counterparts (Fig. 4F). To delineate the mechanisms behind CDH1 inactivation, we employed bisulfite sequencing (38) to determine DNA methylation status of CDH1 promoter (−251 to +139) in both decitabineR and parental cells. Consistent with an increase of global DNA methylation in resistant cells, the bisulfite sequencing disclosed a remarkable increase of DNA methylation from 2% in parental to 13% in decitabineR cells in the tested region (Fig. 4G), suggesting that long term exposure to decitabine induces further epigenetic CDH1 silencing, in agreement with the contribution of CDH1 loss to the acquired drug resistance (44, 45). Given the positive association between DNMT1 expression and DNA methylation levels, our findings suggest that DNA hypermethylation in decitabineR results from DNMT1 up-regulation, thus providing an explanation for why genomic DNA is remethylated in decitabine-treated patients.
FIGURE 4.

DNA is remethylated in decitabineR. A, decitabineR or parental H1975 cells were recovered in drug-free medium for 96 h and subjected to Western blot (left panel) or qPCR (right panel). B, the measurement of DNMT1 expression by Western blot (left panel) and its quantification (right panel) in tumors from decitabineR or parental H1975 cells (n = 6 tumors/group). C, the genomic DNA was isolated from decitabineR or parental H1975 cells recovered in drug-free medium for 96 h and subjected to dot-blot. The representative images are shown (lower panel), and the graphs (upper panel) indicate the relative densitometric intensities expressed as means of the dot from three independent experiments. D, Western blot (top two panels) and dot-blot (bottom panel) for the whole cell lysates and the genomic DNA from decitabineR or parental A549 cells recovered in drug-free medium for 96 h. E, the genomic DNA was isolated from H1975 decitabineR or parental tumors and subjected to dot-blot. The graphs indicate the relative densitometric intensities expressed as means of the dot. F, qPCR (left) and Western blot (right) to show the change of CDH1 in H1975 decitabineR and parental cells recovered in drug-free medium for 96 h. G, DNA methylation status of CpG sites in the CDHI promoter region (transcription start site −251 to +139) obtained from bisulfite sequencing of H1975 decitabineR or parental cells growing in drug-free medium for 96 h. Upper panel, distribution of CpG dinucleotides is indicated as small vertical lines. The numbers indicate the position of cytosine residues of CpG relative to the transcriptional starting site of CDH1. Lower panels, open circles represent unmethylated cytosines; filled circles represent methylated cytosines. The results of 10 clones are presented. **, p < 0.01.
Hyperactivated Kinase Signaling Appears in Cells Resistant to Decitabine
The Sp1-mediated DNMT1/KIT cross-talk in lung cancer cells hints at their cooperation in the development of decitabine-resistant phenotype. Indeed, Sp1 was remarkably up-regulated at both protein and RNA levels in decitabineR compared with parental cells (Fig. 5A). As a consequence, decitabineR had higher levels of KIT expression and protein phosphorylation than parental cells (Fig. 5B), which was supported by the results from decitabineR tumors displaying higher KIT expression than parental tumors (Fig. 5C). Such KIT up-regulation in decitabineR cells enhanced AKT and STAT3 phosphorylation (Fig. 5D), suggesting the reactivated KIT-AKT/STAT3 cascade, which was independently verified in A549 resistance to decitabine (Fig. 5E). To determine whether other kinases are likely to be changed after long term exposure to decitabine, we carried out co-immunoprecipitation followed by Western blot using anti-4G10. These experiments revealed that more phosphorylated proteins were pulled down in H1975 decitabineR, compared with parental cells (Fig. 5, F and G), suggesting that the activated kinases by long term decitabine treatment may not be restricted to KIT-AKT/STAT3 cascade but extended to the multiple kinase pathways.
FIGURE 5.

Hyperactive kinases appear in decitabineR. A, Western blot (left panel) and PCR (right panel) to determine the levels of Sp1 expression in H1975 decitabineR and parental cells that were maintained in drug-free medium for 96 h. B, Western blot (left panel) and PCR (right panel) to determine the change of KIT signaling in H1975 decitabineR and parental cells growing in drug-free medium for 96 h. C, Western blot (left panel) and its quantification (right panel) to compare KIT expression in H1975 decitabineR and parental tumors (n = 6 tumors/group). D, Western blot for the levels of AKT and STAT3 phosphorylation in H1975 decitabineR and parental cells growing in drug-free medium for 96 h. E, Western blot for the levels of indicated targets in A549 decitabineR and parental cells growing in drug-free medium for 96 h. F and G, the H1975 decitabineR or parental cells were recovered in drug-free medium for 96 h, and the cell lysates were subjected to immunoprecipitation using anti-4G10 followed by probing with anti-4G10. The representative image is shown (F), and the graph is the quantification of densitometric intensities expressed as means of the whole lane from three independent experiments (G). *, p < 0.05; **, p < 0.01.
To provide further insight into the mechanisms underlying decitabine resistance, we performed gene expression microarray in decitabineR and parental cells. Using a 1.5-fold threshold, in total 8416 genes were found to be differentially expressed in resistant compared with parental cells. Approximately 3957 up-regulated (supplemental Table S1) and 4459 down-regulated (supplemental Table S2) genes were observed. To better understand which biological functions are affected by chronic decitabine treatment, we conducted functional annotations using DAVID bioinformatics resources 6.7. The pathways related to both DNA methylation, such as maintenance of DNA methylation, S-adenosyl methionine-dependent methyltransferase activity, and methyltransferase activity, etc., and kinase signaling, for instance, ATPase activity, ATP binding, phosphatase activity, and phosphoprotein, etc., appear on the top lists of activated networks. The genes involved in these pathways are listed in supplemental Table S3. These findings further support that the activated kinases and DNA methylation program in decitabineR are universal.
DNA Hypermethylation Arises in Resistance to PKC412
To investigate whether DNMT1 and KIT oncogenic pathways have been modulated during continuous PKC412 exposure, we grew H1975 and A549 PKC412R in drug-free medium for 96 h, and the cell lysates were subjected to Western blot. Although short term treatment of PKC412 led to the dephosphorylation of KIT and its downstream effectors in vitro and in vivo (46, 47), PKC412R was strongly associated with hyperphosphorylation of KIT, AKT, and STAT3 compared with parental cells in H1975 and A549 (Fig. 6A), suggesting that KIT signaling becomes reactivated in PKC412R. Notably, KIT expression was highly up-regulated in PKC412R tumors (Fig. 6B), indicative of the enhanced KIT kinase activity in resistant-derived tumors. Co-immunoprecipitation showed that more phosphorylated proteins were precipitated by anti-4G10 in PKC412R than in parental cells (Fig. 6C), similar to decitabineR, suggesting that the acquisition of the PKC412-resistant phenotype involves multiple kinase signaling cascades.
FIGURE 6.

The kinase signaling is reactivated and the DNA methylation is increased in PKC412R. A, H1975 or A549 PKC412R and parental cells were recovered in drug-free medium for 96 h, and the cell lysates were applied to Western blot. B, Western blot (left panel) and its quantification (right panel) to compare KIT expression in H1975 PKC412R and parental tumors (n = 6 tumors/group). C, the H1975 PKC412R and parental cells were recovered in drug-free medium for 96 h, and the cell lysates were subjected to immunoprecipitation using anti-4G10 followed by probing with anti-4G10. The representative image is shown (left panel), and the graph (right panel) indicates the densitometric intensities expressed as means of the whole lane from three independent experiments. D, Western blot (H1975 and A549, left panel) and qPCR (H1975, right panel) to determine the alterations of DNMT1 in PKC412R and parental cells growing in drug-free medium for 96 h. E, Western blot (left panel) and quantification (right panel) to compare DNMT1 expression in PKC412R and parental tumors (n = 6 tumors/group). F, the PKC412R and parental cells were grown in PKC412-free medium for 96 h, and the genomic DNA was subjected to dot-blot. Left panel, representative image (H1975 and A549). Right panel, the graph shows the relative densitometric intensities expressed as means of the dot from three independent experiments (H1975). G, the quantification of dot-blot to compare the 5-methylcytosine (5mC) level in H1975 PKC412R and parental tumors (n = 6 tumors/group). H, qPCR (left panel) and Western blot (right panel) for CDH1 expression in H1975 PKC412R and parental cells growing in drug-free medium for 96 h. I, bisulfite analysis for the change of DNA methylation in CDH1 promoter (transcription start site −251 to +139) in H1975 PKC412R or parental cells growing in drug-free medium for 96 h. Open circles indicate unmethylated CpG sites; filled circles indicate methylated CpG sites. The results of 10 clones are presented. *, p < 0.05; **, p < 0.01.
To investigate whether aberrant DNA methylation contributes to PKC412 resistance, we initially measured DNMT1 expression in PKC412R or parental cells in vitro and in vivo. Strikingly, these experiments identified a significant increase of DNMT1 expression in PKC412R H1975, A549 cells and tumors compared with parental counterparts (Fig. 6, D and E). Because DNMT1 gene abundance positively associates with DNA methylation levels (48), DNMT1 up-regulation in PKC412R raised the possibility that DNA methylation could be modulated under chronic PKC412 incubation. Thus, the genomic DNA was extracted from PKC412R or parental cells, and the dot-blot was undertaken to quantify the changes of DNA methylation. Indeed, a significant enhancement of global DNA methylation was evident in PKC412R cells and tumors (Fig. 6, F and G). Importantly, both mRNA and protein of CDH1 levels were significantly decreased in PKC412R compared with parental cells (Fig. 6H). Bisulfite sequencing uncovered that the selected region of CDH1 promoter displayed low levels of DNA methylation (3%) in parental cells, but the methylation was increased to 11% in PKC412R (Fig. 6I). Finally, to obtain a deeper understanding of PKC412 resistance, we carried out gene expression profiling in PKC412R and parental cells to define the differentially expressed genes. If a 1.5-fold change is the threshold, we identified 2213 up-regulated (supplemental Table S4) and 2309 down-regulated (supplemental Table S5) genes in PKC412R compared with parental cells. In line with the results from decitabineR, the pathway analysis by DAVID bioinformatics resources 6.7 discovered the reactivated DNA methylation-associated and kinase-relevant networks as the top candidate signaling. The molecules involved in these pathways are listed in supplemental Table S6, which required further validation for their clinical implications. Based on these results, we make a conclusion that long term treatment with PKC412 triggers global activation of both protein kinases and DNA methyltransferases.
Increased Activation of DNMT1 Plays a Role in Sustaining the High Proliferative Potential of PKC412R Cells
The observation that DNA hypermethylation appears in PKC412R motivated us to hypothesize that PKC412R could be eliminated by DNA hypomethylating agents. To confirm the role of DNA hypermethylation in the increased proliferative rate of PKC412R, we exposed PKC412R to 0.5 μm decitabine, which was a dose that blocked the growth of parental cell, but failed to suppress decitabineR (Fig. 1), for 48 h. As expected, the wound healing ability in PKC412R was significantly impaired by decitabine (Fig. 7, A and B), indicating that PKC412R are decitabine-sensitive. To delineate the mechanisms behind this, we performed Western blot to assess the changes of KIT signaling and DNMT1. Consistent with the previous report (42), such short term treatment with decitabine significantly decreased DNMT1 expression with a concurrent suppression of KIT signaling (Fig. 7C). Further, the abrogation of DNMT1 activity by decitabine led to a significant reduction of global DNA methylation (Fig. 7D). In addition, the results, which PKC412R displayed higher levels of DNMT1 expression than parental cells, suggested that the more aggressive growth of PKC412 may rely on abnormal DNMT1 expression. For this, we transfected PKC412R with DNMT1 siRNA and initially confirmed that DNMT1 expression was decreased and DNA methylation was reduced, compared with the scramble group (Fig. 7E). In addition to KIT, the siRNA-triggered DNMT1 depletion resulted in a significant dephosphorylation of other tyrosine kinases, like FLT3 and ABL (Fig. 7F), and subsequent suppression of wound healing capability (Fig. 7, G and H). Importantly, in line with the inhibitory effects of decitabine, DNMT1 knockdown sensitized PKC412R cells to the treatment of 0.5 μm PKC412 (Fig. 7, I and J), a dose that had no obvious inhibition on PKC412R cell mobility (Fig. 1). Because Sp1 positively regulates KIT expression and is up-regulated in PKC412R (Fig. 7K), Sp1 could play an important role in PKC412R growth. Indeed, siRNA-triggered Sp1 depletion and subsequent down-regulation of KIT and DNMT1 (Fig. 7L) significantly blocked PKC412R cell migration (Fig. 7M). Thus, Sp1 could serve as a therapeutic target in overcoming PKC412R. These results suggest that the growth and migration of PKC412R are dependent on the Sp1-DNMT1 axis.
FIGURE 7.

The inhibition of DNA methylation program eradicates H1975 PKC412R. A and B, wound healing assays in PKC412R cells treated with 0.5 μm decitabine. Quantitative measurement of cell migration was performed at 24 h post-treatment. A, representative image of wound healing. B, graph is the quantification of gap closure. C, PKC412R cells were treated with 0.5 μm decitabine for 48 h, and the cells were lysed for Western blot. D, the genomic DNA was prepared from the treated cells in C and subjected to dot-blot. The graphs show the quantification of dot intensity. E, PKC412R cells were transfected with DNMT1 siRNA or scramble for 48 h, and the protein lysates were subjected to Western blot (top two panels) and the genomic DNA for dot-blot (bottom panel). F, PKC412R cells were transfected with DNMT1 siRNA or scramble for 48 h. The immunoprecipitation was performed using anti-4G10 followed by probing with anti-p-FLT3 or anti-p-ABL. The equal protein used in immunoprecipitation was indicated by β-actin bands. G and H, wound healing assays in PKC412R cells transfected with DNMT1 siRNA. Quantitative measurement of cell migration was performed at 24 h post-transfection. G, representative image of wound healing. H, graph is the quantification of gap closure. I and J, wound healing assays in PKC412R cells transfected with DNMT1 siRNA for 24 h followed by treatment with 0.5 μm PKC412 for additional 24 h. Quantitative measurement of cell migration was performed at 48 h post-treatment. I, representative image of wound healing. J, graph is the quantification of gap closure. K, Western blot for Sp1 expression in PKC412R growing in drug-free medium for 96 h. L and M, PKC412R were transfected with Sp1 siRNA and subjected to Western blot (L) and wound healing assays (M). Quantitative measurement of cell migration was performed at 24 h post-transfection. The data are represented from three independent experiments. *, p < 0.05; **, p < 0.01. si, siRNA.
Abrogation of KIT Activity Circumvents Decitabine Resistance
Because of the reactivation of KIT kinase signaling in decitabineR and given the role of KIT gene abundance in determining its kinase activity (33), we speculated that genetic or pharmacological disruption of KIT function could be an alternative strategy to impair decitabineR cell expansion. To this end, decitabineR were transfected with KIT siRNA or scramble for 48 h. As shown in Fig. 8A, KIT expression and phosphorylation were significantly decreased in siRNA-transfected cells, leading to a reduction of AKT phosphorylation with a concurrent blockage of decitabineR cell migration (Fig. 8, B and C), suggesting that the faster growth of decitabineR relies on the activated KIT signaling. In agreement with this, exposure of decitabineR to 0.5 μm PKC412, a dose that is sufficient to impair the growth of parental cells with no obvious effects on PKC412R proliferation (Fig. 1), greatly suppressed decitabineR cell wound healing ability (Fig. 8, D and E), which might take place via the inhibition of KIT activity and DNA methylation, because PKC412 treatment in decitabineR resulted in KIT and DNMT1 down-regulation and KIT and AKT dephosphorylation (Fig. 8F). Of note, increasing evidence indicates that DNMT1 has both de novo, which does not require cell cycle and cell division, and maintenance enzyme activity. Thus, the reduction of DNMT1-dependent DNA methylation by PCK412 did not involve cell cycle and DNA replication but was accompanied by the activated caspase-3 (Fig. 8G), in agreement with our previous findings that DNMT1 down-regulation by inhibitors or microRNAs causes a strong DNA hypomethylation followed by cell apoptosis and differentiation (41, 49). Further, PKC412 treatment decreased the expression of DNMT3a and DNMT3b (Fig. 8G), providing an explanation for why the change of DNA methylation is greater than change of DNMT1 expression (data not shown). In addition, KIT depletion sensitized decitabineR cells to 0.5 μm decitabine (Fig. 8, H and I), indicating that KIT dysfunction could synergize with decitabine therapy. Finally, given the positive regulatory role of Sp1 in DNMT1 expression and Sp1 overexpression in decitabineR, we tested the effects of Sp1 knockdown on the expansion of decitabineR. As expected, Sp1 down-regulation resulted in suppression of DNMT1 and KIT (Fig. 8J), leading to a significant reduction of decitabineR cell wound healing capability (Fig. 8K) and supporting the promise of targeting Sp1 for eradicating decitabine resistance. Collectively, these findings provide a sound rationale in clinical trials for using kinase inhibitors, as single agent or in combination forms, to eliminate resistance to DNA hypomethylating agents or vice versa.
FIGURE 8.

The abrogation of KIT kinase activity overcomes H1975 decitabineR. A, the decitabineR were transfected with KIT siRNA or scramble for 48 h, and the protein lysates were subjected to Western blot. B and C, wound healing assays in decitabineR transfected with KIT siRNA or scramble. B, representative image of wound healing. C, graph is the quantification of gap closure. D and E, wound healing assays in decitabineR treated with 0.5 μm PKC412. D, representative image of wound healing. E, graph is the quantification of gap closure. F and G, decitabineR were treated with 0.5 μm PKC412 for 48 h, and the cells were lysed for Western blot. H and I, wound healing assays in decitabineR transfected with KIT siRNA for 24 h followed by exposure to 0.5 μm decitabine for another 24 h. Quantitative measurement of cell migration was carried out at 48 h post-treatment. H, representative image of wound healing. I, graph is the quantification of gap closure. J and K, decitabineR were transfected with Sp1 siRNA and subjected to Western blot (J) and wound healing assays (K). Quantitative measurement of cell migration was performed at 24 h post-treatment. The data are represented from three independent experiments. *, p < 0.05; **, p < 0.01. si, siRNA.
Discussion
Because aberrant DNA methylation (41) and abnormal KIT function critically contribute to lung tumorigenesis (23–25, 50), as independent practice, KIT and DNMT1 have been extensively used for therapeutic targets, and their inhibitors have been tested in various pre- and clinical models. However, resistance of tumor cells to PKC412 or decitabine poses huge limitations to their use in treatment. To understand the molecular rules behind resistant phenotypes, we generated resistant cells to PKC412 or decitabine and examined their growth characteristics and the oncogenic pathways that may be involved in. We showed that both decitabineR and PKC412R displayed higher proliferative rate in vitro and possessed stronger tumorigenic potential in vivo. We proved that KIT kinase signaling was reactivated, the global and gene-specific DNA was remethylated, and CDH1 became further epigenetically silenced, importantly, in resistance not only to decitabine, but also to PKC412. Thus, decitabine was shown to overcome resistance to PKC412 that could reciprocally eliminate decitabine resistance. Theoretically, our findings unravel a cross-talk between KIT and DNMT1 pathways in controlling lung cancer cell fate under the selective pressure from PKC412 or decitabine, thus shedding a light on the molecular biology of drug resistance; practicably, our studies provide a rationale in clinical trials for overriding decitabineR by kinase inactivation or vice versa.
PKC412 is a staurosporine-derived tyrosine kinase inhibitor and exerts anti-proliferative activity in different tumor-derived cell types (51), including NSCLC, as a single or combination form (52–54). It is widely accepted that the mechanisms underlying PKC412-triggered cell death or apoptosis after short term exposure involve the dephosphorylation of STAT, AKT, and ERK that are the KIT downstream effectors, thereby resulting in the disruption of such aberrant kinase signaling that favors cell survival (47, 55), leading to cell apoptosis and the blockage of cell growth. However, through the characterization of H1975 and A549 resistant cells, we experimentally demonstrated that PKC412R had higher tumorigenic potential than parental cells, in line with the previous finding that resistant cells had stronger growing rate in vitro and in vivo (56). Although the molecular rules underlying drug resistance are thought to involve the acquired mutations on kinase domains or up-regulation of anti-apoptotic genes (57, 58), our results argue that PKC412R had increased KIT expression and the activated downstream signaling, supporting the previous notion that resistance to treatment of NSCLC is often associated with an aberrantly activated protein kinase B/AKT and protein kinase C (59, 60). Importantly, we present evidence for the first time that prolonged exposure to PKC412 resulted in an up-regulation of DNMT1, an increase of global DNA methylation, and a further silencing of CDH1 through the augmented promoter methylation. Further, the proliferation of PKC412R depends on DNMT1 overexpression, because DNMT1 knockdown impaired PKC412R cell expansion and sensitized PKC412R cells to PKC412 treatment. Given that decitabine is able to suppress PKC412R growth, these results demonstrated the contribution of DNA methylation machinery to the development of PKC412 resistant phenotypes. Thus, decitabine or other DNA hypomethylating agents could be beneficial to lung cancer patient subgroups with unresponsiveness/relapse from PKC412 therapy.
Decitabine is a conventional DNMT1 inhibitor and has been tested in many types of cancers, including myelodysplastic syndrome and lung cancer (17, 61), but the clinical outcomes are disappointing in general, because many patients do not respond to decitabine initially, and most patients who initially respond to decitabine eventually relapse, even with continuous decitabine administration. It is postulated that the causes of decitabine resistance likely involve metabolic pathways (62, 63), including deoxycytidine kinase deficiency or increased deamination by cytidine deaminase. However, we found that in contrast to the short term decitabine treatment that induces DNA demethylation (42), decitabineR had an increase of global DNA methylation, a further down-regulation of CDH1 expression, and an enhancement of CDH1 promoter methylation, in line with previous observations that DNA remethylation was seen in DNMT inhibitor-treated patients at the end of treatment cycle (64). Further, DNMT1 gene was significantly up-regulated in decitabineR cells and tumors compared with parental counterparts, thus providing a reason for why DNA becomes remethylated after decitabine administration. Unexpectedly, our results show that chronic exposure to decitabine-activated protein kinase signaling as indicated by the increased KIT expression and autophosphorylation followed by the enhanced phosphorylation of AKT and STAT3. Further, more phosphorylated kinases were pulled down by anti-4G10 in decitabineR, indicating that the activated kinases are universal. Such hyperactive kinase pathways may contribute to the faster growth of decitabineR cells in vitro and in mice, because KIT dysfunction by siRNA-triggered gene down-regulation or treatment with PKC412 blocked decitabineR proliferation. Also KIT depletion sensitized decitabineR to decitabine. These results document the crucial role of the reactivated kinases in the development of decitabine resistance and suggest that kinase inhibitors could cure lung cancer patients characterized by aberrant epigenetics or unresponsive to decitabine therapy.
Although the critical roles of DNA hypermethylation or hyperactive tyrosine kinase signaling in lung cancer pathogenesis have been well documented, it remains unclear whether these two oncogenic pathways are cooperative in lung cancer. We present evidence that KIT and DNMT1 have regulatory interactions and are significantly up-regulated in both decitabineR and PKC412R. Importantly, these changed pathways are not restricted to KIT and DNMT1 but can be extended to global kinase and DNA methylation networks, as indicated by our gene expression profiling, which simultaneously change the expression of numerous genes that are involved in cancer cell survival and proliferation. Mechanistically, the resistant-associated Sp1 overexpression is essential for the up-regulation of DNMT1 and KIT in PKC412R and decitabineR cells, given that Sp1 is a transactivator of both KIT and DNMT1. Moreover, Sp1 knockdown decreased the expression of KIT and DNMT1 in decitabineR and PKC412R. Although the roles of Sp1 in the development of decitabineR and PKC412R remain to be validated, Sp1 could critically determine lung cancer cell fate under PKC412 and decitabine selection, because Sp1 knockdown significantly impaired the migration of decitabineR and PKC412R, thus supporting Sp1 dysfunction as an alternative strategy to override both decitabineR and PKC412R cells. Notably, the outcomes of long and short term treatment with decitabine and PKC412 are contrasting, including the change of molecular targets (DNMT1 and KIT) and the growth of treated cancer cells. Clearly, short term treatment led to the dysfunction of DNA methylation machinery and kinase signaling followed by the blockage of cell growth and induction of cell apoptosis. In the long term exposure, the inhibited DNA methylation and tyrosine kinases in long- and short-term treatment become simultaneously reactivated, because of cross-talk between these two pathways, in a subpopulation of heterogeneous lung cancer cells that survive and even proliferate faster. These findings provide a rationale for applying kinase inhibitors to decitabine-relapsed patients at the early stage of treatment. Finally, both resistant lines displayed DNMT1 up-regulation, but with different sensitivity to decitabine, although the survival and proliferation of PKC412R were significantly blocked by physiological relevant decitabine, which did not obviously affect the growth of decitabineR. This phenomenon could result from higher levels of DNMT1 expression in decitabineR than in PKC412R, as demonstrated by showing that the expansion of decitabineR could be overcome by higher doses of decitabine, collectively, further appreciating DNMT1 up-regulation in decitabineR survival and proliferation.
In conclusion, we found that cells resistant to decitabine and PKC412 had a higher proliferative rate in vitro and in the xenograft model. We demonstrated that the stronger tumorigenicity of decitabineR and PKC412R strongly associates with the simultaneous reactivation of both kinase signaling and DNA methylation machinery. We proved that cross-application of decitabine or PKC412 could circumvent the resistance to their targeted therapies. These findings identify the communication between epigenetics and kinases in the development of drug resistance, thereby providing a fundamental strategy in overcoming resistance to kinase inhibitors by DNA hypomethylating agents or vice versa.
Author Contributions
S. L. designed the research; F. Y., N. S., and J. P. performed the research; F. Y., J. R. M., P. Y., and S. L. analyzed the data and wrote the paper; F. Y. carried out the biostatistical analysis; and S. L. conceived the idea and supervised the whole project. All authors discussed the results and commented on the manuscript.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants R01CA149623 and R21CA155915 (to S. L.). This work was also supported in part by funds from the Hormel Foundation (to S. L.). The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S6.
- NSCLC
- non-small cell lung cancer
- decitabineR
- decitabine-resistant cell
- PKC412R
- PKC412-resistant cell
- RTK
- receptor tyrosine kinase
- DNMT
- DNA methyltransferase
- H&E
- hematoxylin and eosin
- IHC
- immunohistochemistry
- qPCR
- quantitative PCR.
References
- 1. Fabbri M., Garzon R., Cimmino A., Liu Z., Zanesi N., Callegari E., Liu S., Alder H., Costinean S., Fernandez-Cymering C., Volinia S., Guler G., Morrison C. D., Chan K. K., Marcucci G., Calin G. A., Huebner K., Croce C. M. (2007) MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. U.S.A. 104, 15805–15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duy C., Hurtz C., Shojaee S., Cerchietti L., Geng H., Swaminathan S., Klemm L., Kweon S. M., Nahar R., Braig M., Park E., Kim Y. M., Hofmann W. K., Herzog S., Jumaa H., Koeffler H. P., Yu J. J., Heisterkamp N., Graeber T. G., Wu H., Ye B. H., Melnick A., Müschen M. (2011) BCL6 enables Ph+ acute lymphoblastic leukaemia cells to survive BCR-ABL1 kinase inhibition. Nature 473, 384–388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dufies M., Jacquel A., Belhacene N., Robert G., Cluzeau T., Luciano F., Cassuto J. P., Raynaud S., Auberger P. (2011) Mechanisms of AXL overexpression and function in Imatinib-resistant chronic myeloid leukemia cells. Oncotarget 2, 874–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Easwaran H., Tsai H. C., Baylin S. B. (2014) Cancer epigenetics: tumor heterogeneity, plasticity of stem-like states, and drug resistance. Mol. Cell 54, 716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Iliopoulos D., Guler G., Han S. Y., Johnston D., Druck T., McCorkell K. A., Palazzo J., McCue P. A., Baffa R., Huebner K. (2005) Fragile genes as biomarkers: epigenetic control of WWOX and FHIT in lung, breast and bladder cancer. Oncogene 24, 1625–1633 [DOI] [PubMed] [Google Scholar]
- 6. Kurakawa E., Shimamoto T., Utsumi K., Hirano T., Kato H., Ohyashiki K. (2001) Hypermethylation of p16(INK4a) and p15(INK4b) genes in non-small cell lung cancer. Int. J. Oncol. 19, 277–281 [PubMed] [Google Scholar]
- 7. Fabbri M., Iliopoulos D., Trapasso F., Aqeilan R. I., Cimmino A., Zanesi N., Yendamuri S., Han S. Y., Amadori D., Huebner K., Croce C. M. (2005) WWOX gene restoration prevents lung cancer growth in vitro and in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 15611–15616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin R. K., Hsu H. S., Chang J. W., Chen C. Y., Chen J. T., Wang Y. C. (2007) Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer 55, 205–213 [DOI] [PubMed] [Google Scholar]
- 9. Kim H., Kwon Y. M., Kim J. S., Han J., Shim Y. M., Park J., Kim D. H. (2006) Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer 107, 1042–1049 [DOI] [PubMed] [Google Scholar]
- 10. Shen N., Yan F., Pang J., Wu L. C., Al-Kali A., Litzow M. R., Liu S. (2014) A nucleolin-DNMT1 regulatory axis in acute myeloid leukemogenesis. Oncotarget 5, 5494–5509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Saito Y., Kanai Y., Nakagawa T., Sakamoto M., Saito H., Ishii H., Hirohashi S. (2003) Increased protein expression of DNA methyltransferase (DNMT) 1 is significantly correlated with the malignant potential and poor prognosis of human hepatocellular carcinomas. Int. J. Cancer 105, 527–532 [DOI] [PubMed] [Google Scholar]
- 12. Girault I., Tozlu S., Lidereau R., Bièche I. (2003) Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 9, 4415–4422 [PubMed] [Google Scholar]
- 13. Ding W. J., Fang J. Y., Chen X. Y., Peng Y. S. (2008) The expression and clinical significance of DNA methyltransferase proteins in human gastric cancer. Dig. Dis. Sci. 53, 2083–2089 [DOI] [PubMed] [Google Scholar]
- 14. Etoh T., Kanai Y., Ushijima S., Nakagawa T., Nakanishi Y., Sasako M., Kitano S., Hirohashi S. (2004) Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am. J. Pathol. 164, 689–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Alazzouzi H., Davalos V., Kokko A., Domingo E., Woerner S. M., Wilson A. J., Konrad L., Laiho P., Espín E., Armengol M., Imai K., Yamamoto H., Mariadason J. M., Gebert J. F., Aaltonen L. A., Schwartz S., Jr., Arango D. (2005) Mechanisms of inactivation of the receptor tyrosine kinase EPHB2 in colorectal tumors. Cancer Res. 65, 10170–10173 [DOI] [PubMed] [Google Scholar]
- 16. Blum W., Garzon R., Klisovic R. B., Schwind S., Walker A., Geyer S., Liu S., Havelange V., Becker H., Schaaf L., Mickle J., Devine H., Kefauver C., Devine S. M., Chan K. K., Heerema N. A., Bloomfield C. D., Grever M. R., Byrd J. C., Villalona-Calero M., Croce C. M., Marcucci G. (2010) Clinical response and miR-29b predictive significance in older AML patients treated with a 10-day schedule of decitabine. Proc. Natl. Acad. Sci. U.S.A. 107, 7473–7478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Momparler R. L. (2013) Epigenetic therapy of non-small cell lung cancer using decitabine (5-aza-2′-deoxycytidine). Front. Oncol. 3, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jüttermann R., Li E., Jaenisch R. (1994) Toxicity of 5-aza-2′-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc. Natl. Acad. Sci. U.S.A. 91, 11797–11801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Santi D. V., Norment A., Garrett C. E. (1984) Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl. Acad. Sci. U.S.A. 81, 6993–6997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Müller C. I., Rüter B., Koeffler H. P., Lübbert M. (2006) DNA hypermethylation of myeloid cells, a novel therapeutic target in MDS and AML. Curr. Pharm. Biotechnol. 7, 315–321 [DOI] [PubMed] [Google Scholar]
- 21. Wang R., Kobayashi R., Bishop J. M. (1996) Cellular adherence elicits ligand-independent activation of the Met cell-surface receptor. Proc. Natl. Acad. Sci. U.S.A. 93, 8425–8430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giordano S., Ponzetto C., Di Renzo M. F., Cooper C. S., Comoglio P. M. (1989) Tyrosine kinase receptor indistinguishable from the c-met protein. Nature 339, 155–156 [DOI] [PubMed] [Google Scholar]
- 23. Jafri N. F., Ma P. C., Maulik G., Salgia R. (2003) Mechanisms of metastasis as related to receptor tyrosine kinases in small-cell lung cancer. J. Environ. Pathol. Toxicol. Oncol. 22, 147–165 [DOI] [PubMed] [Google Scholar]
- 24. Tsuura Y., Hiraki H., Watanabe K., Igarashi S., Shimamura K., Fukuda T., Suzuki T., Seito T. (1994) Preferential localization of c-kit product in tissue mast cells, basal cells of skin, epithelial cells of breast, small cell lung carcinoma and seminoma/dysgerminoma in human: immunohistochemical study on formalin-fixed, paraffin-embedded tissues. Virchows Arch. 424, 135–141 [DOI] [PubMed] [Google Scholar]
- 25. Boldrini L., Ursino S., Gisfredi S., Faviana P., Donati V., Camacci T., Lucchi M., Mussi A., Basolo F., Pingitore R., Fontanini G. (2004) Expression and mutational status of c-kit in small-cell lung cancer: prognostic relevance. Clin. Cancer Res. 10, 4101–4108 [DOI] [PubMed] [Google Scholar]
- 26. Carlomagno F., Santoro M. (2005) Receptor tyrosine kinases as targets for anticancer therapeutics. Curr. Med. Chem. 12, 1773–1781 [DOI] [PubMed] [Google Scholar]
- 27. Rossi A., Maione P., Colantuoni G., Ferrara C., Rossi E., Guerriero C., Nicolella D., Falanga M., Palazzolo G., Gridelli C. (2009) Recent developments of targeted therapies in the treatment of non-small cell lung cancer. Curr. Drug Discov. Technol. 6, 91–102 [DOI] [PubMed] [Google Scholar]
- 28. Ansari J., Palmer D. H., Rea D. W., Hussain S. A. (2009) Role of tyrosine kinase inhibitors in lung cancer. Anticancer Agents Med. Chem 9, 569–575 [DOI] [PubMed] [Google Scholar]
- 29. Monnerat C., Henriksson R., Le Chevalier T., Novello S., Berthaud P., Faivre S., Raymond E. (2004) Phase I study of PKC412 (N-benzoyl-staurosporine), a novel oral protein kinase C inhibitor, combined with gemcitabine and cisplatin in patients with non-small-cell lung cancer. Ann. Oncol. 15, 316–323 [DOI] [PubMed] [Google Scholar]
- 30. Fischer B., Marinov M., Arcaro A. (2007) Targeting receptor tyrosine kinase signalling in small cell lung cancer (SCLC): what have we learned so far? Cancer Treat. Rev. 33, 391–406 [DOI] [PubMed] [Google Scholar]
- 31. Fabbro D., Ruetz S., Bodis S., Pruschy M., Csermak K., Man A., Campochiaro P., Wood J., O'Reilly T., Meyer T. (2000) PKC412–a protein kinase inhibitor with a broad therapeutic potential. Anticancer Drug. Des. 15, 17–28 [PubMed] [Google Scholar]
- 32. Heinrich M. C., Blanke C. D., Druker B. J., Corless C. L. (2002) Inhibition of KIT tyrosine kinase activity: a novel molecular approach to the treatment of KIT-positive malignancies. J. Clin. Oncol. 20, 1692–1703 [DOI] [PubMed] [Google Scholar]
- 33. Liu S., Wu L. C., Pang J., Santhanam R., Schwind S., Wu Y. Z., Hickey C. J., Yu J., Becker H., Maharry K., Radmacher M. D., Li C., Whitman S. P., Mishra A., Stauffer N., Eiring A. M., Briesewitz R., Baiocchi R. A., Chan K. K., Paschka P., Caligiuri M. A., Byrd J. C., Croce C. M., Bloomfield C. D., Perrotti D., Garzon R., Marcucci G. (2010) Sp1/NFκB/HDAC/miR-29b regulatory network in KIT-driven myeloid leukemia. Cancer Cell 17, 333–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sierra J. R., Cepero V., Giordano S. (2010) Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol. Cancer 9, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lockwood W. W., Coe B. P., Williams A. C., MacAulay C., Lam W. L. (2007) Whole genome tiling path array CGH analysis of segmental copy number alterations in cervical cancer cell lines. Int. J. Cancer 120, 436–443 [DOI] [PubMed] [Google Scholar]
- 36. Mishra M. V., Bisht K. S., Sun L., Muldoon-Jacobs K., Awwad R., Kaushal A., Nguyen P., Huang L., Pennington J. D., Markovina S., Bradbury C. M., Gius D. (2008) DNMT1 as a molecular target in a multimodality-resistant phenotype in tumor cells. Mol. Cancer Res. 6, 243–249 [DOI] [PubMed] [Google Scholar]
- 37. Jelinek J., Gharibyan V., Estecio M. R., Kondo K., He R., Chung W., Lu Y., Zhang N., Liang S., Kantarjian H. M., Cortes J. E., Issa J. P. (2011) Aberrant DNA methylation is associated with disease progression, resistance to imatinib and shortened survival in chronic myelogenous leukemia. PLoS One 6, e22110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yan F., Shen N., Pang J., Xie D., Deng B., Molina J. R., Yang P., Liu S. (2014) Restoration of miR-101 suppresses lung tumorigenesis through inhibition of DNMT3a-dependent DNA methylation. Cell Death Dis. 5, e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Propper D. J., McDonald A. C., Man A., Thavasu P., Balkwill F., Braybrooke J. P., Caponigro F., Graf P., Dutreix C., Blackie R., Kaye S. B., Ganesan T. S., Talbot D. C., Harris A. L., Twelves C. (2001) Phase I and pharmacokinetic study of PKC412, an inhibitor of protein kinase C. J. Clin. Oncol. 19, 1485–1492 [DOI] [PubMed] [Google Scholar]
- 40. Daskalakis M., Nguyen T. T., Nguyen C., Guldberg P., Köhler G., Wijermans P., Jones P. A., Lübbert M. (2002) Demethylation of a hypermethylated P15/INK4B gene in patients with myelodysplastic syndrome by 5-Aza-2′-deoxycytidine (decitabine) treatment. Blood 100, 2957–2964 [DOI] [PubMed] [Google Scholar]
- 41. Liu S., Liu Z., Xie Z., Pang J., Yu J., Lehmann E., Huynh L., Vukosavljevic T., Takeki M., Klisovic R. B., Baiocchi R. A., Blum W., Porcu P., Garzon R., Byrd J. C., Perrotti D., Caligiuri M. A., Chan K. K., Wu L. C., Marcucci G. (2008) Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-κB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 111, 2364–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu Z., Liu S., Xie Z., Blum W., Perrotti D., Paschka P., Klisovic R., Byrd J., Chan K. K., Marcucci G. (2007) Characterization of in vitro and in vivo hypomethylating effects of decitabine in acute myeloid leukemia by a rapid, specific and sensitive LC-MS/MS method. Nucleic Acids Res. 35, e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ohira T., Gemmill R. M., Ferguson K., Kusy S., Roche J., Brambilla E., Zeng C., Baron A., Bemis L., Erickson P., Wilder E., Rustgi A., Kitajewski J., Gabrielson E., Bremnes R., Franklin W., Drabkin H. A. (2003) WNT7a induces E-cadherin in lung cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 10429–10434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jiang L., Chan J. Y., Fung K. P. (2012) Epigenetic loss of CDH1 correlates with multidrug resistance in human hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 422, 739–744 [DOI] [PubMed] [Google Scholar]
- 45. Wang Q., Zhou H. S., Huang K. K., Jiang X. J., Wu F. Q., Cao R., Yin C. X., Liao L. B., Zheng Z. X., He H., Lin R., Yi Z. S., Xu D., Yang M., Meng F. Y. (2012) Imatinib and bortezomib induce the expression and distribution of anaphase-promoting complex adaptor protein Cdh1 in blast crisis of chronic myeloid leukemia. Int. J. Oncol. 40, 418–426 [DOI] [PubMed] [Google Scholar]
- 46. Gleixner K. V., Mayerhofer M., Aichberger K. J., Derdak S., Sonneck K., Böhm A., Gruze A., Samorapoompichit P., Manley P. W., Fabbro D., Pickl W. F., Sillaber C., Valent P. (2006) PKC412 inhibits in vitro growth of neoplastic human mast cells expressing the D816V-mutated variant of KIT: comparison with AMN107, imatinib, and cladribine (2CdA) and evaluation of cooperative drug effects. Blood 107, 752–759 [DOI] [PubMed] [Google Scholar]
- 47. Growney J. D., Clark J. J., Adelsperger J., Stone R., Fabbro D., Griffin J. D., Gilliland D. G. (2005) Activation mutations of human c-KIT resistant to imatinib mesylate are sensitive to the tyrosine kinase inhibitor PKC412. Blood 106, 721–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Meissner A., Gnirke A., Bell G. W., Ramsahoye B., Lander E. S., Jaenisch R. (2005) Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 33, 5868–5877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Garzon R., Liu S., Fabbri M., Liu Z., Heaphy C. E., Callegari E., Schwind S., Pang J., Yu J., Muthusamy N., Havelange V., Volinia S., Blum W., Rush L. J., Perrotti D., Andreeff M., Bloomfield C. D., Byrd J. C., Chan K., Wu L. C., Croce C. M., Marcucci G. (2009) MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 113, 6411–6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Went P. T., Dirnhofer S., Bundi M., Mirlacher M., Schraml P., Mangialaio S., Dimitrijevic S., Kononen J., Lugli A., Simon R., Sauter G. (2004) Prevalence of KIT expression in human tumors. J. Clin. Oncol. 22, 4514–4522 [DOI] [PubMed] [Google Scholar]
- 51. Fabbro D., Buchdunger E., Wood J., Mestan J., Hofmann F., Ferrari S., Mett H., O'Reilly T., Meyer T. (1999) Inhibitors of protein kinases: CGP 41251, a protein kinase inhibitor with potential as an anticancer agent. Pharmacol. Ther. 82, 293–301 [DOI] [PubMed] [Google Scholar]
- 52. Ikegami Y., Yano S., Nakao K. (1996) Effects of the new selective protein kinase C inhibitor 4′-N-benzoyl staurosporine on cell cycle distribution and growth inhibition in human small cell lung cancer cells. Arzneimittelforschung 46, 201–204 [PubMed] [Google Scholar]
- 53. Courage C., Budworth J., Gescher A. (1995) Comparison of ability of protein kinase C inhibitors to arrest cell growth and to alter cellular protein kinase C localisation. Br. J. Cancer 71, 697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tenzer A., Zingg D., Rocha S., Hemmings B., Fabbro D., Glanzmann C., Schubiger P. A., Bodis S., Pruschy M. (2001) The phosphatidylinositide 3′-kinase/Akt survival pathway is a target for the anticancer and radiosensitizing agent PKC412, an inhibitor of protein kinase C. Cancer Res. 61, 8203–8210 [PubMed] [Google Scholar]
- 55. Steelman L. S., Pohnert S. C., Shelton J. G., Franklin R. A., Bertrand F. E., McCubrey J. A. (2004) JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia 18, 189–218 [DOI] [PubMed] [Google Scholar]
- 56. Wu J. J., Zhang X. D., Gillespie S., Hersey P. (2005) Selection for TRAIL resistance results in melanoma cells with high proliferative potential. FEBS Lett. 579, 1940–1944 [DOI] [PubMed] [Google Scholar]
- 57. Weisberg E., Roesel J., Bold G., Furet P., Jiang J., Cools J., Wright R. D., Nelson E., Barrett R., Ray A., Moreno D., Hall-Meyers E., Stone R., Galinsky I., Fox E., Gilliland G., Daley J. F., Lazo-Kallanian S., Kung A. L., Griffin J. D. (2008) Antileukemic effects of the novel, mutant FLT3 inhibitor NVP-AST487: effects on PKC412-sensitive and -resistant FLT3-expressing cells. Blood 112, 5161–5170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Stölzel F., Steudel C., Oelschlägel U., Mohr B., Koch S., Ehninger G., Thiede C. (2010) Mechanisms of resistance against PKC412 in resistant FLT3-ITD positive human acute myeloid leukemia cells. Ann. Hematol. 89, 653–662 [DOI] [PubMed] [Google Scholar]
- 59. Brognard J., Clark A. S., Ni Y., Dennis P. A. (2001) Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Res. 61, 3986–3997 [PubMed] [Google Scholar]
- 60. Kandasamy K., Srivastava R. K. (2002) Role of the phosphatidylinositol 3′-kinase/PTEN/Akt kinase pathway in tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in non-small cell lung cancer cells. Cancer Res. 62, 4929–4937 [PubMed] [Google Scholar]
- 61. Hopfer O., Komor M., Koehler I. S., Schulze M., Hoelzer D., Thiel E., Hofmann W. K. (2007) DNA methylation profiling of myelodysplastic syndrome hematopoietic progenitor cells during in vitro lineage-specific differentiation. Exp. Hematol. 35, 712–723 [DOI] [PubMed] [Google Scholar]
- 62. Galmarini C. M., Mackey J. R., Dumontet C. (2001) Nucleoside analogues: mechanisms of drug resistance and reversal strategies. Leukemia 15, 875–890 [DOI] [PubMed] [Google Scholar]
- 63. Stam R. W., den Boer M. L., Meijerink J. P., Ebus M. E., Peters G. J., Noordhuis P., Janka-Schaub G. E., Armstrong S. A., Korsmeyer S. J., Pieters R. (2003) Differential mRNA expression of Ara-C-metabolizing enzymes explains Ara-C sensitivity in MLL gene-rearranged infant acute lymphoblastic leukemia. Blood 101, 1270–1276 [DOI] [PubMed] [Google Scholar]
- 64. Garcia-Manero G., Gore S. D., Cogle C., Ward R., Shi T., Macbeth K. J., Laille E., Giordano H., Sakoian S., Jabbour E., Kantarjian H., Skikne B. (2011) Phase I study of oral azacitidine in myelodysplastic syndromes, chronic myelomonocytic leukemia, and acute myeloid leukemia. J. Clin. Oncol. 29, 2521–2527 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.