

Background: The effects of inositol 1,4,5-trisphosphate (IP3)-linked hormones are determined by the frequency, amplitude, and duration of Ca2+ oscillations.
Results: Comparison of IP3 uncaging and hormone stimulation showed that PKC has distinct effects on IP3 formation, metabolism, IP3 receptor function, and Ca2+ wave propagation.
Conclusion: PKC modulates Ca2+ oscillation frequency, duration, and wave velocity.
Significance: PKC feedback shapes Ca2+ oscillations and provides signal versatility.
Keywords: calcium; calcium intracellular release; hepatocyte; inositol 1,4,5-trisphosphate (IP3); inositol trisphosphate receptor (IP3R); PKC; calcium oscillations
Abstract
How Ca2+ oscillations are generated and fine-tuned to yield versatile downstream responses remains to be elucidated. In hepatocytes, G protein-coupled receptor-linked Ca2+ oscillations report signal strength via frequency, whereas Ca2+ spike amplitude and wave velocity remain constant. IP3 uncaging also triggers oscillatory Ca2+ release, but, in contrast to hormones, Ca2+ spike amplitude, width, and wave velocity were dependent on [IP3] and were not perturbed by phospholipase C (PLC) inhibition. These data indicate that oscillations elicited by IP3 uncaging are driven by the biphasic regulation of the IP3 receptor by Ca2+, and, unlike hormone-dependent responses, do not require PLC. Removal of extracellular Ca2+ did not perturb Ca2+ oscillations elicited by IP3 uncaging, indicating that reloading of endoplasmic reticulum stores via plasma membrane Ca2+ influx does not entrain the signal. Activation and inhibition of PKC attenuated hormone-induced Ca2+ oscillations but had no effect on Ca2+ increases induced by uncaging IP3. Importantly, PKC activation and inhibition differentially affected Ca2+ spike frequencies and kinetics. PKC activation amplifies negative feedback loops at the level of G protein-coupled receptor PLC activity and/or IP3 metabolism to attenuate IP3 levels and suppress the generation of Ca2+ oscillations. Inhibition of PKC relieves negative feedback regulation of IP3 accumulation and, thereby, shifts Ca2+ oscillations toward sustained responses or dramatically prolonged spikes. PKC down-regulation attenuates phenylephrine-induced Ca2+ wave velocity, whereas responses to IP3 uncaging are enhanced. The ability to assess Ca2+ responses in the absence of PLC activity indicates that IP3 receptor modulation by PKC regulates Ca2+ release and wave velocity.
Introduction
Calcium oscillations and waves generated by the activation of PLC-linked2 GPCRs regulate a multitude of mechanisms from gene transcription to secretion (1–3). In many cell types, including hepatocytes, stimulus strength is encoded by the frequency of Ca2+ oscillations, with interspike intervals ranging from >250 s at low hormone concentrations to <30 s when challenged with higher hormone levels (1, 4–6). These Ca2+ signals are generated by PLC-mediated hydrolysis of phosphatidylinositol bisphosphate (PIP2) to yield IP3 and the subsequent activation of IP3R Ca2+ release channels in the ER (7). However, the mechanisms driving the subsequent repetitive Ca2+ oscillations have yet to be fully resolved (8–12). Many studies have aimed to determine whether these oscillations arise solely because of the biphasic effects of cytosolic [Ca2+] on IP3R gating (13–16), i.e. Ca2+-induced Ca2+ release (CICR), or whether regenerative PLC activation and/or cyclical protein phosphorylation events are also required (17–19). We have demonstrated recently that intracellular buffering of IP3, using a recombinant protein containing the ligand binding domain of rat IP3R type I, results in an inhibition of Ca2+ oscillations, a decrease in the rates of Ca2+ rise, and a slowing of Ca2+ wave propagation speed (17, 20). These data demonstrate that IP3 levels dynamically regulate Ca2+ oscillations, providing evidence that cross-coupling between IP3 and Ca2+ is required to maintain hormone-induced Ca2+ oscillations in non-excitable cells such as hepatocytes.
The Ca2+ oscillation frequency increases with agonist concentration in hepatocytes (1, 5, 6), but the individual Ca2+ spikes have a constant amplitude and rate of rise and propagate as intracellular Ca2+ waves at a constant velocity independent of agonist dose. Nevertheless, the falling phase of the [Ca2+] spikes shows greater diversity (1, 6), and different agonists can give characteristically distinct shapes of [Ca2+] spikes that vary only in the decay phase, even when observed in the same individual cell (2, 17, 19, 20). Moreover, the duration of cytosolic Ca2+ elevation, in addition to spike frequency, has been demonstrated to regulate transcription (21–23). It is therefore important to determine not only how PLC-dependent Ca2+ oscillations are generated but also how spike and wave kinetics are further modulated to account for the versatility of Ca2+ signaling.
Long interspike periods between Ca2+ transients at low hormone doses and the broad dynamic range of frequency modulation suggest that Ca2+ interspike periods and spike kinetics are dynamically controlled by feedback loops that regulate IP3 generation and metabolism as well as IP3R function (17, 20). PLC-dependent signal transduction activates PKC (24), which, in turn, has the potential to phosphorylate and regulate multiple proteins in the Ca2+ signaling cascade, including GPCRs (25, 26), PLC (27), IP3R (28, 29), and IP3 kinase (30). Importantly, concurrent with Ca2+ oscillations, repetitive translocation of PKC isoforms, both conventional and novel, to the plasma membrane have been reported (31, 32), indicating cyclic activation of these enzymes. Furthermore, previous studies in hepatocytes have shown that both activation and inhibition of PKC can affect hormone-induced Ca2+ oscillation kinetics (33, 34).
In this study, we compared Ca2+ signals elicited by hormone and photorelease of caged IP3 and examined how the ensuing Ca2+ oscillations are regulated in response to each stimulus. Our data reveal that Ca2+ oscillations elicited by direct release of caged IP3 are graded, with the transient amplitude, frequency, and wave velocity dependent on the amount of IP3 released. Moreover, these Ca2+ responses were independent of PLC activity, indicating that IP3 uncaging generates Ca2+ oscillations solely through CICR. This is in contrast to hormone-induced Ca2+ oscillations, which depend on IP3 oscillations cross-coupled with Ca2+ spiking (17, 20) (i.e. regenerative PLC activation) and have characteristic spike properties independent of agonist dose. Therefore, uncaging of IP3 provides a tool to assess modulators of Ca2+ transients in the absence of PLC activity and other hormone-dependent signaling cascades. We show that Ca2+ oscillations elicited by IP3 uncaging persist in the absence of extracellular Ca2+, demonstrating that reloading of ER Ca2+ stores does not entrain these periodic Ca2+ signals. Modulation of Ca2+ signaling by PKC was assessed in both PLC- and CICR-dependent paradigms. Paradoxically, both activation and inhibition of PKC decreased the frequency of hormone-induced Ca2+ oscillations but via different mechanisms. Activation of PKC inhibited regenerative IP3 generation by the GPCR/PLC, whereas inhibition of PKC relieved this negative feedback, allowing more prolonged and sustained IP3 generation and, therefore, Ca2+ release. By contrast, CICR oscillations elicited by uncaging IP3 were potentiated by PKC activation. Furthermore, PKC down-regulation decreased Ca2+ wave velocity in agonist-stimulated cells, whereas it actually increased Ca2+ wave velocity after direct IP3 release. These data demonstrate that PKC activity regulates IP3 levels via effects on GPCR coupling, PLC activity, and/or IP3 metabolism while also effecting IP3R sensitivity to regulate Ca2+ spike frequency, width, and Ca2+ wave velocity.
Experimental Procedures
Primary Cell Culture
Isolated hepatocytes were prepared by collagenase perfusion of livers obtained from male Sprague-Dawley rats. Cells were maintained in Williams E medium for 2–6 h for experiments using freshly isolated cells or 16–24 h for experiments using overnight cultured cells, as described previously (1, 4). Animal studies were approved by the Institutional Animal Care and Use Committee at Rutgers, New Jersey Medical School.
Cytosolic Ca2+ Measurements in Response to Hormones
Calcium imaging experiments were performed in HEPES-buffered physiological saline solution comprised of 25 mm HEPES (pH 7.4), 121 mm NaCl, 5 mm NaHCO3, 10 mm glucose, 4.7 mm KCl, 1.2 mm KH2PO4, 1.2 mm MgSO4, 2.0 mm CaCl2, and 0.25% (w/v) fatty acid-free BSA and supplemented with the organic anion transport inhibitors sulfobromophthalein (100 μm) or probenecid (200 μm). Hepatocytes were loaded with Fura-2 by incubation with 2–5 μm Fura-2/AM and Pluronic acid® F-127 (0.02% v/v) for 20–40 min. Cells were transferred to a thermostatically regulated microscope chamber (37 °C). Fura-2 fluorescence images (excitation, 340 and 380 nm, emission 420–600 nm) were acquired at 1.5- to 3-s intervals with a cooled charge-coupled device camera coupled to an epifluorescent microscope, as described previously (35).
Hormone-induced PLC Activity and IP3 Detection
Intracellular IP3 levels or PLC activity were measured using FRET-based genetically engineered probes. Isolated hepatocytes were transfected by electroporation with the Amaxa rat/mouse hepatocyte nucleofector kit according to the instructions of the manufacturer (Lonza). PLC activity was determined by cotransfection with PLCδ4YFP and PLCδ4CFPPH domains (cDNA was a gift from Dr. Balla, National Institutes of Health), which yield a FRET signal while bound to membrane PIP2 that declines as PIP2 is hydrolyzed. IP3 measurements were determined with the IP3 sensor IRIS-1. IRIS-1 cDNA was a gift from Dr. Mikoshiba (RIKEN Brain Science Institute, Japan) (36). FRET images were acquired at 3-s intervals by illumination with 436 ± 20 nm using a 455-nm-long band pass dichroic filter. FRET donor and acceptor fluorescence images were separated with a 505-nm-long band pass dichroic mirror and directed to 480 ± 30 nm (CFP) or 535 ± 40 nm (YFP/Venus) emission filters using an image beamsplitter (Optical InsightsTM). The FRET ratio was calculated on a cell-by-cell basis and averaged from all expressing cells in the microscope field. FRET signal changes between PLCδ4YFP and PLCδ4CFP PH domains were corrected for YFP bleach using linear regression analysis.
Photorelease of Caged IP3
Overnight cultured hepatocytes were loaded in HEPES-buffered physiological saline solution with the membrane-permeant form of caged IP3 (2 μm; d-2, 3-O-isopropylidene-6-O-(2-nitro-4,5-dimethoxy)benzyl-myo-inositol 1,4,5-trisphosphate-hexakis(propionoxymethyl) ester; Sichem GmbH) for 45 min at room temperature, followed by 30-min loading with the calcium indicator dye Fluo-4/AM (5 μm). Cells were transferred to the microscope chamber of a spinning disc confocal microscope. Fluo-4 images (excitation, 488 nm; emission, 510-nm-long band pass filter) were acquired at 10 Hz. Photorelease of caged IP3 was achieved by light pulses from a nitrogen-charged UV laser (Photon Technology International). The cell-permeant caged IP3 is synthesized with the 2- and 3-hydroxyl groups of myo-inositol protected by an isopropylidene group to ensure that the phosphate groups remain in the 1,4 and 5 positions (37). Of note, when released from the cage, this modified form of IP3 is metabolized at a slower rate, in the order of minutes, compared with natural IP3, which is metabolized in seconds (37). Cell viability was assessed by the addition of maximal hormone concentrations at the end of each experiment. Only cells responsive to hormone stimulation are included in the presented data.
Western Blotting
Hepatocyte lysates were prepared in a buffer comprised of 150 mm NaCl, 10 mm Tris-HCl, 1 mm EDTA, and 0.2 mm PMSF supplemented with 1% (w/v) SDS, 0.5% (v/v) Nonidet P-40, 10 μg/ml aprotinin, and 1 μg/ml leupeptin (pH 7.4). Lysates were resolved by SDS-PAGE on a 10% polyacrylamide gel and transferred to a PVDF membrane. Membranes were blocked for 1 h in Tris-buffered saline (pH 7.5) containing 5% (w/v) nonfat dry milk and 0.1% (v/v) Tween 20. The membranes were incubated with anti-PKCα, (Cell Signaling Technology), anti-PKCϵ, and anti-PKCζ (Santa Cruz Biotechnology) overnight at 4 °C. The protein loading control was determined by stripping PVDF membranes and reprobing with anti-α-tubulin (Cell Signaling Technology).
[3H]Inositol Phosphate Accumulation
Total [3H]inositol phosphate accumulation was determined as described previously (31). In brief, primary hepatocytes were labeled overnight with 2.5 μCi ml−1 myo-[3H]inositol (American Radiolabeled Chemicals, Inc.) in 6-well plates. In some studies, cultures were treated overnight with phorbol-12-myristate-13-acetate (PMA) or 4α-PMA (1 μm) to assess the effect of down-regulating PKC. Cultures were washed with HEPES-buffered physiological saline solution and incubated for 20 min at 37 °C, followed by an additional 10-min treatment with 10 mm LiCl to block inositol monophosphatase activity. Cells were treated with 100 nm vasopressin for 15 min in the absence or presence of PMA (1 nm) or bisindolylmaleimide I (BIM, 5 μm) to assess the acute effects of PKC activation and inhibition, respectively. Incubations were terminated by addition of ice-cold tricholoroacetic acid. Water soluble [H3]inositol-containing components were extracted by addition of tri-n-octylamine:1,1,2-trichlorofluoroethane (1:1 ratio). [H3]inositol phosphates were separated by ion exchange chromatography using Dowex resin in the formate form. Lower-order inositols and glycerophospholipids were removed by elution with 0.4 m ammonium formate/0.1 M formic acid. IP3 and higher-order inositols were then eluted with 1.2 m ammonium formate/0.1 M formic acid. Ultima-Flo (PerkinElmer Life Sciences) was added to the eluate, and disintegrations per minute was determined using liquid scintillation counting. Data are expressed as a -fold increase over basal (inositol phosphate turnover levels in the absence of hormone).
Data Analysis
Image analysis was performed using in-house customized software and ImageJ (National Institutes of Health). Graph plotting and data analysis were performed with GraphPad Prism software. Statistical analysis was performed using Student's t test or one-way analysis of variance where indicated.
Results
Photorelease of Caged IP3 Elicits Ca2+ Oscillations in Hepatocytes
Photorelease of caged IP3 in hepatocytes induced cytosolic Ca2+ increases, with Ca2+ oscillations observed in many cells (Fig. 1A). Similar to hormone-induced Ca2+ oscillations (1, 9), the frequency and number of cells responding to IP3 uncaging increased with stimulus strength, as determined by the number of UV flashes, and tended toward sustained Ca2+ increases with the strongest stimulation (Fig. 1, B and C). A single pulse from the UV laser resulted in Ca2+ responses in only 22.5 ± 10.2% of cells. Incrementally increasing the number of UV flashes (applied as a rapid burst) increased the percentage of cells responding (2× UV, 44.2 ± 14.8%; 3× UV, 84 ± 9.2%; 4× UV, 86.7 ± 8.6%; mean ± S.E. from >100 cells in three independent experiments). In addition, increasing the number of UV flashes shifted the Ca2+ signature from predominantly no response and single Ca2+ transients at low illumination toward oscillatory and sustained (peak/plateau) Ca2+ increases at higher illumination, presumably reflecting increased levels of IP3 (Fig. 1C). Therefore, the Ca2+ signals induced by uncaging IP3 appear to mimic hormone-induced Ca2+ responses, the proportion of responsive cells, and the oscillatory and saturated Ca2+ responses increasing with stimulus strength.
FIGURE 1.

Photorelease of caged IP3 elicits Ca2+ oscillations in primary rat hepatocytes. Isolated hepatocytes were cultured overnight and then loaded with caged IP3 and Fluo-4. A, representative trace showing cytosolic Ca2+ responses to photolysis of caged IP3. Rapid trains of one, two, three, or four UV pulses were applied as indicated (arrows). B, the percentage of cells responding to one to four UV flash events. C, comparison of the types of Ca2+ responses observed after each train of UV pulses: no response, single spike, oscillations, or a sustained Ca2+ increase (peak/plateau). Data shown are mean ± S.E. from ≥100 cells from five independent experiments. D and E, summary of Ca2+ transient amplitude (D) and Ca2+ spike width measured at half-peak height (E) in cells in which oscillations were observed after one, two, and three UV flashes. Data are mean ± S.E. of the first three Ca2+ transients for each individual cell (n = 15). F, Ca2+ wave propagation rates as a function of number of UV flashes. Data are mean ± S.E. from cells in which Ca2+ waves were observed after one, two, and three UV flashes (n = 13). *, p < 0.05; **, p < 0.01; ***, p < 0.001; Student's t test.
A characteristic of hormone-induced Ca2+ oscillations is that although the frequency increases with agonist dose, Ca2+ spike kinetics, including amplitude, rate of rise, and peak width are constant for all agonist doses (1, 5, 6, 9). By contrast, photorelease of caged IP3 resulted in Ca2+ peak heights and widths that increased with stimulus strength (Fig. 1, D and E). These data are the mean ± S.E. calculated from the average of the first three Ca2+ transients from cells in which oscillations were observed at each level of UV exposure. Of particular note, the mean duration of Ca2+ spikes elicited by a single UV flash was 3.8 ± 0.24 s at half-peak height, which is much shorter than hormone-stimulated Ca2+ transients measured in this work (Figs. 4 and 7) and previous studies (1, 38). Broader Ca2+ spike widths were achieved with multiple UV flashes (11.5 ± 2.2 s at half-peak height for 3× UV flashes), but this is still a shorter duration than the Ca2+ spike widths of >20 s typically observed with hormone stimulation. The velocity of Ca2+ waves elicited by IP3 uncaging was also dependent on the number of UV flashes (Fig. 1F). Ca2+ waves induced by hormones propagate at 15–25 μm/s independent of hormone dose (5), whereas Ca2+ waves induced by IP3 uncaging rose from 8.2 μm/s ± 0.7 at 1× UV to 23.9 μm/s ± 4.8 with 3× UV.
FIGURE 4.

Down-regulation of PKC enhances hormone-stimulated Ca2+ signals in cultured hepatocytes. Cultured hepatocytes were treated overnight with the inactive analogue 4α-PMA (1 μm, control) or PMA (1 μm, PKC-DR) to down-regulate classical and novel PKC isoforms. Cells were loaded with Fura-2 and then stimulated with phenylephrine (PE, 20 μm). A—C, typical agonist-induced Ca2+ responses are shown for control (A) and PKC-DR (B and C) cells. D and E, summary data showing the effects of PKC down-regulation on the type of Ca2+ responses and the width of the Ca2+ spikes induced by PE stimulation. Data are mean ± S.E. from ≥50 cells from three independent experiments. **, p < 0.01; ***, p < 0.001; Student's t test. F, Western blots showing PKCα, PKCϵ, and PKCζ protein levels in control and PKC-DR hepatocyte lysates. Levels of α-tubulin are shown as loading controls.
FIGURE 7.

Effects of acute activation and inhibition of PKC on hormone-induced Ca2+ oscillations. The effects of PMA (1 nm) and BIM (5 μm) on phenylephrine (PE)-induced Ca2+ oscillations were examined in hepatocytes cultured for 1 h. Representative traces of PMA- (A) and BIM-treated (B) hepatocytes show either a decrease in oscillation frequency (top panel) or suppression of oscillations (bottom panel). The effect of drug treatment on phenylephrine-induced Ca2+ oscillation frequency (C) and Ca2+ spike width at half-peak height (D) are summarized. The frequencies of agonist-induced Ca2+ oscillations were calculated from 5-min periods in the absence or presence of the drugs. Ca2+ spike widths were calculated from the three oscillations prior to and after drug treatment. The drugs were present at least 1 min prior to carrying out the analysis. Data are mean ± S.E. from ≥15 cells from three independent experiments. **, p < 0.01; ***, p < 0.001; Student's t test. E, the effect of acute activation or inhibition of PKC and PKC-DR on total inositol phosphate production in response to 100 nm vasopressin (VP) stimulation for 15 min. Data are expressed as -fold increase over basal and are the mean ± S.E. from three independent experiments. *, p < 0.05; **, p < 0.01; analysis of variance.
IP3-induced Ca2+ Oscillations Do Not Require PLC Activity
To address whether IP3 regeneration through Ca2+ activation of PLC is required to elicit Ca2+ oscillations in response to photolysis of IP3, we performed experiments in the presence of the aminosteroid PLC inhibitor U71322. Hepatocytes were loaded with caged IP3 and concurrently treated for 75 min with 20 μm U73122, the inactive analogue U73433, or vehicle (dimethyl sulfoxide). The cells were first exposed to a rapid train of 4× UV flashes to uncage IP3, followed by 10 nm vasopressin (Fig. 2, A and B). The percentage of cells eliciting a Ca2+ increase and oscillatory Ca2+ responses to each stimulus are summarized in Fig. 2, C and D. In our hands, non-toxic concentrations of U73122 were insufficient to completely block hormone-induced Ca2+ increases in all cells (higher concentrations perturbed Ca2+ release by thapsigargin, indicating off-target effects). Nevertheless, a significant inhibition of vasopressin-induced Ca2+ transients was observed after U73122 treatment. U73122 reduced the percentage of cells responding to hormone stimulation from 81 ± 6 to 33 ± 9 and the percentage of cells displaying Ca2+ oscillations from 60 ± 5 to 22 ± 6. By contrast, the Ca2+ increases and oscillatory responses elicited by photolysis of caged IP3 were not significantly different between treatment groups (Fig. 2, C and D).
FIGURE 2.

IP3-induced Ca2+ oscillations do not depend upon the activation of PLC. Hepatocytes cultured overnight and loaded with caged IP3 and Fluo-4 were treated with the PLC inhibitor U73122, the inactive analogue U74344 (20 μm for each drug, 75 min), or vehicle (dimethyl sulfoxide (DMSO), 0.1% v/v). Cells were then stimulated with UV illumination followed by 3 nm vasopressin (VP) as indicated. A and B, representative Ca2+ responses in dimethyl sulfoxide-treated (A) or U73122-treated (B) cells are shown. C and D, the effect of PLC inhibition on the percentage of responsive (C) and oscillating cells (D) to each stimulus are summarized. Data are representative of ≥60 cells from four independent experiments. E and F, summary of the effect of apyrase (30 units/ml, 5 min) on the proportion of cells responding (E) or the percentage of cells giving oscillatory Ca2+ responses (F). Data are mean ± S.E. of 45 cells from three independent experiments. *, p < 0.05; **, p < 0.01; Student's t test.
We also considered the possibility that ATP released from the hepatocytes in culture might act in a paracrine fashion to cause tonic subthreshold activation of PLC, the activity of which could be amplified upon direct photorelease of IP3. Pretreatment of hepatocytes with 30 units/ml (5 min) of apyrase to hydrolyze extracellular ATP was without effect on the number of cells responding to photolysis of caged IP3 or the proportion of cells displaying oscillatory changes in cytosolic Ca2+ (Fig. 2, E and F). Taken together, these data indicate that positive feedback of Ca2+ on PLC does not contribute to the Ca2+ signals elicited by uncaging IP3, and, when IP3 is sufficiently elevated, Ca2+ oscillations are driven primarily by CICR at the IP3R.
Plasma Membrane Ca2+ Entry Is Not a Requirement for IP3-driven Ca2+ Oscillations
Store-operated Ca2+ entry plays a fundamental role in maintaining Ca2+ homeostasis and to replete internal Ca2+ stores when cells respond to Ca2+-mobilizing hormones (39). However, there is a continuing debate regarding the importance of store-operated Ca2+ entry and Ca2+ store load in the generation and feedback control of hormone-stimulated Ca2+ oscillations (40–42). To determine whether extracellular Ca2+ entry regulates IP3R activation or sensitivity to IP3, we assessed Ca2+ signals elicited by photorelease of caged IP3 in the absence and presence of extracellular Ca2+. Hepatocytes were maintained in either HEPES-buffered physiological saline solution containing 2 mm CaCl2 or switched to Ca2+-free buffer 5–10 min prior to uncaging IP3 with 3× UV flashes (Fig. 3). A somewhat higher proportion of cells did not respond to photorelease of caged IP3 in Ca2+-free (37.3 ± 0.7%) compared with Ca2+-replete conditions (19.3 ± 3.1%) (Fig. 3C), which may reflect an effect of partial Ca2+ store depletion. Nevertheless, Ca2+ oscillations were still observed in response to IP3 uncaging in the presence or absence of extracellular Ca2+ (Fig. 3, A and B), with no impact on oscillation frequency over a 5-min period (Fig. 3D). These data indicate that plasma membrane Ca2+ entry is not a requirement to sustain repetitive Ca2+ release from the ER. However, the width of the Ca2+ spike, measured at half-peak height for the first three Ca2+ transients, was decreased significantly in the absence of extracellular Ca2+ (5.5 ± 0.2 s compared with 9.3 ± 0.4 s in the presence of extracellular Ca2+). Therefore, Ca2+ entry and ER Ca2+ load may contribute to IP3R-induced Ca2+ transients by prolonging spike duration (Fig. 3E).
FIGURE 3.

Extracellular Ca2+ is not required for IP3-induced Ca2+ oscillations. Isolated hepatocytes loaded with caged IP3 and Fluo-4 were stimulated with 3 UV flashes (UV). A and B, representative traces of Ca2+ responses are shown in the presence (A) and absence (B) of extracellular Ca2+. C–E, summary of the Ca2+ signatures observed (C), oscillation frequency (D), and Ca2+ peak width at half-height (E) in the presence and absence of extracellular Ca2+. Data are mean ± S.E. of 25 cells from two independent experiments. *, p < 0.05, Student's t test.
PKC Down-regulation Perturbs Negative Feedback Inhibition of Ca2+ Mobilization
Previous studies have highlighted the complexity of PKC regulation of hormone-induced Ca2+ oscillations in hepatocytes, reporting that both activators and inhibitors of PKC suppressed the responses to phenylephrine (33, 34). We examined the effect of chronic down-regulation of conventional and novel PKCs (PKC-DR) by overnight treatment (16–24 h) with 1 μm PMA or the inactive analogue 4α-PMA. A comparison of responses in control 4α-PMA-treated and PMA-treated hepatocytes revealed a dramatic shift in the type of Ca2+ responses elicited by phenylephrine (20 μm). Under control conditions, 65 ± 6.5% of cells responded with an oscillatory Ca2+ signature (Fig. 4A shows a representative trace of a Ca2+ response in control cells), whereas PKC-DR resulted in predominately sustained Ca2+ rises in 73 ± 3% of cells (see Fig. 4B for a representative trace) compared with only 11 ± 0.6% of sustained responses in control cells. In PKC-DR cells, only 16.2 ± 2.2% responded with oscillatory Ca2+ signatures (Fig. 4C shows a representative trace of Ca2+ oscillations after PKC-DR). The proportion of cells responding with oscillatory or sustained responses for each treatment group is summarized in Fig. 4D. The small population of PKC-DR cells in which Ca2+ oscillations were observed displayed responses characteristically different from the stereotypic Ca2+ oscillations in control cells. The oscillation frequency was reduced, and the spike widths were very substantially prolonged. The individual Ca2+ spike widths measured at half-peak height in phenylephrine-stimulated control cells were very consistent, with a mean value of 24 ± 0.6 s, whereas the spike durations in PKC-DR cells were almost 3-fold longer, with a width at half-peak height of 68 ± 3.3 s (Fig. 4E). We confirmed by Western blotting that treating cells overnight with phorbol ester leads to the down-regulation and degradation of the conventional, PKCα, and novel PKCϵ (phorbol ester-activated PKC isoenzymes in hepatocytes) without affecting the atypical isoform PKCζ1 (Fig. 4F).
PKC phosphorylation of the plasma membrane Ca2+ pump and of Orai channels has been reported (43, 44), indicating that PKC activity may regulate plasma membrane Ca2+ efflux and/or entry. Indeed, Orai1 has been shown to be basally phosphorylated by PKC, and inhibition of PKC leads to enhanced Ca2+ entry (44). To determine whether PKC-DR affects Ca2+ transport across the plasma membrane in hepatocytes, we measured Ca2+ influx and efflux rates in cells treated overnight with PMA or 4α-PMA (Fig. 5). ER Ca2+ stores were depleted with thapsigargin (Fig. 5, A–C) or ATP (to assess agonist-dependent effects on Ca2+ influx) (Fig. 5, D--F) in the absence of extracellular Ca2+ to induce Store-operated Ca2+ entry pathways. The PKC-DR protocol did not affect the rates of Ca2+ influx upon Ca2+ readdition (Fig. 5, B and E) or the rates of plasma membrane Ca2+ pump-mediated Ca2+ efflux from the cells after removal of extracellular Ca2+ (Fig. 5, C and F). These data suggest that PKC activity does not play a major role in regulating Ca2+ entry or Ca2+ extrusion at the plasma membrane in hepatocytes.
FIGURE 5.

Measurement of Ca2+ influx and efflux rates in control and PKC-DR hepatocytes. Hepatocytes were treated overnight with 4α-PMA (1 μm, Control) or PMA (1 μm, PKC-DR) and then loaded with Fura-2. Cultures were washed into Ca2+-free buffer prior to data acquisition. A—F, internal Ca2+ stores were depleted with thapsigargin (Thaps, 4 μm) (A–C) or by stimulation with the purinergic agonist ATP (200 μm) (D–F), followed by repletion of extracellular Ca2+ (2 mm) to initiate Ca2+ entry. Where indicated, the buffer was switch to Ca2+-free medium plus 5 mm BAPTA to stop Ca2+ influx and measure the rates of Ca2+ efflux from the cell. Ca2+ influx (B and E) and efflux (C and F) were plotted, and exponential rate constants (tau) were calculated using non-linear regression analysis. Similar results were obtained when the initial rates were measured (data not shown).
Negative feedback regulation by PKC on both GPCRs and PLC isoenzymes has been implicated in the regulation of Ca2+ oscillations (27, 31, 45). Therefore, we assessed the effect of PKC-DR on hormone-stimulated PLC activity and IP3 generation in hepatocytes using FRET-based molecular indicators. To monitor PLC activity, CFP and YFP proteins conjugated to PLCδ4PH domain were coexpressed. Hormone-stimulated PIP2 breakdown leads to a decrease in FRET between the CFP and YFP moieties, as described previously for CFP and YFP PLCδ1PH (46). The pleckstrin homology (PH) domain of PLCδ4 has a lower affinity for IP3 compared with PLCδ1, providing a more selective readout of PLC activity (PIP2 hydrolysis) over intracellular [IP3]. Dynamic changes in cytosolic [IP3] were determined with the IRIS-1 molecular probe containing a mutated version of the ligand binding domain of IP3R type 1 flanked by CFP and Venus (20, 36). PLC activity elicited by agonist stimulation (ATP, 200 μm) was potentiated in PKC-DR cells more than 2-fold compared with control 4α-PMA-treated cells (Fig. 6A). Similar effects on ATP-induced IP3 increases were also observed (Fig. 6B). Therefore, loss of PKC enhances PLC activity and increases the overall level of cellular [IP3], resulting in sustained and prolonged Ca2+ responses. These data indicate that negative feedback inhibition of IP3 generation by PKC is a key element in shaping agonist-induced Ca2+ oscillations.
FIGURE 6.

Down-regulation of PKC potentiates hormone-stimulated PLC activity and IP3 levels. Hepatocytes were transfected with eCFP-PH-PLCδ4 and eYFP-PH-PLCδ4 to monitor PIP2 levels (PLC activity) or IRIS-1 to monitor IP3 production (see “Experimental Procedures”) and then cultured overnight in the presence of 2 μm 4α-PMA (Control) or 2 μm PMA (PKC-DR). Cells were stimulated with ATP (200 μm), and maximal FRET changes were determined. A, representative traces showing the mean PLC response in control and PKC-DR cells. Traces are averaged from 27 and 25 cells, respectively, and are normalized to the basal FRET level (mean basal FRET values were 0.85 ± 0.011 for control and 1.028 ± 0.066 for PKC-DR cells, p = 0.033, Student's t test). B, mean peak FRET change (absolute values) ± S.E. for ≥70 cells from five independent experiments. **, p < 0.01, Student's t test. C, representative experiment showing the effect of PKC down-regulation on ATP-induced increases in IP3 levels for control and PKC-DR cells. Traces are averaged from five and four cells, respectively, and are normalized to the basal FRET level (mean basal FRET values were 0.66 ± 0.016 for control and 0.70 ± 0.013 for PKC-DR cells, p = 0.053, Student's t test). D, mean peak FRET change (absolute values) ± S.E. for ≥25 cells from 10 independent experiments. ***, p < 0.001, Student's t test.
Acute Effect of PKC Activation and Inhibition on Hormone-evoked Ca2+ Signaling
In view of results with PKC-DR, we investigated the effects of acute activation or inhibition of PKC on Ca2+ signals evoked by hormone. Hepatocytes were treated with phenylephrine at a dose that elicited repetitive Ca2+ oscillations (1–20 μm), and then the acute effects of PMA (1 nm) or BIM (5 μm) on the Ca2+ response was determined in each cell. Changes in Ca2+ oscillation frequency and Ca2+ spike width were calculated in hepatocytes that displayed continuous Ca2+ spiking for at least 5 min after application of drugs. Activation of PKC by PMA caused either a decrease in oscillation frequency (Fig. 7A, top panel) or a halt in oscillations (Fig. 7A, bottom panel). PMA treatment reduced the oscillation frequency in 32 ± 5% of the cell population and terminated the response in the remaining 68 ± 5%. This negative regulatory effect of PKC activation is consistent with the enhanced PLC/IP3 and Ca2+ responses observed in PKC-DR cells described above. However, counterintuitively, inhibition of PKC with BIM also decreased Ca2+ oscillation frequency. Following BIM treatment, the Ca2+ oscillation frequency was reduced in a majority of cells (63 ± 11% of cells, Fig. 7B, top panel), and there was a complete loss of Ca2+ oscillations in a smaller proportion of cells (37 ± 11% of cells, Fig. 7B, bottom panel).
Although the effects of PMA and BIM on the frequency of agonist-induced Ca2+ oscillations both manifest as frequency decreases, there were quantitative and qualitative differences in the responses to activation and inhibition of PKC with these agents. PMA treatment caused a 50% reduction in oscillation frequency (Fig. 7C) but only a small change in spike width (Fig. 7D). By contrast, BIM caused a modest 20% reduction in Ca2+ oscillation frequency (Fig. 7C) but dramatically prolonged the duration of the Ca2+ spikes (Fig. 7D). Furthermore, comparison of the effects of PMA and BIM revealed qualitative differences with respect to the termination of the Ca2+ oscillations. PKC activation with PMA caused an abrupt termination of the response or one to three blunted Ca2+ spikes prior to cessation, as shown in Fig. 7A, bottom panel. The termination of Ca2+ oscillations following PKC inhibition with BIM was quite different. There was a final sustained or peak/plateau Ca2+ increase (Fig. 7B) similar to those typically observed with a maximum hormone dose (1, 47, 48). Therefore, the effects of PKC inhibition with BIM are compatible with the enhanced Ca2+ signaling observed with PKC-DR. There is a broadening of the Ca2+ oscillations and shift from oscillatory to sustained Ca2+ signals (compare Fig. 7B with Fig. 4). Moreover, the apparent reduction in Ca2+ oscillation frequency with BIM can be ascribed to the prolongation of the Ca2+ spike widths because the interspike interval actually decreased from 56.7 ± 5.7 s to 39.6 ± 4.6 s (p < 0.01) after BIM addition. The decreased Ca2+ oscillation frequency and complete termination of Ca2+ signals with PMA treatment is also consistent with the PKC-DR data, where negative feedback effects of PKC are ablated.
We also compared the effects of PMA, BIM, and PKC-DR on total [3H]inositol phosphate accumulation (Fig. 7E). PKC-DR and acute BIM treatment both potentiated inositol phosphate accumulation in comparison with vasopressin alone. This corroborates our single-cell Ca2+ and IP3 imaging data, indicating that elimination of PKC activity results in elevated IP3 generation because of loss of negative feedback. At the single cell level, acute PMA treatment reduced Ca2+ oscillation frequency (Fig. 7C), but there was no comparable effect on [3H]inositol phosphate accumulation (Fig. 7E). This finding may reflect multiple opposing effects of PKC, including negative regulation of PLC activation and positive regulation of IP3 5-phosphatase (49), because the assay measures total inositol phosphate formation in the presence of Li+. This assay was used because the low sensitivity of the [3H]inositol labeling approach in hepatocytes precludes measurement of individual IP3 isomers. Taken together, the data described above demonstrate that acute inhibition of PKC or PKC-DR eliminates an important negative feedback pathway at the level of hormone-stimulated PLC activity and/or IP3 metabolism, leading to sustained or significantly broader Ca2+ transients. Consistent with this, acute activation of PKC blunts hormone-induced Ca2+ responses because of activation of these negative feedback loops. Therefore, PKC acts at multiple targets to modulate the frequency and shape of hormone-induced Ca2+ oscillations.
Effects of PKC on Ca2+ Oscillations Induced by Uncaging IP3
To further elucidate targets of PKC, we examined the effect of PKC-DR and acute activation or inhibition of PKC on Ca2+ oscillations triggered by uncaging IP3. Significantly, comparison of IP3-induced Ca2+ transients in control 4α-PMA-treated cells and PKC-DR cells (representative traces are shown in Fig. 8A) revealed no differences in the Ca2+ signals elicited by increasing exposure to UV. PKC-DR had no effect on the proportion of cells responding (Fig. 8B) or the type of Ca2+ signature observed (Fig. 8C). Furthermore, no significant effects were observed on Ca2+ oscillation frequency (Fig. 8D) or Ca2+ spike width (Fig. 8E). Therefore, elimination of phorbol ester-sensitive PKC activity through PKC down-regulation does not affect IP3R function in the absence of hormone.
FIGURE 8.

Effects of PKC on IP3 release-induced Ca2+ oscillations. A–E, hepatocytes were treated overnight with 4α-PMA (1 μm, Control) or PMA (1 μm, PKC-DR) then loaded with caged IP3 and Fluo-4. Shown are representative traces of single control and PKC-DR hepatocytes stimulated with increasing UV light flashes (A), the percentage of cells responding to one, two, and three UV light flashes (B), the percentage of cells with no response, single spike, oscillations, or saturated (peak/plateau) Ca2+ responses (C), the oscillation frequency (D), and the spike width half-peak height (E). F—H, overnight cultured hepatocytes loaded with caged IP3 and Fluo-4 were treated with PMA (1 μm), 4α-PMA (1 μm), or BIM (5 μm) for 5 min prior to IP3 uncaging (F) representative traces. Data summarize (G) the percentage of cells responding to two and four UV light flashes and (H) the oscillation frequency after four UV flashes. Data are mean ± S.E. from ≥25 cells from four independent experiments. *, p < 0.05, analysis of variance.
Acute treatment of cells with PMA or BIM (representative traces are shown in Fig. 8F) was also without effect on the proportion of cells responding to photorelease of caged IP3 (Fig. 8G) or the Ca2+ spike width for oscillations resulting from IP3 uncaging (data not shown). However, PKC activation with PMA causes a 2-fold increase in the Ca2+ oscillation frequency elicited by photo-released IP3, from 1.1 ± 0.26 spikes min−1 in control cells to 2.35 ± 0.30 min−1 in PMA-treated cells (Fig. 8H). This is in clear contrast to the inhibitory effect of PMA to reduce the frequency of phenylephrine-induced Ca2+ oscillations (Fig. 7C). This result suggests that there is a direct modulation of IP3Rs by PKC, which enhances channel activity and excitability. With global activation of PKC by PMA, the negative feedback mediated by PLC inhibition presumably predominates during hormone stimulation, whereas the positive feedback effect on IP3-induced Ca2+ release becomes apparent during IP3 uncaging when PLC is not activated. Therefore, under physiological conditions, activation of PKC by specific hormone receptors may differentially target negative feedback regulation of IP3 generation (or degradation) and positive feedback on Ca2+ release to shape the resulting Ca2+ transients.
PKC Activity Modulates Ca2+ Wave Velocity in Response to Both Hormone and Photorelease of Caged IP3
In the liver, hepatocyte and whole organ function is regulated not only by Ca2+ spike frequency but by Ca2+ wave propagation across individual cells (intracellular waves) and between cells (intercellular waves) within the liver lobule (5, 50–52). The propagation rates for hormone-induced intracellular Ca2+ waves are fixed over a wide range of agonist doses (52, 53). This lack of dependence on stimulus strength has led to the assumption that Ca2+ wave propagation is driven by a saltatory CICR processes (5, 54). However, we reported recently that the cytosolic expression of an intracellular IP3 buffer slows Ca2+ wave velocity in a stimulus strength-dependent fashion (20). Those findings are consistent with a role for regeneration of IP3 via positive feedback of Ca2+ on PLC, either globally or locally, which yields a cross-coupling between IP3 and Ca2+ that maximizes the CICR process, leading to stereotypic waves of IP3R activation.
In this study, we examined whether PKC has the potential to regulate Ca2+ wave propagation rates in addition to Ca2+ oscillation frequency and kinetics. First we examined the effect of PKC activation and down-regulation on intracellular Ca2+ waves elicited by IP3 uncaging. As shown in Fig. 9A, acute activation of PKC with PMA led to a 2-fold increase in the rates of Ca2+ waves elicited by photorelease of IP3 (16 ± 3.8 μm/s in control 4α-PMA treated cells versus 35.5 ± 5.4 μm/s after PMA treatment). This enhancement of Ca2+ wave propagation likely reflects the potentiation of IP3R activity that also manifests as an increased Ca2+ oscillation frequency during IP3 uncaging (Fig. 8F). As with IP3-induced Ca2+ oscillations, the effect of PKC down-regulation on Ca2+ wave propagation was not significant (Fig. 9B), presumably because there is little basal activity even without PKC-DR in the absence of hormone stimulation.
FIGURE 9.
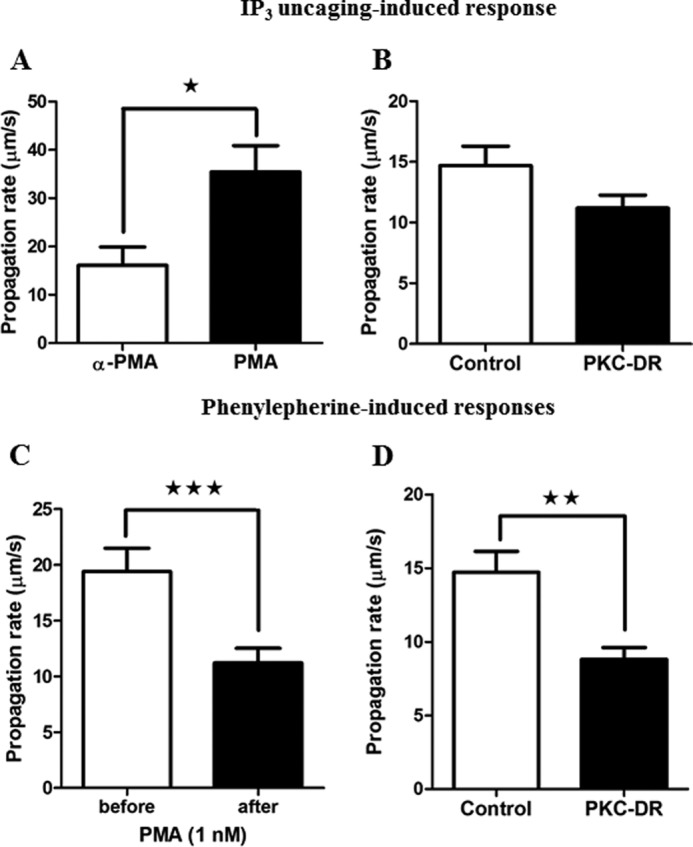
Ca2+ wave velocity is regulated by PKC activity. Isolated hepatocytes were treated overnight with 4α-PMA (1 μm, Control) or PMA (1 μm, PKC-DR) or treated acutely with 1 nm PMA to assess the effect of PKC on Ca2+ waves initiated by phenylephrine or caged IP3. Ca2+ wave propagation rates were calculated in micrometers per second by determining time at half-peak height from regions of interest from the wave initiation site and the opposite pole of the hepatocyte. A and B, the effect of acute PKC activation (1 μm PMA, A) and PKC-DR down-regulation (B) on caged IP3 (three UV flashes) induced Ca2+ wave velocity (micrometers per second ± S.E. for ≥16 cells from three independent experiments). C and D, the effect of acute PKC activation (1 nm) (C) and PKC-DR (D) on Ca2+ wave propagation rate in response to phenylephrine. Data are mean wave velocity (micrometers per second) ± S.E. for ≥18 cells from four independent experiments and from ≥20 cells from three independent experiments, respectively. *, p < 0.05; **, p < 0.01; ***, p < 0.001; Student's t test.
The effect of PKC on hormone-induced Ca2+ waves is more complex because it affects both IP3 generation and IP3R function. In phenylephrine-stimulated hepatocytes, acute activation of PKC with PMA caused a decrease in Ca2+ wave velocity from a control rate of 19.4 ± 2.0 μm/s to 11.2 ± 1.3 μm/s (Fig. 9C). The negative effect of acute PMA on Ca2+ wave velocity presumably results from suppression of IP3 production via enhanced negative feedback inhibition at the level of the hormone receptor or PLC. Therefore, the inhibitory effect of PMA predominates just as it does for the generation of Ca2+ oscillations (Fig. 7). However, despite the fact that PKC-DR prevents this inhibitory effect on PLC activation and greatly enhances IP3 generation, its predominant effect at the level of Ca2+ waves was also to slow the rate of propagation. The wave propagation rate in control 4α-PMA treated cells was 14.7 ± 1.4 μm/s, and this decreased to 8.8 ± 0.8 μm/s after PKC down-regulation (Fig. 9D). Therefore, in the presence of hormone, the effects of PKC-DR are also manifest in a reduced level of IP3R excitability. This provides further evidence that hormone-activated PKC positively regulates Ca2+ release and wave propagation by enhancing IP3R function, either directly or indirectly. Taken together, these data demonstrate that PKC activation during hormone stimulation of the GPCR/PLC signaling system has positive (targeting the IP3R) and negative feedback (IP3 generation and metabolism) mechanisms that regulate Ca2+ spike width, oscillation frequency, and wave velocity.
Discussion
IP3-dependent Ca2+ oscillations and waves are a major class of Ca2+ signals, and understanding the mechanisms that drive the oscillatory behavior and shape the kinetics of individual Ca2+ spikes is key to elucidating how Ca2+-regulated targets are modulated. There is a substantial stochastic component to IP3R-dependent Ca2+ oscillations (55), but the Ca2+ responses to different hormones have distinct stereotypic shapes with hormone-specific kinetic properties in hepatocytes (1, 34). Therefore, there must be further deterministic regulation of the Ca2+ signaling machinery beyond IP3R isoform expression and subcellular distribution. A combination of modeling and experimental data demonstrate that hormone-induced Ca2+ oscillations in hepatocytes depend on positive feedback of Ca2+ on PLC and consequent cross-coupling of Ca2+ and IP3 oscillations (17, 20).
In this study, we characterized Ca2+ responses induced by IP3 uncaging in hepatocytes and show that, in the absence of a GPCR ligand, Ca2+ oscillations are driven by CICR and do not require PLC activation. This is on the basis of a number of lines of evidence. First, PLC inhibition failed to suppress Ca2+ oscillations elicited by direct release of IP3. Second, graded steps of IP3 uncaging with increasing numbers of UV flashes in the absence of hormone resulted in stimulus strength-dependent effects on Ca2+ spike amplitude, width, and wave velocity. This is in clear contrast to the constant Ca2+ spike amplitude and kinetic properties for GPCR-dependent Ca2+ oscillations and waves over a wide range of hormone doses (1, 5, 6, 9, 56). Third, Ca2+ spike widths elicited by caged IP3 were of substantially shorter duration than hormone-induced Ca2+ oscillations reported in this study and previously (1, 9). Significantly, these data demonstrate that, although Ca2+ oscillations can be generated by CICR at the IP3R independent of PLC activity, this is not sufficient to recapitulate the oscillatory Ca2+ signals elicited by hormones. Regenerative PLC activation and cyclical fluctuations in IP3 levels are essential features of hormone-generated baseline-separated Ca2+ oscillations. The ability to compare IP3 uncaging with GPCR-generated Ca2+ signals has enabled us to further dissect how Ca2+ oscillations are shaped and regulated.
It has been suggested that hormone-induced Ca2+ oscillations may rely in part on positive Ca2+ feedback regulation of the Ca2+ sensitive, but hormone-insensitive, PLC isoforms, i.e. δ and η (57, 58). However, we found that global Ca2+ increases induced by photolysis of caged IP3 do not increase PLC activity in hepatocytes, even though this causes similar [Ca2+]i increases to those observed with hormone, in a range (0.1–10 μm) sufficient to activate PLCδ isoforms (59). On the basis of these data, we conclude that GPCR stimulation is a prerequisite for regenerative PLC activation and, presumably, depends on the PLCβ isoforms.
We found that photorelease of IP3 caused Ca2+ oscillations with similar frequency in the presence or absence of extracellular Ca2+. This provides evidence that plasma membrane Ca2+ entry pathways and the associated refilling of intracellular Ca2+ stores is not an intrinsic component of IP3-dependent Ca2+ oscillations in hepatocytes (39). In addition, these data suggest that the Ca2+ filling state of the ER does not determine Ca2+ oscillation frequency in hepatocytes. Nevertheless, we observed a reduction in Ca2+ spike width in the absence of Ca2+ entry, providing evidence that store-operated Ca2+ entry can play a role in shaping Ca2+ transients.
PKC isoenzymes are key mediators of GPCR/PLC signaling, acting to decode complex spatiotemporal Ca2+ changes and regulate cell function (31, 32). However, because many of the proteins involved in generating Ca2+ signals are also PKC substrates, this family of enzymes may also dynamically regulate Ca2+ signaling (25, 29). Indeed, multiple and sometimes opposing effects of PKC on PLC, IP3, and Ca2+ release are highlighted in this study, revealing targets both upstream and downstream of IP3 generation. Down-regulation of phorbol ester-sensitive PKC isoforms had the most dramatic effect on the hormone-induced Ca2+ oscillations, potentiating PLC activity and the intracellular levels of IP3 and Ca2+.
The effects of acute PKC inhibition with BIM were similar to PKC-DR, evoking broader Ca2+ spike widths and maximal Ca2+ responses in the presence of hormone. By contrast, inhibition or elimination of PKC activity had no effect on the Ca2+ responses elicited by direct photorelease of caged IP3. These data demonstrate a fundamental role of PKC in the termination of Ca2+ transients via negative feedback regulation of IP3 levels. Indeed, differences in the declining phase of each Ca2+ spike during Ca2+ oscillations elicited by activation of distinct GPCRs (1, 6) may reflect differential sensitivity to PKC or specific pools of PKC associated with each hormone receptor type (61).
The effects of acute PKC activation were more complex. PMA treatment modestly decreased Ca2+ oscillation frequency and spike width in the presence of hormone, whereas the frequency of oscillations after direct release of IP3 was increased. These data can be explained by dual opposing actions of PKC to suppress IP3 generation while enhancing IP3R activity. Most interesting is our observation that PKC down-regulation decreases Ca2+ wave velocity in the presence of hormone, despite increasing IP3 generation. Although these results may appear contradictory, they can also be explained by the dual actions of PKC to inhibit IP3 generation and enhance IP3-induced Ca2+ release. Specifically, even though PKC-DR suppresses the negative feedbacks that limit IP3 generation, allowing for more prolonged Ca2+ release in response to hormone, PKC-DR also eliminates the positive actions of PKC to enhance IP3R excitability and, thereby, slows Ca2+ wave propagation. This is supported by the very different effects of PKC-DR and PMA on the velocity of Ca2+ waves initiated by photorelease of caged IP3. Therefore, PKC-DR has no effect on IP3-induced Ca2+ waves because there is no role for negative feedback of PKC on IP3 generation and no sensitization of the IP3R (this would require PLC activation and diacylglycerol generation). Similarly, PMA dramatically enhances IP3-induced Ca2+ waves because it directly sensitizes the IP3R but has no negative feedback effect on IP3 generation. This modulation of Ca2+ wave propagation rates by PKC action on IP3R sensitivity provides an important, hitherto unrecognized, level of regulation of intracellular Ca2+ signaling.
Taken together, the data presented here show that PKC regulates multiple and sometimes counteracting steps in the IP3-dependent Ca2+ signaling pathway. Our data identify a number of potential PKC targets capable of Ca2+ signal modulation, but further work is required to elucidate which PKC isoforms regulate each target and whether these are cell type/receptor-specific. Translocation of GFP-tagged PKC isoenzymes have provided some insight into receptor-specific effects (62) or differential subcellular distributions of the enzymes upon hormone stimulation (60). However, whether the endogenous PKCs behave in a similar fashion or whether overexpressed PKC protein buffers cellular responses leave the data open to interpretation.
We conclude that, in the presence of sufficient cytosolic IP3, Ca2+ oscillations and waves can be generated in hepatocytes simply by biphasic regulation of the IP3R by Ca2+. However, at physiologically relevant hormone levels, Ca2+ oscillations depend on positive feedback of Ca2+ on PLCβ and are driven by cross-coupling between Ca2+ and IP3, and these elements of the Ca2+ signaling pathway can be specifically tuned and modulated by PKC. Therefore, physiological activation and deactivation of different PKC isoforms with distinct temporal and spatial profiles has the ability to profoundly shape Ca2+ oscillation kinetics, wave propagation rates, and the balance between positive and negative feedback mechanisms.
Author Contributions
A. P. T., P. J. B., and L. D. G. conceived and designed the study. P. J. B. wrote the manuscript. P. J. B. and W. M. designed and performed experiments and analyzed data. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK082954 and AI099277 (to A. P. T.). This work was also supported by the Thomas P. Infusion Endowed Chair (to A. P. T.). The authors declare that they have no conflicts of interest with the contents of this article.
- PLC
- phospholipase C
- GPCR
- G protein-coupled receptor
- PIP2
- phosphatidylinositol bisphosphate
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- inositol 1,4,5-trisphosphate receptor
- ER
- endoplasmic reticulum
- CICR
- Ca2+-induced Ca2+ release
- CFP
- cyan fluorescent protein
- YFP
- yellow fluorescent protein
- PMA
- phorbol-12-myristate-13-acetate
- BIM
- bisindolylmaleimide I
- DR
- down-regulation
- PH
- pleckstrin homology.
References
- 1. Rooney T. A., Sass E. J., Thomas A. P. (1989) Characterization of cytosolic calcium oscillations induced by phenylephrine and vasopressin in single fura-2-loaded hepatocytes. J. Biol. Chem. 264, 17131–17141 [PubMed] [Google Scholar]
- 2. Bootman M. D., Berridge M. J., Lipp P. (1997) Cooking with calcium: the recipes for composing global signals from elementary events. Cell 91, 367–373 [DOI] [PubMed] [Google Scholar]
- 3. Berridge M. J., Lipp P., Bootman M. D. (2000) The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 1, 11–21 [DOI] [PubMed] [Google Scholar]
- 4. Thomas A. P., Renard D. C., Rooney T. A. (1991) Spatial and temporal organization of calcium signalling in hepatocytes. Cell Calcium 12, 111–126 [DOI] [PubMed] [Google Scholar]
- 5. Gaspers L. D., Thomas A. P. (2005) Calcium signaling in liver. Cell Calcium 38, 329–342 [DOI] [PubMed] [Google Scholar]
- 6. Bartlett P. J., Gaspers L. D., Pierobon N., Thomas A. P. (2014) Calcium-dependent regulation of glucose homeostasis in the liver. Cell Calcium 55, 306–316 [DOI] [PubMed] [Google Scholar]
- 7. Irvine R. F. (2003) 20 years of Ins(1,4,5)P3, and 40 years before. Nat. Rev. Mol. Cell Biol. 4, 580–585 [DOI] [PubMed] [Google Scholar]
- 8. Bootman M. D., Young K. W., Young J. M., Moreton R. B., Berridge M. J. (1996) Extracellular calcium concentration controls the frequency of intracellular calcium spiking independently of inositol 1,4,5-trisphosphate production in HeLa cells. Biochem. J. 314, 347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thomas A. P., Bird G. S., Hajnóczky G., Robb-Gaspers L. D., Putney J. W., Jr. (1996) Spatial and temporal aspects of cellular calcium signaling. FASEB J. 10, 1505–1517 [PubMed] [Google Scholar]
- 10. Dupont G., Combettes L., Bird G. S., Putney J. W. (2011) Calcium oscillations. Cold Spring Harb. Perspect. Biol. 3, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sneyd J., Tsaneva-Atanasova K., Reznikov V., Bai Y., Sanderson M. J., Yule D. I. (2006) A method for determining the dependence of calcium oscillations on inositol trisphosphate oscillations. Proc. Natl. Acad. Sci. U.S.A. 103, 1675–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meyer T., Stryer L. (1988) Molecular model for receptor-stimulated calcium spiking. Proc. Natl. Acad. Sci. U.S.A. 85, 5051–5055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bezprozvanny I., Watras J., Ehrlich B. E. (1991) Bell-shaped calcium-response curves of lns(l,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 351, 751–754 [DOI] [PubMed] [Google Scholar]
- 14. Thurley K., Falcke M. (2011) Derivation of Ca2+ signals from puff properties reveals that pathway function is robust against cell variability but sensitive for control. Proc. Natl. Acad. Sci. U.S.A. 108, 427–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Young G. W., Keizer J. (1992) A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc. Natl. Acad. Sci. U.S.A. 89, 9895–9899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marchant J. S., Parker I. (2001) Role of elementary Ca(2+) puffs in generating repetitive Ca(2+) oscillations. EMBO J. 20, 65–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Politi A., Gaspers L. D., Thomas A. P., Höfer T. (2006) Models of IP3 and Ca2+ oscillations: frequency encoding and identification of underlying feedbacks. Biophys. J. 90, 3120–3133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Salazar C., Politi A. Z., Höfer T. (2008) Decoding of calcium oscillations by phosphorylation cycles: analytic results. Biophys. J. 94, 1203–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dupont G., Erneux C. (1997) Simulations of the effects of inositol 1,4,5-trisphosphate 3-kinase and 5-phosphatase activities on Ca2+ oscillations. Cell Calcium 22, 321–331 [DOI] [PubMed] [Google Scholar]
- 20. Gaspers L. D., Bartlett P. J., Politi A., Burnett P., Metzger W., Johnston J., Joseph S. K., Höfer T., Thomas A. P. (2014) Hormone-induced calcium oscillations depend on cross-coupling with inositol 1,4,5-trisphosphate oscillations. Cell Rep. 9, 1209–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dolmetsch R. E., Xu K., Lewis R. S. (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392, 933–936 [DOI] [PubMed] [Google Scholar]
- 22. Li W., Llopis J., Whitney M., Zlokarnik G., Tsien R. Y. (1998) Cell-permeant caged InsP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature 392, 936–941 [DOI] [PubMed] [Google Scholar]
- 23. Zhu L., Song S., Pi Y., Yu Y., She W., Ye H., Su Y., Hu Q. (2011) Cumulated Ca2+ spike duration underlies Ca2+ oscillation frequency-regulated NFκB transcriptional activity. J. Cell Sci. 124, 2591–2601 [DOI] [PubMed] [Google Scholar]
- 24. Newton A. C. (2001) Protein kinase C: structural and spatial regulation by phosphorylation, cofactors, and macromolecular interactions. Chem. Rev. 101, 2353–2364 [DOI] [PubMed] [Google Scholar]
- 25. Nash M. S., Young K. W., Challiss R. A., Nahorski S. R. (2001) Intracellular signalling. Receptor-specific messenger oscillations. Nature 413, 381–382 [DOI] [PubMed] [Google Scholar]
- 26. Young K. W., Nash M. S., Challiss R. A., Nahorski S. R. (2003) Role of Ca2+ feedback on single cell inositol 1,4,5-trisphosphate oscillations mediated by G-protein-coupled receptors. J. Biol. Chem. 278, 20753–20760 [DOI] [PubMed] [Google Scholar]
- 27. Strassheim D., Williams C. L. (2000) P2Y2 purinergic and M3 muscarinic acetylcholine receptors activate different phospholipase C-β isoforms that are uniquely susceptible to protein kinase C-dependent phosphorylation and inactivation. J. Biol. Chem. 275, 39767–39772 [DOI] [PubMed] [Google Scholar]
- 28. Foskett J. K., White C., Cheung K.-H., Mak D.-O. D. (2007) Inositol trisphosphate receptor Ca2+ release channels. Physiol. Rev. 87, 593–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vanderheyden V., Devogelaere B., Missiaen L., De Smedt H., Bultynck G., Parys J. B. (2009) Regulation of inositol 1,4,5-trisphosphate-induced Ca2+ release by reversible phosphorylation and dephosphorylation. Biochim. Biophys. Acta 1793, 959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Woodring P. J., Garrison J. C. (1997) Expression, purification, and regulation of two isoforms of the inositol 1,4,5-trisphosphate 3-kinase. J. Biol. Chem. 272, 30447–30454 [DOI] [PubMed] [Google Scholar]
- 31. Bartlett P. J., Young K. W., Nahorski S. R., Challiss R. A. (2005) Single cell analysis and temporal profiling of agonist-mediated inositol 1,4,5-trisphosphate, Ca2+, diacylglycerol, and protein kinase C signaling using fluorescent biosensors. J. Biol. Chem. 280, 21837–21846 [DOI] [PubMed] [Google Scholar]
- 32. Oancea E., Meyer T. (1998) Protein kinase C as a molecular machine for decoding calcium and diacylglycerol signals. Cell 95, 307–318 [DOI] [PubMed] [Google Scholar]
- 33. Berrie C. P., Cobbold P. H. (1995) Both activators and inhibitors of protein kinase C promote the inhibition of phenylephrine-induced [Ca2+]i oscillations in single intact rat hepatocytes. Cell Calcium 18, 232–244 [DOI] [PubMed] [Google Scholar]
- 34. Sanchez-Bueno A., Dixon C. J., Woods N. M., Cuthbertson K. S., Cobbold P. H. (1990) Inhibitors of protein kinase C prolong the falling phase of each free-calcium transient in a hormone-stimulated hepatocyte. Biochem. J. 268, 627–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hajnóczky G., Thomas A. P. (1997) Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO J. 16, 3533–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mikoshiba K. (2007) IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J. Neurochem. 102, 1426–1446 [DOI] [PubMed] [Google Scholar]
- 37. Dakin K., Li W.-H. (2007) Cell membrane permeable esters of d-myo-inositol 1,4,5-trisphosphate. Cell Calcium. 42, 291–301 [DOI] [PubMed] [Google Scholar]
- 38. Rooney T. A., Thomas A. P. (1991) Organization of intracellular calcium signals generated by inositol lipid-dependent hormones. Pharmacol. Ther. 49, 223–237 [DOI] [PubMed] [Google Scholar]
- 39. Smyth J. T., Hwang S. Y., Tomita T., DeHaven W. I., Mercer J. C., Putney J. W. (2010) Activation and regulation of store-operated calcium entry. J. Cell. Mol. Med. 14, 2337–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Putney J. W., Bird G. S. (2008) Cytoplasmic calcium oscillations and store-operated calcium influx. J. Physiol. 586, 3055–3059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamasaki-Mann M., Parker I. (2011) Enhanced ER Ca2+ store filling by overexpression of SERCA2b promotes IP3-evoked puffs. Cell Calcium. 50, 36–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fraiman D., Dawson S. P. (2004) A model of the IP3 receptor with a luminal calcium binding site: stochastic simulations and analysis. Cell Calcium. 35, 403–413 [DOI] [PubMed] [Google Scholar]
- 43. Brini M., Carafoli E. (2011) The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 3, a004168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kawasaki T., Ueyama T., Lange I., Feske S., Saito N. (2010) Protein kinase C-induced phosphorylation of Orai1 regulates the intracellular Ca2+ level via the store-operated Ca2+ channel. J. Biol. Chem. 285, 25720–25730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dupont G., Lokenye E. F., Challiss R. A. (2011) A model for Ca2+ oscillations stimulated by the type 5 metabotropic glutamate receptor: an unusual mechanism based on repetitive, reversible phosphorylation of the receptor. Biochimie 93, 2132–2138 [DOI] [PubMed] [Google Scholar]
- 46. van der Wal J., Habets R., Várnai P., Balla T., Jalink K. (2001) Monitoring agonist-induced phospholipase C Activation in live cells by fluorescence resonance energy transfer. J. Biol. Chem. 276, 15337–15344 [DOI] [PubMed] [Google Scholar]
- 47. Hajnóczky G., Robb-Gaspers L. D., Seitz M. B., Thomas A. P. (1995) Decoding of cytosolic calcium oscillations in the mitochondria. Cell 82, 415–424 [DOI] [PubMed] [Google Scholar]
- 48. Robb-Gaspers L. D., Rutter G. A., Burnett P., Hajnóczky G., Denton R. M., Thomas A. P. (1998) Coupling between cytosolic and mitochondrial calcium oscillations: role in the regulation of hepatic metabolism. Biochim. Biophys. Acta 1366, 17–32 [DOI] [PubMed] [Google Scholar]
- 49. Connolly T. M., Lawing W. J., Jr., Majerus P. W. (1986) Protein kinase C phosphorylates human platelet inositol trisphosphate 5′-phosphomonoesterase, increasing the phosphatase activity. Cell 46, 951–958 [DOI] [PubMed] [Google Scholar]
- 50. Nathanson M. H., Burgstahler A. D., Mennone A., Fallon M. B., Gonzalez C. B., Saez J. C. (1995) Ca2+ waves are organized among hepatocytes in the intact organ. Am. J. Physiol. Gastrointest. Liver Physiol. 269, G167–G171 [DOI] [PubMed] [Google Scholar]
- 51. Tordjmann T., Berthon B., Jacquemin E., Clair C., Stelly N., Guillon G., Claret M., Combettes L. (1998) Receptor-oriented intercellular calcium waves evoked by vasopressin in rat hepatocytes. EMBO J. 17, 4695–4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robb-Gaspers L. D., Thomas A. P. (1995) Coordination of Ca signaling by intercellular propagation of Ca waves in the intact liver. J. Biol. Chem. 270, 8102–8107 [DOI] [PubMed] [Google Scholar]
- 53. Rooney T. A., Sass E. J., Thomas A. P. (1990) Agonist-induced cytosolic calcium oscillations originate from a specific locus in single hepatocytes. J. Biol. Chem. 265, 10792–10796 [PubMed] [Google Scholar]
- 54. Nathanson M. H., Burgstahler A. D., Fallon M. B. (1994) Multistep mechanism of polarized Ca2+ wave patterns in hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 267, G338–G349 [DOI] [PubMed] [Google Scholar]
- 55. Thurley K., Tovey S. C., Moenke G., Prince V. L., Meena A., Thomas A. P., Skupin A., Taylor C. W., Falcke M. (2014) Reliable encoding of stimulus intensities within random sequences of intracellular Ca2+ spikes. Sci. Signal. 7, ra59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rooney T. A., Renard D. C., Sass E. J., Thomas A. P. (1991) Oscillatory cytosolic calcium waves independent of stimulated inositol 1,4,5-trisphosphate formation in hepatocytes. J. Biol. Chem. 266, 12272–12282 [PubMed] [Google Scholar]
- 57. Banno Y., Okano Y., Nozawa Y. (1994) Thrombin-mediated phosphoinositide hydrolysis in Chinese hamster ovary cells overexpressing phospholipase C-δ 1. J. Biol. Chem. 269, 15846–15852 [PubMed] [Google Scholar]
- 58. Kim J. K., Choi J. W., Lim S., Kwon O., Seo J. K., Ryu S. H., Suh P.-G. (2011) Phospholipase C-η1 is activated by intracellular Ca2+ mobilization and enhances GPCRs/PLC/Ca2+ signaling. Cell. Signal. 23, 1022–1029 [DOI] [PubMed] [Google Scholar]
- 59. Allen V., Swigart P., Cheung R., Cockcroft S., Katan M. (1997) Regulation of inositol lipid-specific phospholipase cδ by changes in Ca2+ ion concentrations. Biochem. J. 327, 545–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Collazos A., Diouf B., Guérineau N. C., Quittau-Prévostel C., Peter M., Coudane F., Hollande F., Joubert D. (2006) A spatiotemporally coordinated cascade of protein kinase C activation controls isoform-selective translocation. Mol. Cell. Biol. 26, 2247–2261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shin D. M., Luo X., Wilkie T. M., Miller L. J., Peck A. B., Humphreys-Beher M. G., Muallem S. (2001) Polarized expression of G protein-coupled receptors and an all-or-none discharge of Ca2+ pools at initiation sites of [Ca2+] i waves in polarized exocrine cells. J. Biol. Chem. 276, 44146–44156 [DOI] [PubMed] [Google Scholar]
- 62. Uchino M., Sakai N., Kashiwagi K., Shirai Y., Shinohara Y., Hirose K., Iino M., Yamamura T., Saito N. (2004) Isoform-specific phosphorylation of metabotropic glutamate receptor 5 by protein kinase C (PKC) blocks Ca2+ oscillation and oscillatory translocation of Ca2+-dependent PKC. J. Biol. Chem. 279, 2254–2261 [DOI] [PubMed] [Google Scholar]