

Background: The infectious salmon anemia virus (ISAV) fusion (F) protein displays pH-dependent host fusion activity.
Results: Thermal stability of ISAV F is inversely correlated with pH.
Conclusion: ISAV F exhibits class I viral fusion architecture that is stabilized at fusion pH by carboxyl-carboxylate electrostatics.
Significance: This analysis contributes new model principles to our understanding of the diversity of viral entry strategies.
Keywords: animal virus, circular dichroism (CD), crystal structure, virus entry, X-ray crystallography, Class I viral fusion protein, ISAV, infectious salmon anemia virus, Isavirus, carboxyl-carboxylate
Abstract
Segment 5, ORF 1 of the infectious salmon anemia virus (ISAV) genome, encodes for the ISAV F protein, which is responsible for viral-host endosomal membrane fusion during a productive ISAV infection. The entry machinery of ISAV is composed of a complex of the ISAV F and ISAV hemagglutinin esterase (HE) proteins in an unknown stoichiometry prior to receptor engagement by ISAV HE. Following binding of the receptor to ISAV HE, dissociation of the ISAV F protein from HE, and subsequent endocytosis, the ISAV F protein resolves into a fusion-competent oligomeric state. Here, we present a 2.1 Å crystal structure of the fusion core of the ISAV F protein determined at low pH. This structure has allowed us to unambiguously demonstrate that the ISAV entry machinery exhibits typical class I viral fusion protein architecture. Furthermore, we have determined stabilizing factors that accommodate the pH-dependent mode of ISAV transmission, and our structure has allowed the identification of a central coil that is conserved across numerous and varied post-fusion viral glycoprotein structures. We then discuss a mechanistic model of ISAV fusion that parallels the paramyxoviral class I fusion strategy wherein attachment and fusion are relegated to separate proteins in a similar fashion to ISAV fusion.
Introduction
Infectious salmon anemia (ISA)3 was first identified in Norway in 1984 (1) in farmed Atlantic salmon (Salmo salar). Since then, outbreaks have spread to both sides of the North American continent, as well as in South America, resulting in the culling of entire salmon stocks and causing considerable economic losses. The disease appears as a systemic condition characterized by severe anemia and hemorrhages in several organs with an average mortality rate of 30%, ranging from 15 to 90% over a period of several months.
In the mid-1990s, the causative agent of ISA was identified as an orthomyxovirus. ISAV is a pleomorphic, negative-strand enveloped virus of the Isavirus genus that primarily targets endothelial and leukocytic cells of fish, such as salmon, rainbow trout, brown trout, Atlantic herring, and Arctic char (2). The genome of ISAV consists of eight single-stranded RNA segments (14.3 kb) that encode for at least 10 proteins (3). ISAV targets endothelial, gill epithelial, and erythrocyte cells, and can easily spread throughout fish populations through contaminated water or equipment, which makes ISAV difficult to contain and eradicate. Contamination with ISAV requires the immediate quarantine and destruction of infected fish, extensive disinfection activities for the affected production facilities, and a fallow period prior to restocking.
For most enveloped viruses, a single virus-encoded glycoprotein facilitates both host cell attachment and membrane fusion. These viral fusion glycoproteins are classified into three categories (class I, II, and III) by their structural features (reviewed in Ref. 4). Orthomyxoviruses, such as influenza A, B, and C viruses, utilize a class I viral glycoprotein for entry, and as such, the viral glycoprotein irreversibly catalyzes the fusion of viral and host membrane through a series of conformational rearrangements. Following receptor-driven endocytosis and a subsequent drop in pH, these rearrangements culminate in the formation of an energetically stable bundle of anti-parallel helical hairpins that juxtapose the host membrane embedded fusion peptide and the transmembrane domain of the viral fusion protein, thereby catalyzing fusion of lipid bilayers.
ISAV entry is similar to other orthomyxoviruses, where receptor binding to sialic acids initiates an internalization process into endosomes, followed by low pH activation of the fusion machinery (5). However, a major difference exists in the functional organization of the key viral glycoproteins involved in entry. ISAV segment 6, ORF 1, encodes for a viral hemagglutinin esterase (HE) glycoprotein, which is required for initial host cell attachment and has additional receptor-destroying activity that is required to prevent the virus from self-aggregating and to promote the release of viral progenies from the infected cell (6). ISAV segment 5, ORF 1, encodes for a single-chain (∼50 kDa) type Ia transmembrane fusion (F) protein that is necessary and nominally sufficient to catalyze the merger of the viral and host endosomal membrane for entry (7). Both of these viral proteins are targeted to the plasma membrane during assembly and constitute the majority of the virus-encoded components in the membrane of infectious particles (6). It is unclear how receptor binding occurs and how this information is transmitted to the F glycoprotein to initiate viral fusion. ISAV is the only known member of the Orthomyxoviridae genera with attachment and fusion activities on different proteins, and understanding this process offers a unique opportunity to contribute new model principles and to understand the diversity of viral entry strategies.
Currently, most hypotheses regarding ISAV viral entry are based on poor homology models with putative counterparts in influenza viruses. Here we present a crystal structure of the ISAV F core fusion protein at a resolution of 2.1 Å. This represents the first structure of any ISAV entry glycoprotein. Our model clearly illustrates that the ISAV F protein fusion glycoprotein adopts a traditional class I viral fusion architecture. Extensive structural characterization and analyses of orthomyxoviral fusion proteins exemplify the exquisite regulatory role that particular ionic residues play in viral fusion. Our structure has allowed us to identify electrostatic requirements for the stability of the ISAV F protein at the pH of fusion and comment on some of the unique features of this aquatic pathogen. Comparisons between orthomyxoviral fusion proteins and the ISAV F protein allow for more sophisticated hypotheses to be generated regarding the unique mechanism of ISAV entry.
Experimental Procedures
In Silico Characterization of Orthomyxoviral Fusion Glycoproteins
Pairwise alignment of the ISAV (isolate Atlantic salmon/Norway/810/9/99) F protein sequence (UniProt accession number: Q8V3T9) with influenza A virus (IAV) (strain A/Brevig Mission/1/1918 H1N1 HA (UniProt accession number: Q9WFX3), influenza B virus (IBV) (strain B/Lee/1940) HA (UniProt accession number: P03460), and influenza C virus (ICV) (strain C/Johannesburg/1/1966) hemagglutinin esterase fusion (HEF) (UniProt accession number: P07975) was performed using the CLUSTALΩ program (8). Following primary sequence alignment, secondary structure and location or presence of the putative fusion peptide, transmembrane, and coiled-coil domains were predicted using the NPS@CONSENSUS (9), TMPRED (10), and COILS suite of programs (11), respectively.
Construct Design and Protein Production
DNA corresponding to the full-length ISAV F (residues 1–444) and IAV HA (residues 382–519) were codon-optimized and commercially synthesized. ISAV F294–404 and IAV HA382–519 were subcloned into pET46 Ek/LIC. To avoid nonspecific intermolecular disulfide-mediated aggregation, cysteine to serine mutations (ISAV F, C382S,C388S,C390S; IAV HA, C481S) were generated by site-directed mutagenesis.
ISAV F and IAV HA2 fusion glycoproteins were expressed in BL21 (DE3) Escherichia coli cells. Cell cultures were grown to A600 = 0.6 at 37 °C and induced with a final concentration of 0.5 mm isopropyl-1-thio-β-d-galactopyranoside for 20 h at 18 °C. Cells were resuspended in 50 mm Tris-HCl, pH 8.0, 300 mm NaCl, and 20 mm imidazole. E. coli cells were lysed using a hydraulic cell disruption system (Constant Systems) and purified by standard nickel-nitrilotriacetic acid affinity chromatography. Prior to crystallization trials, recombinant ISAV F294–404 protein was dialyzed against 10 mm Tris-HCl, pH 7.5, and 50 mm NaCl and digested with thrombin at 4 °C over 48 h to remove the polyhistidine tag and generate the ISAV F294–383 fragment. Cleavage reactions were stopped with a final concentration of 2 mm PMSF and applied onto an anion exchange column (MonoQ 5/50 GL). Prior to biophysical assays or crystallization trials, the fusion proteins were further purified by size exclusion chromatography using a Superdex 200 prep grade 10/300 column (GE Healthcare) equilibrated in 10 mm Tris-HCl, pH 7.5, and 150 mm NaCl. Protein concentration was determined by absorbance at λ = 280 nm, and purity was monitored by SDS-PAGE and mass spectrometry.
Crystallization and Structure Determination
Initial sparse matrix crystallization screening of ISAV F294–383 (∼30 mg/ml) was performed by sitting drop vapor diffusion using the Douglas Instruments Oryx 8 liquid handling system. ISAV F294–383 was crystallized by sitting drop vapor diffusion in 0.2 m lithium sulfate, 0.1 m sodium acetate, pH 4.5, 2 mg/ml tetramethylthionine chloride, and 50% (v/v) PEG 400. Data for ISAV F294–383 were collected on beamline 08ID-1 at the Canadian Light Source (Saskatoon, Saskatchewan). All data were reduced using XDS (12), and scaling was performed using programs from the CCP4 program suite, Pointless and Aimless (13, 14). The structure of ISAV F294–383 was determined by molecular replacement, using the program PHENIX.phaser (15) with a polyalanine model of the HIV-1 gp41 inner helix (Protein Data Bank (PDB) number 3UIA; residues 15–48) and a polyalanine model of the pre-fusion parainfluenza virus 5 F protein (PDB number 2B9B; residues 450–465) as search models. Initial attempts at molecular replacement phasing using existing post-fusion structures of orthomyxoviral HA as search models failed (PDB numbers 1HTM; 1QU1; and 4NKJ). The polyalanine HIV-1 gp41 inner helix model was sufficient to find a molecular replacement solution; however, the density was appreciably clearer once the polyalanine parainfluenza 5 F core was included as an ensemble. Following molecular replacement, PHENIX.autobuild (16) was used to build in side chains and extend the placed search model. Iterative rounds of model rebuilding and refinement were performed using the program Coot (17) and PHENIX.refine (18), respectively. 10% of unique reflections were held back during refinement as the test set. Clear electron density was seen for residues 311–377 in ISAV F294–383; however, only weak or no electron density was observed for the first 14 residues. The tetramethylthionine ligand was located along the 3-fold axis, with ∼30% occupancy. Following structural determination of the post-fusion ISAV F294–383, a search for its closest structural neighbor was performed using the DaliLite version 3 server Dali (19), and heptad repeat analysis was performed using SOCKET (20). All data collection and refinement statistics are presented in Table 1.
TABLE 1.
Data collection and refinement statistics
| ISAV F (294–383) | |
|---|---|
| Data collection | |
| Wavelength (Å) | 0.9795 |
| Resolution range (Å) | 38.7–2.1 (2.16–2.10)a |
| Space group | H 3 2 |
| Unit cell | |
| a, b, c (Å) | 42.9 42.9 232.2 |
| α, β, γ (°) | 90.0 90.0 120.0 |
| Total reflections | 23,796 (1,453) |
| Unique reflections | 5,037 (361) |
| Multiplicity | 4.7 (4.0) |
| Completeness (%) | 97.6 (88.6) |
| Mean I/σ(I) | 13.7 (3.2) |
| Wilson B-factor | 28.7 |
| Rmeasb | 0.073 (0.56) |
| Refinement | |
| Rwork | 0.20 |
| Rfree | 0.25 |
| Number of non-hydrogen atoms | 562 |
| Macromolecules | 534 |
| Ligands | 22 |
| Water | 6 |
| Protein residues | 70 |
| RMSD (bonds; Å) | 0.011 |
| RMSD (angles; °) | 1.23 |
| Ramachandran favored (%) | 100 |
| Ramachandran outliers (%) | 0 |
| Average B-factor | 41.2 |
| Macromolecules | 41.3 |
| Ligands | 36.7 |
| Solvent | 47.9 |
a Statistics for the highest-resolution shell are shown in parentheses.
b Rmeas: multiplicity-independent r = Sum(√(N/(N − 1))(|Ihl − 〈Ih〉|))/Sum(〈Ih〉).
Circular Dichroism and Thermal Melts
CD spectral scans and thermal melting curves of ISAV F294–404 and IAV HA2382–519 were acquired on a Jasco J-810 spectropolarimeter in quartz cuvettes (Helma) with a 1-mm path length. Assays were conducted using protein concentrations ranging from 10 to 25 μm. All ISAV F294–404 and IAV HA2382–519 fusion proteins were buffer-exchanged by extensive dialysis into the following buffer conditions: pH 4.5–5.5, sodium acetate (NaOAc) buffer (20 mm NaOAc, 150 mm NaCl, or 500 mm NaCl); pH 6.0–7.5, potassium phosphate buffer (20 mm K2HPO4/KH2PO4, 150 mm NaCl, or 500 mm NaCl). Following dialysis, CD wavelength scans were collected between 200 and 260 nm and averaged over three accumulations at 20 °C. Thermal denaturation assays were carried out at a single wavelength (217 nm) by increasing the temperature from 20 to 80 °C for all ISAV F constructs or from 20 to 95 °C for IAV HA2382–519 and monitoring the resultant change in ellipticity. After each denaturation scan, the cuvette was allowed to return to 20 °C and an additional wavelength scan was collected. All thermal denaturation data were baseline-subtracted, normalized between 0 (folded) and 1 (unfolded), and fit to a nonlinear biphasic sigmoidal curve using GraphPad Prism (version 5.01). Apparent Tm values were determined from the peak value of the first derivative of the thermal melt curves as calculated by the Jasco J-810 software suite. All melts were performed in triplicate with independently purified recombinant protein. Data are represented as the mean of the triplicate experiments ± S.D.
Results and Discussion
In Silico Characterization of the ISAV F Protein Draws Parallels to Other Orthomyxoviral Fusion Proteins
ClustalΩ pairwise alignment between full-length ISAV F and IAV HA, IBV HA, and ICV HEF showed 13.9% (87 identical and 138 similar positions), 13.7% (87 identical and 141 similar positions), and 13.4% (94 identical and 123 similar positions), respectively. Pairwise alignment between IAV and IBV fusion proteins results in a 26.5% identity (157 identical and 206 similar positions) and a 15.1% identity (108 identical and 192 similar positions) between IAV and ICV fusion proteins. However, upon comparison of secondary structure predictions between ISAV F and the other orthomyxoviral fusion proteins using NPS@:CONSENSUS (9), a trend is observed (Fig. 1A). In each viral fusion protein, a high propensity for helical secondary structure is observed for the last ∼120 amino acids of the primary sequence. These helices are sandwiched between two regions predicted by TMPRED (10) to be transmembrane domains. Within influenza virus glycoproteins, these predicted regions have been experimentally determined to be the fusion peptide and transmembrane anchor, respectively (Fig. 1B). In IAV and IBV, this α-helical region corresponds with the post-fusion core trimeric bundle of anti-parallel helical hairpins. Although the sequence alignment indicates that there is minimal conservation between the ISAV F primary protein sequence and the other orthomyxoviral fusion proteins, we were able to demonstrate that all of our orthomyxoviral fusion protein primary sequences, including ISAV F, were predicted to contain a coiled-coil domain at this region (Fig. 1C) using the program COILS (11). Our in silico characterization results have indicated that the ISAV F protein uses a dissimilar primary amino acid sequence from that of its orthomyxoviral cousins to achieve similar tertiary architecture for host-viral fusion.
FIGURE 1.

Computational characterization of orthomyxovirus fusion proteins. A–C, representative protein sequences of IAV HA (H1N1), IBV HA, ICV HEF, and ISAV F proteins were used for protein secondary structure (A), transmembrane propensity (B), and coiled-coil propensity (C). Numerals along the x-axis of the graphs correspond to the primary sequence of a given orthomyxoviral fusion glycoprotein.
ISAV F294–383 Core Fusion Protein Exhibits Class I Fusion Machinery
Crystal structures of the fusion proteins from IAV, IBV, and ICV viruses have been experimentally determined (21–23), and their fusion proteins have been largely annotated; however, ISAV is the only orthomyxovirus to isolate its fusion activity from receptor binding and/or destroying activities. In silico comparisons between the ISAV F protein and other orthomyxoviral glycoproteins with fusion activity predict similar secondary structural elements that correspond with the core fusion protein of previously annotated orthomyxoviral fusion proteins (Fig. 1). Therefore, we designed our initial E. coli expression constructs within the bounds of the predicted fusion peptide at ISAV F residues 277–293 and the predicted transmembrane domain at residues 413–435. Recombinant ISAV F294–404 expressed as a soluble protein in milligram quantities in E. coli. Purified ISAV F294–404 was subjected to enzymatic digestion by thrombin protease to remove our His6 purification tag. However, ISAV F294–404 contains a naturally occurring thrombin cleavage site at residue 383, and this site was also cleaved during proteolysis (confirmed by MALDI-TOF mass spectrometry). We have termed this product ISAV F294–383. The crystal structure of post-fusion ISAV F294–383 was determined at 2.1 Å resolution in space group H 3 2 (Table 1). Each asymmetric unit contains one ISAV F294–383 molecule, and the biological trimeric bundle was generated through the crystallographic symmetry operators (Fig. 2, A and B).
FIGURE 2.

ISAV F294–383 exhibits typical architecture of class I fusion machinery. A, stereo diagram of the structure of the trimeric ISAV F294–383 core fusion protein. ISAV F294–383 is composed of an extended central coiled-coil that abruptly breaks at Tyr357, facilitating the positioning of the C-terminal helix against the neighboring protein chain. Two chloride ions coordinated by Asn340 and Asn347, respectively, are shown as green spheres. B, structure of the trimeric ISAV F294–383 core fusion protein as generated by crystallographic symmetry. C, the chain reversal region of ISAV F294–383 forms a packing interface with the adjacent chain of the trimer. The packing generates layers of hydrophobic residues followed by polar interactions and then culminates in a hydrophobic core. Black residue labels denote hydrophobic residues, red labels correspond to negatively charged residues, blue labels are for positively charged residues and a green label indicates a polar uncharged residue. D, in IAV HA2 (PDB code: 1HTM), the C-terminal helix packs against the same chain and employs an almost entirely hydrophobic packing arrangement. Of interest, His106 is situated at the apex of the chain reversal region and is hypothesized to act as a stabilizing residue at low pH.
The overall architecture of ISAV F294–383 is reminiscent of previously determined post-fusion orthomyxoviral fusion proteins. The ISAV F fusion core is fully α-helical and trimeric, as is characteristic of class I fusion machinery. It is composed of a central 14-turn extended coiled-coil with hydrophobic residues packed into the core, away from the bulk solvent. The central coiled-coil region abruptly breaks at Tyr357, forming a helical hairpin that switches back and continues for another four turns. This four-turn helix packs against the adjacent helix of the coiled-coil core, generating the hallmark class I viral protein post-fusion conformation (Fig. 2B). Interestingly, the outer helix on ISAV F spirals back to pack against an adjacent ISAV chain. This spiral architecture appears to be a result of alternating layers of polar and hydrophobic intermolecular interactions (Fig. 2C). This is unlike the IAV hemagglutinin, which folds directly back onto itself to form a true hairpin (Fig. 2D).
Chloride coordination is common to most class I fusion proteins; however, ISAV is the first orthomyxovirus to our knowledge where this has been experimentally observed. There are two chloride coordination sites within the central core. The trimeric nature of the central coiled-coil allows the formation of two asparagine layers (Asn340 and Asn347) to coordinate the two chloride ions. Interestingly, the polar layer formed by Asn347 found approximately four helical turns before the chain reversal region is conserved in the post-fusion structures of both IAV HA2 at residue Asn95 and IBV HA2 at residue Asn65 (22–24). In our ISAV F294–383 crystallographic model, the chloride ion is flanked by residue Ser344, whereas in the IBV post-fusion HA2, the corresponding residue is a threonine (Thr61). This arrangement on IBV HA2 places a methyl group into the space that the chloride ion occupies in ISAV F294–383. On IAV HA2, this region is occluded by Leu91 and Trp92.
Thermal Stability of pH-dependent Orthomyxoviral Fusion Core Proteins Correlates with Biological Fusion Requirements
The thermal stability of ISAV F294–404 and IAV HA382–519 core fusion protein was determined by CD spectroscopy over a wide pH range. Both IAV and ISAV host-viral envelope fusion events are pH-dependent, and an inverse correlation between class I core fusion protein thermal stability and pH has previously been demonstrated (25, 26). Additionally, it is known that the IAV HA fusion core is stabilized as the pH is adjusted to that of viral fusion in a biological system (27). As expected, our recombinant IAV HA fusion core was stabilized as it was subjected to an increasingly acidic environment (Fig. 3A). This stability gradient was also observed for the ISAV F core fusion protein (Fig. 3B), albeit the IAV HA fusion core (Tm = 86.5 °C; pH 4.5) demonstrated greater thermal stability overall than ISAV F294–404 (Tm = 67.5 °C; pH 5.5), and below the ISAV pH of fusion, ISAV F294–404 became increasingly unstable (Tm = 57 °C; pH 5.0, Tm =48 °C; pH 4.5) (Fig. 4A). In vivo, there is an extensive conformational change that occurs between the pre- and post-fusion species of the IAV HA protein. Notably, the B-loop of the trimeric IAV HA protein becomes α-helical and generates an extended coiled-coil core that begins at the N-cap region that encompasses the residues that previously formed the B-loop and terminates at a helical hairpin nearly 100 Å away (22). This conformation is nearly identical for the IAV HA protein (23), and the triggers for these changes are receptor engagement and decreasing pH. However, in the absence of the receptor binding subunit, the HA proteins adopt a post-fusion conformation even at neutral pH. Indeed, our orthomyxoviral fusion core protein constructs represent these proteins in the post-fusion conformation as CD wavelength spectral scans are superimposable between pH 7.5 and that of fusion for both IAV HA382–519 and ISAV F294–404 (Fig. 4, B and C), but importantly, their respective thermal stabilities increase dramatically in response to acidification (IAV HA382–519 ΔTm = +8 °C; ISAV F294–404 ΔTm = +16 °C).
FIGURE 3.
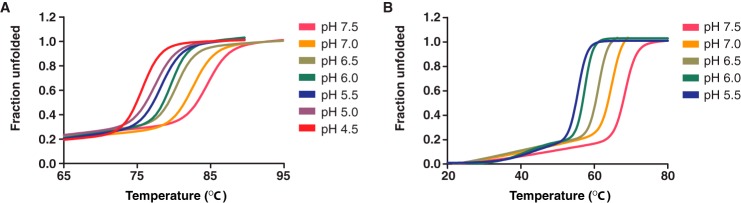
Thermal stability of orthomyxovirus fusion proteins as a function of pH. A and B, the molar ellipticity of 12 μm IAV HA383–519 (A) and 20 μm ISAV F294–404 (B) was measured at 217 nm from 20 to 95 °C in buffers with pH values ranging from 4.5 through 7.5 and 5.5 through 7.5, respectively. All thermal melts were performed in triplicate with independently purified recombinant protein. Data were normalized between 0 (folded) and 1 (unfolded) and fit to a nonlinear biphasic sigmoidal curve in GraphPad Prism (version 5.01).
FIGURE 4.
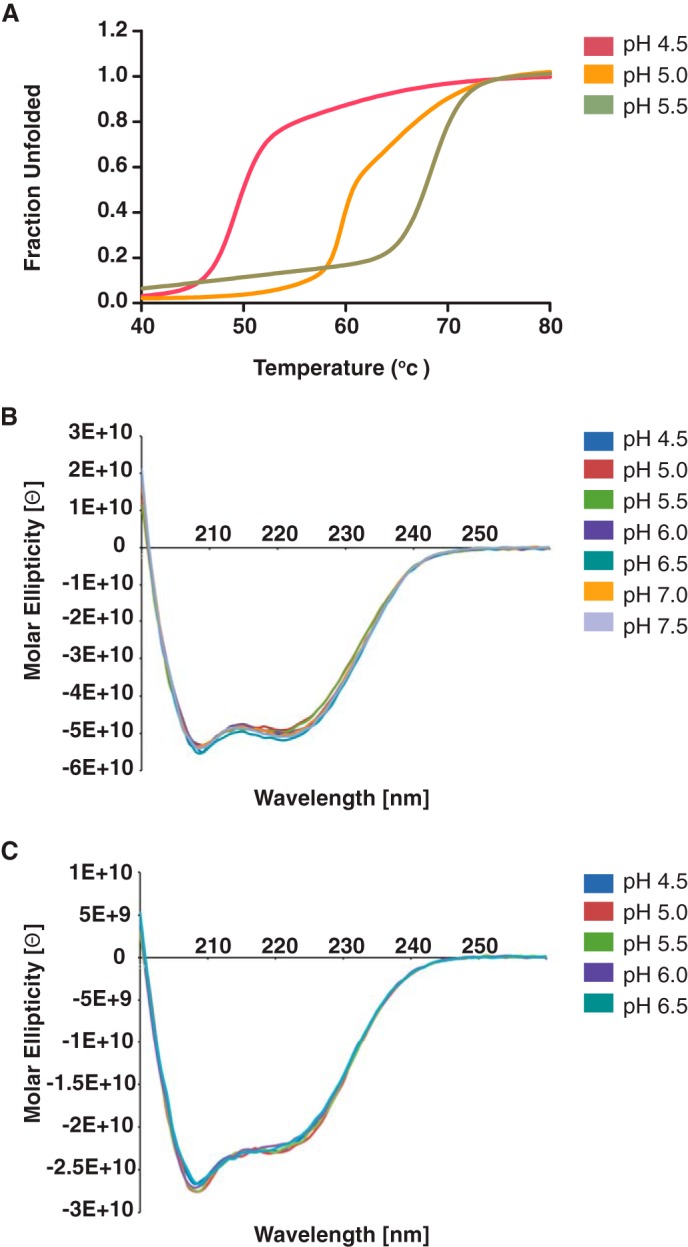
Thermal stability and wavelength spectra of orthomyxovirus fusion proteins as a function of pH. A, the molar ellipticity of 20 μm ISAV F294–404 was measured at 217 nm from 20 to 95 °C in acetate buffers with pH values ranging from 4.5 through 5.5. All thermal melts were performed in triplicate with independently purified recombinant protein. Data were normalized between 0 (folded) and 1 (unfolded) and fit to a nonlinear biphasic sigmoidal curve in GraphPad Prism (version 5.01). B and C, the molar ellipticity of 12 μm IAV HA383–519 (B) and 20 μm ISAV F294–404 (C) was measured from 260 to 190 nm at 20 °C in buffers with pH values ranging from 4.5 through 7.5 and from 5.5 through 7.5, respectively.
Electrostatics Control the pH-dependent Stabilization of the ISAV F Core Fusion Protein
Unlike retroviral glycoproteins that contain both an immunosuppressive motif and a CX6CC motif within structured chain reversal regions (28–30), IAV HA, IBV HA, and ISAV F proteins all generate similar post-fusion bundles that lack an appreciable chain reversal region. Previously, we have shown that at low pH, positively charged residues located at the apex of the chain reversal region in various class I fusion proteins can stabilize the helical dipole moment present in the post-fusion conformation (25). A single conservative point mutation (R106H) at the apex of the chain reversal region in the post-fusion conformation of IAV HA can alter the pH of glycoprotein-catalyzed fusion events (31). However, in the ISAV F protein structure, the chain reversal region is capped by neutral polar residues, Tyr357 and Asn359, generating a polar environment that can interact with the bulk solvent, but does nothing to cap the negative helix dipole moment generated by the helical coiled-coil at the pH of fusion or otherwise. As such, the observed thermal stability of ISAV F294–404 is 19 °C lower than that of IAV HA382–519. Upon inspection of our crystallographic model, an alternate stabilization strategy of ISAV involving electrostatic stabilization became apparent.
The ISAV environmental niche demands that it survive drastic shifts in salinity while maintaining a functional glycoprotein on its surface in a poised state for fusion. Between pH 5.4 and pH 5.6, fusion occurs and the metastable fusion protein becomes an irreversible helical bundle as modeled by the ISAV F294–383 crystal structure. Furthermore, changes in the thermal stability of this conformation negatively correlate with the pH requirements of fusion (as illustrated in Fig. 3). In class I viruses that fuse in a pH-independent fashion, multiple intra- and intermolecular electrostatic interactions lock the outer helices into the core helical bundle to generate the post-fusion conformation (28). Viruses that fuse through a pH-dependent mechanism commonly use histidine-cation or anion-anion/carboxyl-carboxylate electrostatic switches to aid in the conformational rearrangement necessary for fusion (26, 32). Carboxyl-carboxylate interactions have long been demonstrated to stabilize cellular proteins at low pH (33, 34) or destabilize proteins at near neutral pH (35).
The structure of ISAV F294–383 at low pH revealed a carboxyl-carboxylate pair that bands the trimeric helical core together. Glu327 participates in a 2.6 Å O–H–O hydrogen bond with Glu329 of the neighboring helix specifically when the molecule is subjected to low pH (Fig. 5A). NMR studies have identified short strong hydrogen bonds (shorter than 2.75 Å) as important mediators of protein stability (36). Through truncations and mutations, we hypothesized that the Glu327–Glu329 carboxyl-carboxylate electrostatic interaction on the ISAV F core fusion protein acts as a pH sensor. Initially, we looked for changes in thermal stability upon increasing the salt content of the protein buffer solution from 150 mm NaCl to 500 mm NaCl. The 500 mm NaCl solution was chosen to approximate the high salinity of seawater, which ranges from 2.5 to 4.0% (w/v) NaCl. The increased NaCl in the protein buffer solution only modestly increased the thermal stability of the protein by ∼3 °C at pH 7.5 and pH 6.5; however, there was no apparent change in the stability of ISAV F294–404 at the pH of fusion (Fig. 5B). Interestingly, when we incubated the protein with 1 m GuHCl, another chaotrope, we observed a striking stabilization of the protein at pH 7.5 (ΔTm = +8 °C) and a decrease in thermal stability at pH 5.5 (ΔTm = −3 °C) (Fig. 5B). This observation immediately suggested to us that free carboxylates were destabilizing our construct at neutral and near neutral pH values. As a control, incubation of ISAV F294–404 with 1 m urea failed to stabilize the fusion subunit and instead resulted in ∼2 °C decrease of the apparent Tm at all pHs assayed (Fig. 5B).
FIGURE 5.

Mutations and truncations in ISAV F core fusion protein indicate that Glu327:Glu329 carboxyl-carboxylate acts as an ISAV F pH sensor. A, a composite omit 2Fo − Fc electron density map of the ISAV F294–383 core fusion protein, contoured at a level of 1.00 RMSD, illustrating the Glu327:Glu329 carboxyl-carboxylate. Protein thermal stability was assayed by CD spectroscopy. The molar ellipticities of recombinant ISAV F core protein constructs were measured at 217 nm from 20 to 95 °C in buffers with pH values of 7.5, 6.5 and 5.5. B–D, ISAV F294–404 (B), ISAV F294–383 (C), and ISAV F294–404 (D) E327A,E329A were incubated with 0.5 m NaCl, 1 m GuHCl, or 1 m urea overnight and melted at pH 7.5, 6.5, and 5.5. The Tm of ISAV F294–404 E327A,E329A could not be accurately determined at pH 7.5 in the absence of chaotrope and is presented in panel E. For panels B–D, the control protein melt was not incubated with any additional salts or chaotropes. All melts were performed in triplicate with independently purified recombinant protein. Data are represented as mean melting temperature ± S.D. Thermal melts were highly reproducible; S.D. <0.6 °C is not discernible in the graphs. E, thermal denaturation data for ISAV F294–404 E327A,E329A with and without chaotrope. All data were normalized between 0 (folded) and 1 (unfolded), fit to a nonlinear biphasic sigmoidal curve in GraphPad Prism (version 5.01), and performed in triplicate with independently purified recombinant protein.
Finally, we generated a double-mutant ISAV F294–404 E327A,E329A construct to test the role of the observed carboxyl-carboxylate in conformational stability at various pH values. We hypothesized that the Glu327:Glu329 carboxyl-carboxylate would generate destabilizing repulsive electrostatics in the fusion protein at neutral and near neutral pH values while generating strong short O–H–O hydrogen bonds at the pH of fusion. In the pre-fusion conformation of IAV HA, much of the N-terminal portion of the extended central coil observed in the post-fusion conformation is folded out and over, much like a peeled banana. Using our understanding of the pre-fusion glycoprotein from IAV as a framework, it is likely that this region of the ISAV F protein makes electrostatic interactions with the vestigial receptor-binding subunit of the ISAV F protein in its pre-fusion conformation (37, 38). IAV HA switches between the electrostatic HA1-Arg120:HA2-Glu69 to a familiar intramolecular Glu69:Glu74 carboxyl-carboxylate as the pH lowers to that of IAV HA-mediated fusion (32). In the current model, we have shown that the putative ISAV F B-loop becomes a part of the extended central coiled-coil and is locked in by the Glu327:Glu329 carboxyl-carboxylate at the pH of fusion by strong short O–H–O bonds. To account for the possibility of stabilization due to the C-terminal extended region, we assayed our thrombin-proteolyzed construct and observed an identical pH-dependent stabilization. Furthermore, when we incubated the protein with 1 m GuHCl, the stabilization observed for the wild-type and ISAV F294–383 truncations at near neutral pH was absent (Fig. 5C), further implicating the Glu327:Glu329 carboxyl-carboxylate as a pH sensor. Stability assays at various pH values indicate that our E327A,E329A double-mutant protein has largely lost the stability profile associated with a pH-dependent class I fusion protein (Fig. 5D). Additionally, when we performed the thermal stability assays on ISAV F294–404 E327A,E329A at pH 7.5, the melting curve became prostrate over an extensive temperature range. We cannot report an exact melting temperature without a clear inflection point to base the calculation on, but this prolonged melting curve was overcome by incubation with chaotrope (Fig. 5E). Regardless, the ablation of the Glu327:Glu329 carboxyl-carboxylate destabilizes the molecule at pH 5.5 and stabilizes the molecule at near neutral pH values.
Our structure does not include the membrane-proximal C-terminal fragment that is visualized as an extended coil packed against the core in the post-fusion structures of IAV HA and IBV HA. Upon proteolysis of ISAV F294–404 to ISAV F294–383, the corresponding region of the protein was removed. Cysteine to serine mutations introduced into ISAV F294–404 did not alter the labile nature of this region (data not shown). However, the lack of the extended coil region affected the overall stability of the protein at pH 7.5, pH 6.5, and pH 5.5, but the relative increase in thermal stability at the pH of fusion was maintained. Incubation of the truncated construct with 1 m GuHCl had the same results as with the full-length ISAV F construct, stabilizing at neutral and near neutral pH and destabilizing at the pH of fusion (Fig. 5C). Evidently, the membrane-proximal C-terminal fragment lends some stability to the post-fusion conformation of the ISAV F protein; however, it is easily removed by limited tryptic or directed thrombin proteolysis, unlike IAV HA2 (22, 39, 40), suggesting that this protein region is more dynamic than in its terrestrial orthomyxoviral counterpart.
The HA subunits are bound together via an inter-subunit disulfide bond, which is maintained from pre- to post-fusion (22, 23). Mutation of the cysteine located in HA2 that participates in this disulfide bond has been employed in crystallographic studies of HA2 to generate well behaved recombinant protein (24). Like IAV and IBV HA, the ISAV F protein is cleaved into two subunits during viral maturation by host proteases (7). The disulfide-linked organization of processed F protein is also thought to be analogous to the influenza A hemagglutinins; however, mutation of individual cysteine residues in recombinant ISAV F294–404 resulted in disulfide-mediated aggregation of our recombinant trimer (data not shown).
The Post-fusion ISAV F Protein Central Coiled-coil Is Structurally Conserved
The post-fusion structure of the ISAV F294–383 reported herein illustrates an apparently simpler chain reversal region when compared with other known post-fusion orthomyxoviral fusion proteins. Despite the qualitative similarities of our post-fusion model to the overall fold of post-fusion IAV and IBV HA2, it is not clear whether the ISAV F gene evolved with the orthomyxoviruses or was acquired through a different path. A comparative analysis of trimeric ISAV F294–383 to structures deposited in the Protein Data Bank using the Dali server (19) revealed that the post-fusion orthomyxovirus structures are not the closest structural neighbors. IAV HA2 was ranked very close to the bottom of the list (rank: 637) with a 7% sequence identity, a Z-score of 2.8, and an RMSD of 4.4 Å. Additionally, the IBV HA2 post-fusion structure was not represented within the first 1000 results. It is also important to note that the chain reversal region of ISAV F294–383 in the post-fusion conformation could not be aligned with any existing post-fusion structures. Nevertheless, the top structural neighbor (rank: 31) to ISAV F294–383 was a fragment of the HIV-1 gp41 heptad repeat 1 (PDB: 3UIA), which comprises the central coiled-coil of the post-fusion structure. This alignment yielded a Z-score of 4.9 and an RMSD of 1.3 Å within the aligned region. In addition, alignment of the central coiled-coil of ISAV F294–383 was observed with numerous viral fusion proteins including both parainfluenza virus 5 F and hemagglutinin-neuraminidase (HN) and various other core structures of HIV gp41 (Fig. 6). Neither the parainfluenza virus 5 F nor the lentiviral results proved surprising as trimmed structures of these proteins were both used as search models during molecular replacement-based phasing of the ISAV F294–383 crystal structure. Table 2 summarizes the nearest structural neighbors to the ISAV F294–383 curated to include structures of viral fusion glycoproteins. The conservation of the trimeric central coiled-coil within post-fusion viral glycoproteins has been previously shown by Igonet et al. (41, 42) and centers around a heptad repeat stutter region within the coiled coil. Parallel to our Dali queries, we analyzed our structure with the program SOCKET (20) and found that a stutter region exists between Leu326 and Glu329, within the carboxyl-carboxylate pH sensor. A similar stutter region was identified in the IAV HA post-fusion structure, albeit more N-terminal than the post-fusion IAV HA2 carboxyl-carboxylate found at Glu69:Glu74 discussed earlier (28). Our observations illustrate that the ISAV F protein is a unique member of the orthomyxoviral fusion protein repertoire and lend further support to a similar yet simpler post-fusion fold than other orthomyxoviral structures. However, the post-fusion structure of the ICV HEF protein has yet to be described and may further stratify our current library of post-fusion orthomyxoviral fusion proteins.
FIGURE 6.

Nearest neighbor comparisons of ISAV F294–383 with various fusion protein structures illustrates a conserved α-helical core. The trimeric ISAV F F294–383 crystal structure was aligned to structures within the PDB using the Dali server. Structures of aligned viral proteins that catalyze the fusion of membranes are depicted here. Colored in blue are fragments that were not aligned by the Dali server, and fragments colored in pink were aligned. The arrows point to the heptad repeat stutter regions identified by Igonet et al. (41, 42) and are presented in green. Accession codes for the structural models herein can be found in Table 2.
TABLE 2.
Structural conservation of the central coiled-coil of ISAV F294–383
Abbreviations used: MuV, mumps virus; PIV, parainfluenza virus; GTOV, Guanarito virus; LCMV, lymphocytic choriomeningitis virus; MHV, mouse hepatitis virus; MERS-CoV, Middle East respiratory syndrome coronavirus; EBOV, Ebola virus; ASLV, avian sarcoma leukosis virus; CASV, California Academy of Sciences virus; HTLV, human T-cell leukemia virus; SARS-CoV, severe acute respiratory syndrome coronavirus; HRSV, human respiratory syncytial virus; IAV, influenza A virus; MoMuLV, Moloney murine leukemia virus.
| PDB No.a | Dali rank | RMSD | Z-Score | % of identity | |
|---|---|---|---|---|---|
| Å | |||||
| HIV-1 gp41 | 3UIA-a | 31 | 1.3 | 4.9 | 20 |
| MuV F0 | 2FYZ-e | 70 | 2.6 | 4.7 | 4 |
| HIV-1 gp41 | 1CZq-a | 111 | 1.0 | 4.6 | 9 |
| HIV-1 gp41 | 3VGY-c | 125 | 1.5 | 4.6 | 2 |
| HIV-1 gp41 | 3P7K-a | 152 | 1.2 | 4.5 | 22 |
| HIV-1 gp41 | 2X7R-n | 179 | 2.2 | 4.4 | 12 |
| PIV 5 F | 2B9B-b | 201 | 1.9 | 4.3 | 8 |
| HIV-1 gp41 | 2ZFC-a | 209 | 1.7 | 4.3 | 11 |
| PIV 5 HN | 3TSI-d | 238 | 1.7 | 4.2 | 11 |
| HIV-1 gp41 | 3G9R-a | 240 | 1.2 | 4.2 | 7 |
| GTOV GP2 | 4C53-b | 324 | 1.8 | 3.8 | 10 |
| HIV-1 gp41 | 1ENV-a | 325 | 2.8 | 3.8 | 8 |
| LCMV GP2 | 3MKO-a | 326 | 2.7 | 3.8 | 10 |
| MHV S | 1WDF-b | 360 | 1.5 | 3.7 | 13 |
| HIV-1 gp41 | 4DZU-b | 382 | 1.3 | 3.7 | 11 |
| Henipavirus F | 3N27-b | 397 | 2.2 | 3.6 | 10 |
| MERS-CoV S | 4MOD-a | 404 | 3.2 | 3.6 | 10 |
| EBOV GP2 | 1EBO-f | 420 | 3.2 | 3.5 | 11 |
| ASLV TM | 4JPR-a | 433 | 1.4 | 3.5 | 10 |
| CASV GP2 | 4N21-a | 437 | 3.9 | 3.5 | 14 |
| HTLV-1 gp21 | 1MG1-a | 477 | 3.5 | 3.3 | 7 |
| SARS-CoV S | 1WYY | 498 | 1.2 | 3.2 | 20 |
| HRSV F | 1G2C-b | 553 | 2.3 | 3.1 | 8 |
| IAV HA | 1QU1-c | 637 | 4.4 | 2.8 | 7 |
| HIV-1 gp41 | 3K9A-a | 653 | 4.0 | 2.8 | 11 |
| MoMuLV TM | 1MOF-a | 656 | 1.9 | 2.8 | 5 |
a Hyphenated letter following the PDB number refers to the chain used in the Dali alignment with ISAV F294–383.
Triggering of ISAV Fusion
As mentioned previously, ISAV is the only known member of the Orthomyxoviridae genera with attachment and fusion activities on disparate proteins. The physical arrangement of the ISAV entry glycoproteins on the surface of the ISAV virion may provide an explanation for this apparent strategic divergence. ISAV F protein has been observed with ISAV HE protein as co-localized punctae during translocation to, and on the surface of, salmonid cells in culture (43), likely protecting the ISAV F protein from humoral recognition. This is further supported as the HE protein is the main antigen responsible for the neutralization efficiency of the current Centrovet ISAV vaccine (44). Physical dissociation of ISAV HE from ISAV F is observed following receptor engagement, with fusogenic character inversely related to the relative avidity of the association. This avidity is thought to be influenced by the ISAV HE membrane-proximal highly polymorphic region (43); however, nothing is known of the precise structural orientation of this pre-entry complex.
Paramyxoviruses such as measles virus (MeV) present an attractive mechanistic model of fusion that may apply to ISAV fusion. In MeV fusion, binding of the human CD46 or signaling lymphocyte activation molecule (SLAM) surface receptor to the MeV attachment protein (H) triggers a rearrangement of the MeV H head domain. H tetramer reorientation forces the dissociation of the MeV fusion protein (F) and subsequent fusion (45). Interestingly, spontaneous syncytia formation in Chinook head salmon embryo cells expressing ISAV F protein has been observed (7), suggesting that ISAV F protein requires additional factors, such as ISAV HE, to maintain a metastable pre-fusion conformation, analogous to the MeV pre-entry complex.
From a structural standpoint, this is of interest as class I fusion proteins from paramyxoviruses have a large structured domain that intersects the helices that make up the prototypical post-fusion helical bundle (45) instead of the four-residue chain reversal region reported here for the post-fusion ISAV F294–404. It would seem that the ISAV fusion protein is structurally similar to the entry proteins of other orthomyxoviruses, yet has a mechanistic resemblance to that of paramyxoviruses.
Conclusions
Viral fusion following engulfment into endocytic vesicles is a prominent strategy of which both enveloped and non-enveloped viruses make extensive use. There are multiple examples of class I, II, and III fusion glycoproteins where the viral entry machinery consists of pH-sensing residues such as histidine (46–48); however, the ISAV F fusion core does not encode for any readily ionizable histidine residues. We have now shown that stabilization of the ISAV F post-fusion conformation is accomplished largely through a single carboxyl-carboxylate interaction that locks the extended helical core in place, allowing the catalysis of host membrane fusion with the ISAV virion at low pH. Our structure has provided the first experimental insight into the molecular mechanism of ISAV F-mediated low pH fusion and highlights the striking similarities and differences across orthomyxoviral fusion proteins and beyond.
Author Contributions
J. D. C. performed all experiments; H. S-M. subcloned and performed the initial ISAV F expression and crystallization experiments; M. K. K. optimized the expression and purification of IAV HA2382-519; J. D. C. and J. E. L. wrote the manuscript; and J. E. L. conceived and supervised the project. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
This work is based upon data collected by Canadian Light Source (CLS) staff under the instruction of J. D. C. at beamline 08ID-1 at the CLS. The CLS is supported by Natural Sciences and Engineering Research Council of Canada (NSERC), National Research Council of Canada, Canadian Institutes of Health Research (CIHR), the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. In addition, we thank Dr. Azmiri Sultana for extensive discussion and tutelage with regards to the XDS software program and Aiping Dong, Dr. Cheryl Arrowsmith, and Dr. Aled Edwards for access to the Structural Genomics Consortium x-ray diffraction facility. Finally, the authors would like to acknowledge the comments and suggestions made by the reviewers to improve this manuscript.
This work was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) (Grant RGPIN435607-13), Ontario Early Researcher Award (ER-13-09-116), and Canada Research Chair (to J. E. L.) The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4XYP) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ISA
- infectious salmon anemia
- ISAV
- infectious salmon anemia virus
- IAV
- influenza A virus
- IBV
- influenza B virus
- ICV
- influenza C virus
- MeV
- measles virus
- HE
- hemagglutinin esterase
- HEF
- hemagglutinin esterase fusion
- RMSD
- root mean square deviation.
References
- 1. Thorud K. E., Djupvik H. O. (1988) Infectious anaemia in Atlantic salmon (Salmo salar L.). Bull. Eur. Assoc. Fish Pathol. 8, 109–111 [Google Scholar]
- 2. Aamelfot M., Dale O. B., McBeath A., Falk K. (2014) Host tropism of infectious salmon anaemia virus in marine and freshwater fish species. J. Fish. Dis. 10.1111/jfd.12284 [DOI] [PubMed] [Google Scholar]
- 3. Clouthier S. C., Rector T., Brown N. E., Anderson E. D. (2002) Genomic organization of infectious salmon anaemia virus. J. Gen. Virol. 83, 421–428 [DOI] [PubMed] [Google Scholar]
- 4. White J. M., Delos S. E., Brecher M., Schornberg K. (2008) Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43, 189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Edinger T. O., Pohl M. O., Stertz S. (2014) Entry of influenza A virus: host factors and antiviral targets. J. Gen. Virol. 95, 263–277 [DOI] [PubMed] [Google Scholar]
- 6. Falk K., Aspehaug V., Vlasak R., Endresen C. (2004) Identification and characterization of viral structural proteins of infectious salmon anemia virus. J. Virol. 78, 3063–3071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aspehaug V., Mikalsen A. B., Snow M., Biering E., Villoing S. (2005) Characterization of the infectious salmon anemia virus fusion protein. J. Virol. 79, 12544–12553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deléage G., Blanchet C., Geourjon C. (1997) Protein structure prediction: implications for the biologist. Biochimie 79, 681–686 [DOI] [PubMed] [Google Scholar]
- 10. Hofmann K., Stoffel W. (1993) TMbase: a database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler 374, 166 [Google Scholar]
- 11. Lupas A., Van Dyke M., Stock J. (1991) Predicting coiled coils from protein sequences. Science 252, 1162–1164 [DOI] [PubMed] [Google Scholar]
- 12. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. D62, 72–82 [DOI] [PubMed] [Google Scholar]
- 14. Evans P. R. (2011) An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67, 282–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Terwilliger T. C., Grosse-Kunstleve R. W., Afonine P. V., Moriarty N. W., Zwart P. H., Hung L. W., Read R. J., Adams P. D. (2008) Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 64, 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 18. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Walshaw J., Woolfson D. N. (2001) Socket: a program for identifying and analysing coiled-coil motifs within protein structures. J. Mol. Biol. 307, 1427–1450 [DOI] [PubMed] [Google Scholar]
- 21. Rosenthal P. B., Zhang X., Formanowski F., Fitz W., Wong C. H., Meier-Ewert H., Skehel J. J., Wiley D. C. (1998) Structure of the haemagglutinin-esterase-fusion glycoprotein of influenza C virus. Nature 396, 92–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bullough P. A., Hughson F. M., Skehel J. J., Wiley D. C. (1994) Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 371, 37–43 [DOI] [PubMed] [Google Scholar]
- 23. Ni F., Chen X., Shen J., Wang Q. (2014) Structural insights into the membrane fusion mechanism mediated by influenza virus hemagglutinin. Biochemistry 53, 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen J., Skehel J. J., Wiley D. C. (1999) N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA2 subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. U.S.A. 96, 8967–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Aydin H., Smrke B. M., Lee J. E. (2013) Structural characterization of a fusion glycoprotein from a retrovirus that undergoes a hybrid 2-step entry mechanism. FASEB J. 27, 5059–5071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koellhoffer J. F., Dai Z., Malashkevich V. N., Stenglein M. D., Liu Y., Toro R., S Harrison J., Chandran K., DeRisi J. L., Almo S. C., Lai J. R. (2014) Structural characterization of the glycoprotein GP2 core domain from the CAS virus, a novel arenavirus-like species. J. Mol. Biol. 426, 1452–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen J., Wharton S. A., Weissenhorn W., Calder L. J., Hughson F. M., Skehel J. J., Wiley D. C. (1995) A soluble domain of the membrane-anchoring chain of influenza virus hemagglutinin (HA2) folds in Escherichia coli into the low-pH-induced conformation. Proc. Natl. Acad. Sci. U.S.A. 92, 12205–12209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aydin H., Cook J. D., Lee J. E. (2014) Crystal structures of β- and γretrovirus fusion proteins reveal a role for electrostatic stapling in viral entry. J. Virol. 88, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schlecht-Louf G., Renard M., Mangeney M., Letzelter C., Richaud A., Ducos B., Bouallaga I., Heidmann T. (2010) Retroviral infection in vivo requires an immune escape virulence factor encrypted in the envelope protein of oncoretroviruses. Proc. Natl. Acad. Sci. U.S.A. 107, 3782–3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cook J. D., Lee J. E. (2013) The secret life of viral entry glycoproteins: moonlighting in immune evasion. PLoS Pathog. 9, e1003258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu R., Wilson I. A. (2011) Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 85, 5172–5182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Harrison J. S., Higgins C. D., O'Meara M. J., Koellhoffer J. F., Kuhlman B. A., Lai J. R. (2013) Role of electrostatic repulsion in controlling pH-dependent conformational changes of viral fusion proteins. Structure 21, 1085–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sawyer L., James M. N. (1982) Carboxyl-carboxylate interactions in proteins. Nature 295, 79–80 [DOI] [PubMed] [Google Scholar]
- 34. Wohlfahrt G. (2005) Analysis of pH-dependent elements in proteins: geometry and properties of pairs of hydrogen-bonded carboxylic acid side-chains. Proteins 58, 396–406 [DOI] [PubMed] [Google Scholar]
- 35. Lu B., Stubbs G., Culver J. N. (1996) Carboxylate interactions involved in the disassembly of tobacco mosaic tobamovirus. Virology 225, 11–20 [DOI] [PubMed] [Google Scholar]
- 36. Langkilde A., Kristensen S. M., Lo Leggio L., Mølgaard A., Jensen J. H., Houk A. R., Navarro Poulsen J. C., Kauppinen S., Larsen S. (2008) Short strong hydrogen bonds in proteins: a case study of rhamnogalacturonan acetylesterase. Acta Crystallogr. D Biol. Crystallogr. D64, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Carr C. M., Kim P. S. (1993) A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 73, 823–832 [DOI] [PubMed] [Google Scholar]
- 38. Rachakonda P. S., Veit M., Korte T., Ludwig K., Böttcher C., Huang Q., Schmidt M. F., Herrmann A. (2007) The relevance of salt bridges for the stability of the influenza virus hemagglutinin. FASEB J. 21, 995–1002 [DOI] [PubMed] [Google Scholar]
- 39. Ruigrok R. W., Aitken A., Calder L. J., Martin S. R., Skehel J. J., Wharton S. A., Weis W., Wiley D. C. (1988) Studies on the structure of the influenza virus haemagglutinin at the pH of membrane fusion. J. Gen. Virol. 69, 2785–2795 [DOI] [PubMed] [Google Scholar]
- 40. Bullough P. A., Hughson F. M., Treharne A. C., Ruigrok R. W., Skehel J. J., Wiley D. C. (1994) Crystals of a fragment of influenza haemagglutinin in the low pH induced conformation. J. Mol. Biol. 236, 1262–1265 [DOI] [PubMed] [Google Scholar]
- 41. Igonet S., Rey F. A. (2012) SnapShot: Viral and eukaryotic protein fusogens. Cell 151, 1634–1634.e1 [DOI] [PubMed] [Google Scholar]
- 42. Igonet S., Vaney M. C., Vonrhein C., Vonhrein C., Bricogne G., Stura E. A., Hengartner H., Eschli B., Rey F. A. (2011) X-ray structure of the arenavirus glycoprotein GP2 in its postfusion hairpin conformation. Proc. Natl. Acad. Sci. U.S.A. 108, 19967–19972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fourrier M., Lester K., Thoen E., Mikalsen A., Evensen Ø., Falk K., Collet B., McBeath A. (2014) Deletions in the highly polymorphic region (HPR) of infectious salmon anaemia virus HPR0 haemagglutinin-esterase enhance viral fusion and influence the interaction with the fusion protein. J. Gen. Virol. 95, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 44. Falk K. (2014) Vaccination against infectious salmon anemia. in Fish Vaccination (Gudding R., Lillehaug A., Evenson Ø. eds), pp. 313–318, John Wiley & Sons, Ltd, Chichester, UK [Google Scholar]
- 45. Plemper R. K., Brindley M. A., Iorio R. M. (2011) Structural and mechanistic studies of measles virus illuminate paramyxovirus entry. PLoS Pathog. 7, e1002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kalani M. R., Moradi A., Moradi M., Tajkhorshid E. (2013) Characterizing a histidine switch controlling pH-dependent conformational changes of the influenza virus hemagglutinin. Biophys. J. 105, 993–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zheng A., Yuan F., Kleinfelter L. M., Kielian M. (2014) A toggle switch controls the low pH-triggered rearrangement and maturation of the dengue virus envelope proteins. Nat. Commun. 5, 3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stampfer S. D., Lou H., Cohen G. H., Eisenberg R. J., Heldwein E. E. (2010) Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 84, 12924–12933 [DOI] [PMC free article] [PubMed] [Google Scholar]