

Background: The use of nickases may avoid toxicity associated with the NHEJ repair pathway in nuclease-induced gene targeting.
Results: Decoupling the action of the I-DmoI catalytic residues acting on each strand generates a nickase.
Conclusion: An I-DmoI variant was developed for cleaving one of the strands of its DNA target.
Significance: This is the first structure of an engineered nickase in an LAGLIDADG scaffold.
Keywords: genetics, protein engineering, protein-DNA interaction, protein-nucleic acid interaction, x-ray crystallography, gene targeting, nucleases
Abstract
Homing endonucleases are useful tools for genome modification because of their capability to recognize and cleave specifically large DNA targets. These endonucleases generate a DNA double strand break that can be repaired by the DNA damage response machinery. The break can be repaired by homologous recombination, an error-free mechanism, or by non-homologous end joining, a process susceptible to introducing errors in the repaired sequence. The type of DNA cleavage might alter the balance between these two alternatives. The use of “nickases” producing a specific single strand break instead of a double strand break could be an approach to reduce the toxicity associated with non-homologous end joining by promoting the use of homologous recombination to repair the cleavage of a single DNA break. Taking advantage of the sequential DNA cleavage mechanism of I-DmoI LAGLIDADG homing endonuclease, we have developed a new variant that is able to cut preferentially the coding DNA strand, generating a nicked DNA target. Our structural and biochemical analysis shows that by decoupling the action of the catalytic residues acting on each strand we can inhibit one of them while keeping the other functional.
Introduction
Homing endonucleases (HEs),4 also known as meganucleases, are highly sequence-specific enzymes that recognize and cleave long DNA targets (12–45 bp), generating double strand breaks (DSBs). Because of their high specificity, HEs have a low cleavage frequency even in complex genomes, making them powerful tools for genome manipulation in mammalian and plants cells (1, 2). These DSBs can be processed in a conservative way by homologous recombination or in a less accurate manner by non-homologous end joining, which usually results in insertions or deletions leading to genomic instability (3). Recent studies have shown that a single strand nick might stimulate homologous recombination repair (4–7), suggesting that the error-prone non-homologous end joining can be efficiently avoided by customized “nickases” producing single strand breaks, thus reducing the toxicity associated with genome modification. Nickases are used in nature for different natural DNA editing processes (8), but they can be generated artificially by protein engineering on the catalytic sites of zinc finger nucleases, transcription activator-like effector nucleases, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated (CAS), and HEs.
HEs have been classified in five families, the largest of which contains the conserved LAGLIDADG sequence motif (9). The three-dimensional structures of several LAGLIDADG homing endonucleases (LHEs) indicate that these proteins adopt a similar active conformation as homodimers or as monomers with two separate domains. LHEs that contain a single copy of this motif, such as I-CreI and I-CeuI, act as homodimers and recognize a nearly palindromic DNA target. Conversely, LHEs containing two copies, such as I-DmoI, I-SceI, and PI-SceI, act as monomers with two domains, each bearing an LAGLIDADG motif, that recognize non-palindromic DNAs. The interface between the two domains forms the catalytic center, which is composed of two sites, each responsible for cleaving one DNA strand (10).
The I-DmoI catalytic center has two acidic residues located at the end of the two LAGLIDADG α-helixes: Asp-21 (from domain A) and Glu-117 (from domain B) that define two different metal sites (sites A and B, respectively) for cleaving each DNA strand. These sites are separated and need the entrance of a third metal in a central position in the catalytic center (site C) to cleave the DNA strands in a sequential manner by a two-metal ion mechanism (10, 11) (see Fig. 1). In a recent study, we have reported the seven different catalytic intermediates of the I-DmoI cleavage reaction (10). In the present study, we used this information to generate a nickase variant on the I-DmoI scaffold.
FIGURE 1.

I-DmoI active site. A, detailed scheme of the I-DmoI active site at the reactant state (Protein Data Bank code 4UN9) including the 2Fo − Fc map contoured at 1.2σ. The sequence of the DNA target is shown below. Red arrowheads indicate the cleavage sites. B, scheme of the active center contacts. Bases are colored in gray or white depending on whether they contact the protein amino acids or not, respectively. The red arrowheads indicate the positions of the targeted phosphodiester bonds (orange). Catalytic waters are shown in magenta. C, schematic diagram summarizing the catalytic mechanism. The color key is the same as in A. Metal ion occupancy at site C is depicted by the color intensity (brown), revealing its crucial role in catalysis, triggering the consecutive hydrolysis of the targeted phosphodiester bonds in DNA strands and leaving its position once a double strand break is generated.
Experimental Procedures
Protein Expression, Purification, Protein·DNA Complex Formation, and Crystallization
I-DmoI wild type and mutant variants were cloned, expressed, and purified following earlier protocols (11, 12). Protein·DNA complexes were obtained as described in Redondo and co-workers (12), and their crystallization conditions were similar to the wild type I-DmoI·DNA complex crystal structure, ranging from 5 to 6% PEG 4000, 0.07 m sodium acetate, pH 4.6–5.5, and 30% glycerol. The DNA targets were purchased from Proligo and consisted of the following duplexes: 1) 5′-GCCTTGCCGGGTAAGTTCCGGCGCG-3′ and 5′-CGCGCCGGAACTTACCCGGCAAGGC-3′ (wild type; not nicked DNA); 2) 5′-GCCTTGCCGGGTAA-3′, 5′-GTTCCGGCGCG-3′, and 5′-CGCGCCGGAACTTACCCGGCAAGGC-3′ (nicked in strand A); and 3) 5′-CGCGCCGGAACTTACC-3′, 5′-CGGCAAGGC-3′, and 5′-GCCTTGCCGGGTAAGTTCCGGCGCG-3′ (nicked in strand B). Individual strands in Tris-EDTA buffer in the presence of 50 mm NaCl were combined in a 1:1 ratio and annealed by incubating at 95 °C for 10 min and slowly cooling to room temperature to form DNA duplexes. All of them form a 25-bp blunt end duplex after incubation. Sample concentrations were quantified by UV spectroscopy using the calculated extinction coefficients. Protein concentrations were further confirmed by Bradford assay. To discard that differences in mutant cleavage activity could arise from changes in protein stability or DNA binding affinities, the I-DmoI mutants were analyzed by circular dichroism, thermal denaturation, and binding to the 25-bp DNA target in fluorescence binding assays.
Biochemical and Biophysical Characterization of Proteins
Circular dichroism (CD) measurements were performed as described previously (13). Analytical gel filtration chromatography was done at room temperature with an ÄKTA FPLC system (GE Healthcare) using a SuperdexTM 200 10/300 GL column in 20 mm sodium phosphate buffer, pH 6.0 and 1 m NaCl. 100 μl of the I-DmoI protein at 0.3 mg/ml concentration was injected and eluted from the column at a flow rate of 0.2 ml/min. Mass determinations of intact proteins were performed (14), and the intact protein predominantly gave data obtained by deconvoluting the multiply charged ions (data not shown) using MagTran software v.1.02 kindly provided by Zhang and Marshall (15). The mass determination of dissolved crystals was done as described previously (14).
In Vitro DNA Cleavage Assays
Cleavage assays were performed as described previously (13) at 65 °C in 10 mm Tris-HCl, pH 8, 50 mm NaCl, 1–10 mm MgCl2 (or MnCl2), and 1 mm DTT in a 25-μl final volume reaction. Reactions were stopped after 30 min by addition of 5 μl of 45% glycerol, 95 mm EDTA, pH 8, 1.5% (w/v) SDS, 1.5 mg/ml Proteinase K, and 0.048% (w/v) bromphenol blue (6× Buffer Stop); incubated at 37 °C for 30 min; and electrophoresed in a 1% agarose gel (plasmid digestions) or 15% polyacrylamide gel (DNA oligonucleotide duplex digestions). Cleavage assays using nicked DNA targets were performed as described previously (11). Additional duplexes (purchased from Sigma as a racemic mixture) that possess the same 25-bp target sequence but a phosphorothioate bond in place of the scissile bond in the 5′-phosphate of 3GstrandA or the 5′-phosphate of −3CstrandB were used to measure strand cleavage accessibility.
Fluorescence Binding Assay
The dissociation constants of the I-DmoI variants for their cognate DNA were determined using the change in the intrinsic fluorescence intensity of the proteins as the protein·DNA complexes are formed. Binding reactions were performed by mixing 500 nm I-DmoI with various concentrations (0–2000 nm) of the 25-bp-long natural I-DmoI target duplex in binding buffer (10 mm Tris, pH 8, 50 mm NaCl, and 10 mm CaCl2) and incubating for 15 min at 50 °C. Fluorescence emission spectra were recorded at room temperature using a QuantaMaster QM-2000-7 model spectrofluorometer (Photon Technology International) with excitation fixed at 280 nm and emission in the 325–340-nm range. Each spectrum was the average of five consecutive scans. After normalization, the dissociation constants (KD) were determined by non-linear least square fitting (Prism, GraphPad) of the observed change in fluorescence at 334 nm to the following equation.
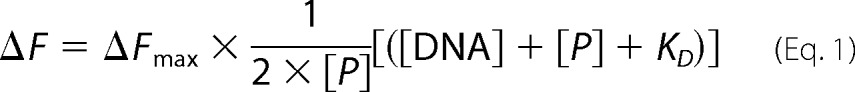 |
where ΔF is the change in protein fluorescence upon complex formation, ΔFmax is the maximum fluorescence change, [P] is the total protein concentration, and [DNA] is the total concentration of the DNA duplex. Three independent titration curves were carried out for each protein and used to determine the average fluorescence values and the standard errors at each titration point.
Data Collection, Structure Solution, Model Building, and Refinement
All data were collected at 100 K using synchrotron radiation at the XALOC beamline (ALBA, Barcelona, Spain) and PXI beamline (Swiss Light Source, Villigen, Switzerland). Diffraction data were recorded on ADSC-Q315r, Mar225 CCD, or Pilatus detectors depending on the beamline. Processing and scaling were accomplished with XDS (16) and Scala (17) software packages. Statistics for the crystallographic data and structure solution are summarized in Table 1. The structures were solved by molecular replacement as implemented in the program Phaser (18). The search models were based on Protein Data Bank codes 2VS7 (I-DmoI·DNA·Ca2+) and 2VS8 (I-DmoI·DNA·Mn2+). The structures were then subjected to iterative cycles of model building and refinement with Coot (19) and PHENIX (20).
TABLE 1.
Data collection and refinement statistics
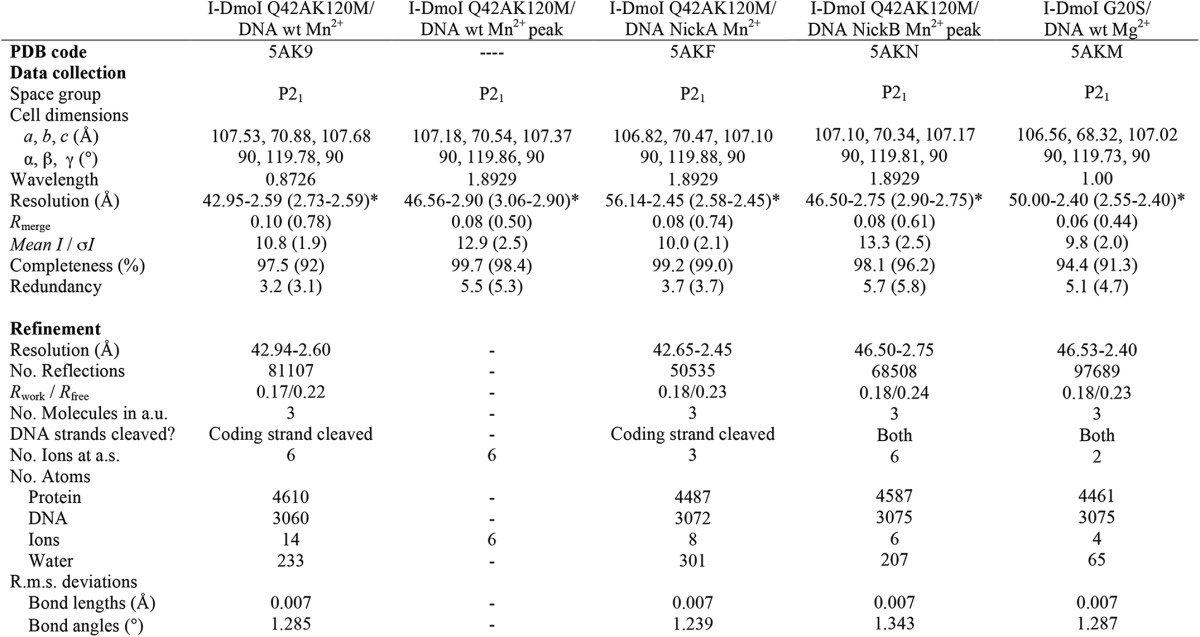
*, values in parentheses are for the highest resolution shell. One crystal was used to solve each structure. a.u., asymmetric unit; a.s., asymmetric surface; R.m.s., root mean square; PDB, Protein Data Bank; -, anomalous diffraction data not deposited.
Quantum Chemical Calculations
A reaction path connecting the pentacoordinate phosphorus state and the non-cleaved DNA for the I-DmoI wild type (Protein Data Bank code 2VS7), I-DmoI G20S (Protein Data Bank code 5AKM), and I-DmoI Q42A/K120M (Protein Data Bank code 5AK9) has been built following the same approach as in Muñoz et al. (21). The reaction paths, obtained by formation of the phosphorus bond with the oxygen of the deoxyribose and cleaving the hydroxyl-phosphorus bond, were calculated for both A and B strands. At each step, all the atoms not directly involved in the reaction were fully relaxed. The system was divided into two parts using ONIOM (22). The quantum mechanical part, formed by the phosphate group, the manganese ion, and its ligands, was modeled via density functional theory using the B3LYP functional and a 6–311G(d,p) basis set. The non-quantum mechanical part, including all the atoms within a distance of 8 Å from the reacting phosphate, interacts with the quantum mechanical subsystem by electronic embedding.
Molecular Dynamics Simulations
The molecular dynamics simulations of the I-DmoI wild type and I-DmoI Q42A/K120M mutant bound to DNA were performed starting from the corresponding crystallographic structures (Protein Data Bank codes 2VS8 and 5AK9, respectively). Version 4 of the molecular dynamics program GROMACS (23) with Amber99sb-ildn* force field (24) was used. After 10 ns of equilibration, a molecular dynamics simulation of 50 ns was performed using a time step of 2 fs. The temperature was kept constant at 343 K using the velocity rescaling algorithm (25). Every 2 ps, the electric field given by the atomic charge distribution was calculated and projected on the phosphorus-oxygen bond of the coding and non-coding strands.
Results
The I-DmoI Active Center
Time course strand cleavage assays showed that in I-DmoI the non-coding strand is cleaved first before the second phosphodiester hydrolysis takes place on the coding strand (10). The catalytic center contains the Asp-21 and Glu-117 residues defining metal sites A and B, respectively (11). Each residue binds a metal ion (Fig. 1A). In addition, we have identified a metal site (site C) that contacts the catalytic acid residues (Asp-21 and Glu-117), the 5′-phosphates of 3GstrandA and −3CstrandB, and the 3′-hydroxyl groups of the riboses of 2AstrandA and −2CstrandB (Fig. 1B).
The I-DmoI cleavage mechanism follows a determined cleavage sequence; the metal sites are occupied in a specific order allowing catalysis (Fig. 1C). Initially, site A is filled followed by site B, and finally a third metal ion enters the central location in site C (10). The metal ion in site C has a pivotal role in the reaction, allowing the proper geometry for two-metal ion catalysis and triggering the digestion of the DNA target. Thus, this metal in the central site has a role equivalent to that of the second metal site in the classical two-metal ion mechanism of phosphodiester bond catalysis. After phosphodiester bond cleavage, the metal in site C abandons the active center, and its position is occupied by a water molecule (10). The metal in site C does not seem to be responsible for the sequential order of cleavage. The active site pocket is lined with several basic residues that contact both DNA and solvent atoms. Thus a lysine residue is essential for proton transfer and solvent activation and does not participate in metal binding but instead is found in a basic pocket near the scissile phosphate and nucleophilic water (26). Crystallographic structure comparison allowed finding similar residues in other HEs (Table 2). The asymmetry between sites A and B in I-DmoI is defined by the presence of Lys-120 on site A. This residue coordinates the water molecule that attacks the scissile phosphate of the non-coding strand (10) (Fig. 1). Although no equivalent residue is found on site B (Table 2), a water molecule can be found at the right position for the second nucleophilic attack (10) (Fig. 1).
TABLE 2.
Summary of conserved motif and active site residues for monomeric LHE structures
PDB, Protein Data Bank; —, no equivalent residue found at this position.

Moreover, the use of phosphorothioates (27), a phosphate modification that affects the hydrolysis reaction, in the 5′-phosphate of 3GstrandA or the 5′-phosphate of −3CstrandB showed that generation of a DSB was much more affected when the modification was present on the coding strand than when it was included in the non-coding strand (Fig. 2), suggesting that the cleavage of the coding strand in site B could be decoupled from the non-coding strand cut in site A. Therefore, after analyzing these data, we searched for different strategies to generate I-DmoI variants that could cut only one of the target strands.
FIGURE 2.
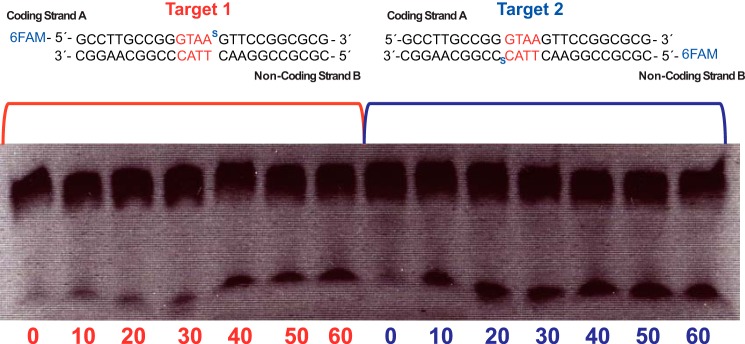
I-DmoI WT strand cleavage preference after phosphorothioate substitution. The figure displays time course titrations using phosphorothioate 6-fluorescein amidite (6FAM) labeling in coding strand A (Target 1) or coding strand B (Target 2). Oligonucleotides and I-DmoI at 750 nm were incubated at 65 °C from 0 to 60 min.
Mutations in Asp-21 and Glu-117
Initially, we attempted to inactivate the acidic metal binding residues in site A or site B. However, mutations such as D44A or D145A in I-SceI, another monomeric member of the LAGLIDADG family (28), and D21N or E117Q in I-DmoI (29) completely abolished the LHE DNA cleavage, thus inactivating the active center. These mutants revealed an intricate coordination between the active sites in the monomeric LHEs, indicating that the mutation of the catalytic residues will not yield nickase variants. This behavior was also observed in other mutants, D21A, D21G, E117A, and E117G, in I-DmoI that lead to totally inactive variants of the enzyme (Table 3). However, in contrast to previous reports (29), the D21N mutation generated a DSB in the target and low nickase activity using a plasmid containing I-DmoI target (Fig. 3A). We also checked whether changes in the length of the side chain of acidic residues could influence the catalysis. We generated the D21E, E117D, and D21E/E117D variants. Both D21E and D21E/E117D variants were inactive (Table 3), most likely due to the fact that site A cannot accommodate a glutamic side chain. However, I-DmoI E117D cleaves both DNA strands although at a reduced rate. These results are consistent with those of Silva and Belfort (30).
TABLE 3.
I-DmoI mutation list and cleavage activity on plasmid DNA
+, low cleavage; ++, medium cleavage; +++, high cleavage.
| I-DmoI mutations | No cleavage | Cleavage | Nick |
|---|---|---|---|
| I-DmoI WT | +++ | ||
| D21A | + | ||
| D21E | + | ||
| D21N | + | + | |
| E117A | + | ||
| E117D | + | ||
| D21E/E117D | + | ||
| Q42E | + | ++ | |
| K120M | + | + | |
| N129D | + | ||
| Q42E/K120M | + | ++ | |
| Q42A/K120M | + | +++ | |
| D21E/K120M | + | ++ | |
| D21E/Q42E/K120M | + | + | |
| D21N/Q42E/K120M | + | ||
| D21E/Q42A/K120M | + | ||
| D21N/Q42A/K120M | + | ||
| G20S | + | ++ | |
| G20S/Q42A/K120M | +++ | ||
| A116S | + | ||
| Q42A/A116S/K120M | + | ||
| G20S/E117D | + | ++ | |
| G20S/K130M | + | ||
| G20S/N129D | + | ||
| G20S/E117D/K130M | + |
FIGURE 3.

I-DmoI and variant cleavage assays. A, these cleavage assays were performed using a plasmid containing the target site. The assay distinguishes between three plasmid states: supercoiled state (no cleavage), linearized state (strands A and B are cleaved), and nicked state (only one strand is cleaved). Plasmid concentrations are 2 nm for the target DNA and 100 nm for I-DmoI WT and I-DmoI variants. The reaction mixture was incubated for 30 min at 65 °C in the presence of 10 mm Mg2+. B, time course cleavage on coding strand A (left panel) and on non-coding strand B (right panel) using I-DmoI Q42A/K120M in complex with its oligonucleotide target DNA. Nicked DNA and I-DmoI Q42A/K120M at 750 nm were incubated at different times ranging from 0 to 35 min at 65 °C in the presence of 10 mm Mg2+. C, time course showing the percentage of coding strand A and non-coding strand B cleavage by I-DmoI Q42A/K120 along time. The gray area represents the differences between strand A and B cleavage. Differences between single and double strand breaks in the target are quantified as a percentage in the chart on the right.
Mutations in the Basic Pocket in Site A
Other essential residues in the basic pocket have been modified in I-SceI and I-AniI (28, 31) (Table 2). These amino acids contribute to organize the solvent network involved in catalysis. For instance in I-CreI, Lys-98 is essential for nucleophilic water activation (26). A comparison of several crystallographic structures has identified the common roles of different residues among several LHEs (26) (Table 2). Mutation of Lys-122 or Lys-223 in the monomeric I-SceI produced variants with strand-specific DNA-nicking activity (28). Similarly, mutations on the Lys-227 residue of the I-AniI HE result in a DNA-nicking activity (31). In I-DmoI, an equivalent residue in the basic pocket is Lys-120, only present in site A (Fig. 1 and Table 2). We generated the I-DmoI K120M variant, and its activity was analyzed using a plasmid assay. After incubation, the substrate was nicked with respect to the I-DmoI wild type or control DNA (Fig. 3A).
Mutations in the Periphery of Sites A and B
Gln-42 in site A and Asn-129 in site B are the polar partners in the basic pocket of I-DmoI (26) (Fig. 1 and Table 3). We mutated these residues to evaluate their role in catalysis. The N129D and Q42E variants were generated to observe whether a charged side chain of a similar size could promote different contacts with solvent surrounding the metal ions in sites A and B. The N129D mutant was inactive; however, Q42E displayed partial nickase activity (Table 3).
Double and Triple Mutants
In contrast with I-AniI where a single mutation, K227M, displayed more than 99% nicking activity (31), different single mutations in the catalytic center do not yield a complete nickase activity. Therefore, we combined double and triple mutants to generate a nickase in I-DmoI (Fig. 3A). The I-DmoI D21E/K120M, I-DmoI Q42E/K120M, and DmoI Q42A/K120M variants were expressed, and their nickase activity was analyzed. The three combinations showed enhanced nicked cleavage with an increase in the ratio of nicked/linearized products. The D21E/K120M and Q42A/K120M displayed the largest nickase activity. Surprisingly, the D21E/K120M mutant cleaved the DNA target, whereas the D21E mutant did not show any activity. Time titration cleavage showed that I-DmoI Q42A/K120M behaves as a nickase, affecting the non-coding strand cleavage rate (Fig. 3B) and generating a nickase activity of almost 70% of the total cleavage reaction (Fig. 3C). This behavior is different compared with I-AniI, another monomeric LHE, where the double mutant of the equivalent residues (Q171K/K227M) is totally inactive (31).
To further improve these nickases, we combined the double mutants with some additional changes, thus generating the triple mutants D21E/Q42E/K120M, D21N/Q42E/K120M, ID21E/Q42A/K120M, and D21N/Q42A/K120M. All these variants were inactive except D21E/Q42E/K120M (Fig. 3A), which displayed substantial nickase activity, but no further improvement was detected when compared with the Q42A/K120M variant.
The Role of Gly-20 in the Active Center
During the reengineering of I-CreI variants targeting the RAG1 gene (21), we observed that the G19S mutation in only one of the monomers improved DNA cleavage. This mutation promoted a new interaction through the hydroxyl side chain, forming a hydrogen bond with the carbonyl group of the Gly-15 backbone from the other monomer. The presence of Ser-19 in both monomers hampered the catalysis probably due to a steric clash that distorts the catalytic center built by both monomers (32). Therefore we introduced this mutation in I-DmoI to check whether a similar effect could happen in monomeric LHEs, mutating the corresponding residues in the I-DmoI scaffold, A116S and G20S, in the neighborhood of sites A and B (Table 3). The A116S variant resulted in a protein with very low double strand cleavage and no nickase activity. However, the G20S mutation generated a protein with reduced double strand cleavage and strong nickase activity (Table 3 and Fig. 4A). Structural analysis of the I-DmoI G20S mutant (Protein Data Bank code 5AKM) showed that both active sites A and B are affected. Superposition of I-DmoI wild type and the G20S structures (Fig. 4B) showed a repulsive interaction between Ser-20 and Ala-116 side chains (affecting site A) and a new hydrogen bond between Ser-20 and Gly-16 (affecting site B) that are not found in the wild type protein. Indeed, quantum chemical analysis of the I-DmoI G20S variant energy profiles (Fig. 5A) provided a static picture of the cleavage barrier (without including any entropic effects), showing that the G20S mutation makes the cleavage of the DNA coding strand more energetically efficient, in about 30 kcal/mol, over the non-coding strand. Thus we generated the I-DmoI Q42A/A116S/K120M variant and the triple mutant I-DmoI G20S/Q42A/K120M without success. The first triple mutant was inactive, whereas the second cleaved both strands (Fig. 4A). The combination of the G20S and the Q42A/K120M mutations did not result in a better nickase activity.
FIGURE 4.
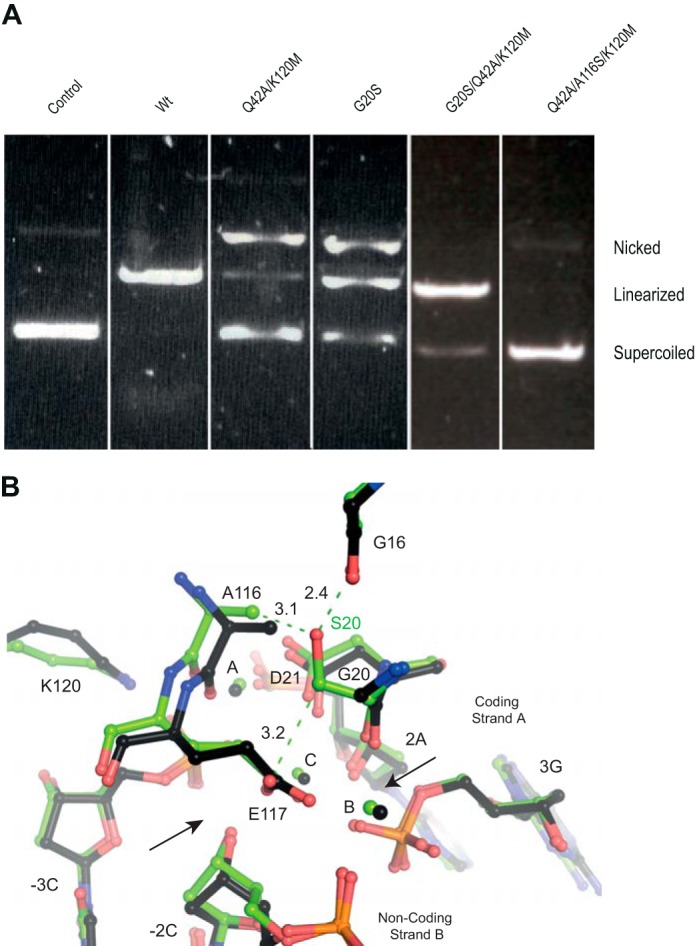
The I-DmoI G20S variant. A, the I-DmoI mutant cleavage assays using a plasmid detect the supercoiled (no strands are cleaved), linearized (strands A and B are cleaved), and nicked states (only one strand is cleaved). Plasmid concentrations are 2 nm for the target DNA and 100 nm for I-DmoI WT and I-DmoI variants. Cleavage incubation was carried out for 50 min at 65 °C in the presence of 10 mm Mg2+. B, superimposition of the active center structures of I-DmoI wild type (black) and the final state of the I-DmoI G20S variant (green).
FIGURE 5.

Quantum mechanics calculations of I-DmoI and mutants. A, cleavage energy barrier difference between coding and non-coding strands in I-DmoI WT (red), I-DmoI Q42A/K120M (green), and I-DmoI G20S (black). Negative energy differences mean that coding strand cleavage is more favorable than non-coding and vice versa. B, distributions of the electric field projections as obtained by molecular dynamics simulations (WT and AM refer to wild type and Q42A/K120M I-DmoI, respectively; CS, coding strand; NCS, non-coding strand; au, atomic units).
To unravel this effect, we solved the I-DmoI G20S crystal structure. We observed that the Ser-20 side chain contacts Ala-116, affecting the catalytic configuration in the active site A. One possibility is that altering the non-coding strand cleavage rate generates the nickase activity. Indeed, quantum chemical calculations showed that the G20S mutation makes the cleavage of the DNA strands energetically different in about 30 kcal/mol (Fig. 5A), supporting this explanation.
After this extensive mutational study, a nickase activity was observed in the I-DmoI Q42A/K120M variant (Fig. 3), although it maintains residual DSB activity. The DNA binding and protein stability of this variant were also checked to discard that any cleavage differences are due to these factors (Fig. 6).
FIGURE 6.
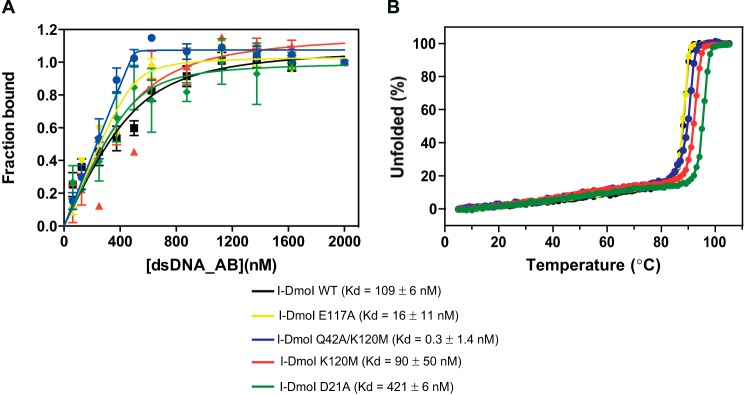
Binding and protein stability of different I-DmoI variants. A, protein-DNA Kd binding of I-DmoI variants. Binding reactions were performed by mixing 500 nm I-DmoI with different concentrations (0–2000 nm) of the 25-bp-long natural I-DmoI target duplex in binding buffer after incubating the mixture for 15 min at 50 °C. Each point was the average of five consecutive measurements. Error bars represent S.D. B, CD thermal denaturation analysis. Thermal denaturation curves were obtained at a protein concentration of 10 μm in a 2-mm cuvette. The ellipticity at 222 nm was recorded at 1 °C/min intervals.
Crystal Structure of a Nicking I-DmoI Q42A/K120M Mutant
The structure of I-DmoI nickase (Q42A/K120M) in complex with its DNA target was solved at 2.6-Å resolution in the presence of Mn2+. The positions of Mn2+ ions in the crystal structure were confirmed by collecting anomalous diffraction data (Table 1 and Fig. 7A). Supporting our activity assays, the enzyme was able to perform catalysis only in the coding strand. We observed in the structure the two mutated residues and the intact non-coding strand. The rest of the catalytic center was very similar to the fully cleaved target structure (Protein Data Bank code 2VS8) (11) (Fig. 7B). The two Mn2+ ions are located in the same position, and the catalytic residues superimpose very well with the wild type structure (Cαactive site root mean square deviation, 0.26 Å). Although the nucleophilic water in site A is present at the active center, the Q42A/K120M mutations alter the catalytic water chemical environment, hampering the phosphodiester bond cleavage in the non-coding strand B (Fig. 7A).
FIGURE 7.

I-DmoI Q42A/K120M characterization. A, detailed view of the I-DmoI Q42A/K120M active site in the presence of 2 mm Mn2+ and its target DNA. B, active center structure superimposition between I-DmoI Q42A/K120M (orange) and I-DmoI WT at the reactant state (green). C, detailed view of the I-DmoI Q42A/K120M active site in the presence of 2 mm Mn2+ and its target DNA nicked only in coding strand A. D, detailed view of the I-DmoI Q42A/K120M active site in the presence of 2 mm Mn2+ and its target DNA nicked in non-coding strand B. Fo − Fc omit maps (colored in gray) around the cleavable phosphodiester bonds are superimposed onto their corresponding refined structures. The omit map shows density contoured at 7σ. Anomalous maps (red) revealing the number and position of ions show density contoured at 5σ. The oligonucleotide sequence for the DNA strands is the same as in Fig. 1A. Non-coding strand B is displayed in yellow, coding strand A is colored in green, and backbone protein is shown in gray. Mn2+ ions are depicted as black spheres, structural waters are in cyan, and catalytic waters are in magenta. The arrows point to the phosphodiester bond cleaved.
To further test our nickase mutant, we crystallized and solved the structures of I-DmoI Q42A/K120M in complex with artificially nicked double strand oligonucleotides, mimicking cleaved coding or non-coding strands, in the presence of Mn2+ (Fig. 7, C and D). The structure of I-DmoI Q42A/K120M in complex with nicked DNA in the coding strand (Fig. 7C) displayed an intact phosphodiester bond in the non-coding strand. Interestingly, in site B, a water molecule is observed instead of the expected Mn2+ ion. The most likely explanation for this finding is that the nick in the coding strand positions the phosphate group in a conformation that disturbs the entrance of the catalytic ion.
Conversely, the catalytic water molecule is found close to the Mn2+ ion at active site A (Fig. 7C), but the mutation K120M seems to avoid the activation of this water for Sn2 nucleophilic attack on the corresponding phosphate of the non-coding strand. In addition, I-DmoI Q42A/K120M crystallized with nicked DNA in the non-coding strand shows a double strand break and two Mn2+ ions (Fig. 7D), one in the active site B (metal site B) and the other one in the active site A (metal site A) resembling the final catalysis step reported previously (10, 11). These three structures (Protein Data Bank codes 5AK9, 5AKF, and 5AKN) show that the I-DmoI Q42A/K120M variant functions primarily as a nickase.
Molecular Dynamics of the Nickase Variant
The electric field projections for the I-DmoI wild type and the Q42A/K120M mutant on the P–O bonds were measured to observe possible differences in the activation energy (Fig. 5B). The Q42A/K120M double mutation produced significant changes in the electric field on the non-coding strand, shifting the electric field distribution ∼0.03 atomic unit compared with I-DmoI wild type. However, the effect of this double mutation on the coding strand is almost negligible as it displays an electric field distribution similar to that of the I-DmoI wild type. These results provide a physical explanation of the experimental data. Thus, assuming that the titration time only depends on the cleavage barriers and these barriers are mainly determined by the perturbation of the catalytic center environment, the nickase effect observed in the Q42A/K120M variant might be explained by the perturbation produced by the mutations in site A dynamics while keeping site B intact. These changes would not allow phase-space configurations compatible with the cleavage on the non-coding strand.
Discussion
Taking advantage of the sequential catalytic mechanism of I-DmoI and the different chemical environments in sites A and B (10) (Fig. 1), we have introduced mutations in the active center to generate a nickase activity. Our biochemical experiments, the crystal structures, and a molecular dynamics analysis have dissected the intricate relationship in the catalytic center, providing the framework to engineer a specific single strand cutter on the I-DmoI scaffold.
Different mutations (Table 3) were carried out in the I-DmoI catalytic center to determine the role of these residues during cleavage. Mutations in the acidic residues involved in metal ion binding have been reported to abolish LHE activity. However, the I-DmoI D21N mutant presented DSB and low nickase activity (Fig. 3A), whereas the I-DmoI E117Q variant showed no activity, supporting that site B is more difficult to modify. Other conservative variants that modified the size of the side chains of these residues (I-DmoI D21E and I-DmoI D21E/E117D) were totally inactive. In contrast, I-DmoI E117D partially cleaved DNA. Therefore, we discarded the possibility of building a nickase by disrupting or altering one of the acidic residues in site A or B.
The basic residues in the periphery of sites A and B contribute to order the network of water molecules involved in catalysis (Table 2). The basic pocket is composed by Lys-120 and Gln-42 in site A and Asn-129 in B. Although we precluded I-DmoI cleavage by the N129D mutation, the Q42E and K120M mutations generated a partial nickase by disturbing non-coding strand cleavage. Finally, the combination of Q42A/K120M generated a nickase variant activity (Figs. 3 and 7). The crystal structure of this mutant was solved, showing only the coding strand cleaved when it was co-crystallized with an intact oligonucleotide (Fig. 7A). The crystals structures with nicked oligonucleotides show that this variant was unable to cut the non-coding strand even when the coding strand was cleaved (Fig. 7, C and D). These structures corroborate that the Q42A/K120M mutations alter the cleavage rate in site A without affecting site B. This is the first time that the crystal structure of an engineered LHE working as a nickase has been observed (Protein Data Bank codes 5AK9, 5AKF, and 5AKN).
These data were supported by in vitro cleavage titration assays (Fig. 3B). Our molecular dynamics studies also supported these experimental observations (Fig. 5), suggesting that the Q42A/K120M protein dynamics disturb the non-coding strand cleavage.
Our attempts to improve this nickase variant by combining the previously reported activity-enhancing mutation G20S (21) with the Q42A/K120M variant did not work. Also modifications in Ala-116, which is similarly located in site B, did not yield the desired results (Figs. 4 and 5A and Table 3). The triple mutant I-DmoI G20S/Q42A/K120M recovered an almost perfect double strand cleavage with no nickase activity. This suggests that a compensatory effect must occur with the G20S mutation over Q42A/K120M to obtain the reversion of the nickase activity created by Q42A/K120M.
The extensive network of interactions in the active center of the I-DmoI catalytic site made the possibility of building a nickase in this enzyme a difficult task. However, our data dissecting the sequential mechanism of DNA target cleavage have provided us with the information to decouple the cleavage of the target strands. Our work shows that the redesign of LHEs can also be attempted in their active centers. The customization of these enzymes by combining new target recognition sequences and single and double strand cutters may be an option to create new genome-modifying tools.
Author Contributions
G. M. and J. P. conceived and coordinated the study and wrote the paper. P. D suggested experiments and commented on the results. R. A. and M. J. M. designed, performed, and analyzed the experiments. P. R. and M. M. provided technical assistance and contributed to the preparation of the figures. M. D. A. performed the molecular dynamics calculations. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank the Swiss Light Source and XALOC beamline staff for support. This research received funding from the European Community's FP7 Grant Agreement 283570 (BioStruct-X; providing access to Swiss Light Source and ALBA synchrotrons). The Novo Nordisk Foundation Center for Protein Research, University of Copenhagen is supported financially by the Novo Nordisk Foundation (Grant agreement NNF14CC0001).
This work was supported by the European Community's Seventh Framework Program (FP7/2007–2013) under BioStruct-X (Grant 283570), Ministerio de Economía y Competitividad Grant BFU2011-23815/BMC (to G. M.), the Fundación Ramón Areces (to G. M.), Comunidad Autónoma de Madrid Grant CAM-S2010/BMD-2305 (to G. M.), and Ministerio de Economia y Competitividad Grant JCI-2011-09308 (to R. M.). The authors declare no conflict of interests with this manuscript.
The atomic coordinates and structure factors (codes 5AK9, 5AKF, 5AKN, and 5AKM) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HE
- homing endonuclease
- DSB
- double strand break
- LHE
- LAGLIDADG homing endonuclease.
References
- 1. Daboussi F., Zaslavskiy M., Poirot L., Loperfido M., Gouble A., Guyot V., Leduc S., Galetto R., Grizot S., Oficjalska D., Perez C., Delacôte F., Dupuy A., Chion-Sotinel I., Le Clerre D., Lebuhotel C., Danos O., Lemaire F., Oussedik K., Cédrone F., Epinat J. C., Smith J., Yáñez-Muñoz R. J., Dickson G., Popplewell L., Koo T., VandenDriessche T., Chuah M. K., Duclert A., Duchateau P., Pâques F. (2012) Chromosomal context and epigenetic mechanisms control the efficacy of genome editing by rare-cutting designer endonucleases. Nucleic Acids Res. 40, 6367–6379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Joshi R., Ho K. K., Tenney K., Chen J. H., Golden B. L., Gimble F. S. (2011) Evolution of I-SceI homing endonucleases with increased DNA recognition site specificity. J. Mol. Biol. 405, 185–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mimitou E. P., Symington L. S. (2009) Nucleases and helicases take center stage in homologous recombination. Trends Biochem. Sci. 34, 264–272 [DOI] [PubMed] [Google Scholar]
- 4. Davis L., Maizels N. (2011) DNA nicks promote efficient and safe targeted gene correction. PLoS One 6, e23981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee G. S., Neiditch M. B., Salus S. S., Roth D. B. (2004) RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell 117, 171–184 [DOI] [PubMed] [Google Scholar]
- 6. Metzger M. J., McConnell-Smith A., Stoddard B. L., Miller A. D. (2011) Single-strand nicks induce homologous recombination with less toxicity than double-strand breaks using an AAV vector template. Nucleic Acids Res. 39, 926–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. van Nierop G. P., de Vries A. A., Holkers M., Vrijsen K. R., Gonçalves M. A. (2009) Stimulation of homology-directed gene targeting at an endogenous human locus by a nicking endonuclease. Nucleic Acids Res. 37, 5725–5736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberts R. J., Vincze T., Posfai J., Macelis D. (2015) REBASE-a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res. 43, D298–D299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Marcaida M. J., Muñoz I. G., Blanco F. J., Prieto J., Montoya G. (2010) Homing endonucleases: from basics to therapeutic applications. Cell. Mol. Life Sci. 67, 727–748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molina R., Stella S., Redondo P., Gomez H., Marcaida M. J., Orozco M., Prieto J., Montoya G. (2015) Visualizing phosphodiester-bond hydrolysis by an endonuclease. Nat. Struct. Mol. Biol. 22, 65–72 [DOI] [PubMed] [Google Scholar]
- 11. Marcaida M. J., Prieto J., Redondo P., Nadra A. D., Alibés A., Serrano L., Grizot S., Duchateau P., Pâques F., Blanco F. J., Montoya G. (2008) Crystal structure of I-DmoI in complex with its target DNA provides new insights into meganuclease engineering. Proc. Natl. Acad. Sci. U. S. A. 105, 16888–16893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prieto J., Redondo P., Padró D., Arnould S., Epinat J. C., Pâques F., Blanco F. J., Montoya G. (2007) The C-terminal loop of the homing endonuclease I-CreI is essential for site recognition, DNA binding and cleavage. Nucleic Acids Res. 35, 3262–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prieto J., Epinat J. C., Redondo P., Ramos E., Padró D., Cédrone F., Montoya G., Pâques F., Blanco F. J. (2008) Generation and analysis of mesophilic variants of the thermostable archaeal I-DmoI homing endonuclease. J. Biol. Chem. 283, 4364–4374 [DOI] [PubMed] [Google Scholar]
- 14. Redondo P., Prieto J., Muñoz I. G., Alibés A., Stricher F., Serrano L., Cabaniols J. P., Daboussi F., Arnould S., Perez C., Duchateau P., Pâques F., Blanco F. J., Montoya G. (2008) Molecular basis of xeroderma pigmentosum group C DNA recognition by engineered meganucleases. Nature 456, 107–111 [DOI] [PubMed] [Google Scholar]
- 15. Zhang Z., Marshall A. G. (1998) A universal algorithm for fast and automated charge state deconvolution of electrospray mass-to-charge ratio spectra. J. Am. Soc. Mass. Spectrom. 9, 225–233 [DOI] [PubMed] [Google Scholar]
- 16. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 18. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Muñoz I. G., Prieto J., Subramanian S., Coloma J., Redondo P., Villate M., Merino N., Marenchino M., D'Abramo M., Gervasio F. L., Grizot S., Daboussi F., Smith J., Chion-Sotinel I., Pâques F., Duchateau P., Alibés A., Stricher F., Serrano L., Blanco F. J., Montoya G. (2011) Molecular basis of engineered meganuclease targeting of the endogenous human RAG1 locus. Nucleic Acids Res. 39, 729–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dapprich S., Komaromi I., Byun K. S., Morokuma K., Frisch M. J. (1999) A new ONIOM implementation in Gaussian 98. 1. The calculation of energies, gradients and vibrational frequencies and electric field derivatives. J. Mol. Struct. 462, 1–21 [Google Scholar]
- 23. Hess B., Kutzner C., van der Spoel D., Lindahl E. (2008) GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theor. Comput. 4, 435–447 [DOI] [PubMed] [Google Scholar]
- 24. Hornak V., Abel R., Okur A., Strockbine B., Roitberg A., Simmerling C. (2006) Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 65, 712–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bussi G., Donadio D., Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101, [DOI] [PubMed] [Google Scholar]
- 26. Chevalier B., Sussman D., Otis C., Noël A. J., Turmel M., Lemieux C., Stephens K., Monnat R. J., Jr., Stoddard B. L. (2004) Metal-dependent DNA cleavage mechanism of the I-CreI LAGLIDADG homing endonuclease. Biochemistry 43, 14015–14026 [DOI] [PubMed] [Google Scholar]
- 27. Armalyte E., Bujnicki J. M., Giedriene J., Gasiunas G., Kosiński J., Lubys A. (2005) Mva1269I: a monomeric type IIS restriction endonuclease from Micrococcus varians with two EcoRI- and FokI-like catalytic domains. J. Biol. Chem. 280, 41584–41594 [DOI] [PubMed] [Google Scholar]
- 28. Niu Y., Tenney K., Li H., Gimble F. S. (2008) Engineering variants of the I-SceI homing endonuclease with strand-specific and site-specific DNA-nicking activity. J. Mol. Biol. 382, 188–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lykke-Andersen J., Garrett R. A., Kjems J. (1997) Mapping metal ions at the catalytic centres of two intron-encoded endonucleases. EMBO J. 16, 3272–3281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Silva G. H., Belfort M. (2004) Analysis of the LAGLIDADG interface of the monomeric homing endonuclease I-DmoI. Nucleic Acids Res. 32, 3156–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McConnell Smith A., Takeuchi R., Pellenz S., Davis L., Maizels N., Monnat R. J., Jr., Stoddard B. L. (2009) Generation of a nicking enzyme that stimulates site-specific gene conversion from the I-AniI LAGLIDADG homing endonuclease. Proc. Natl. Acad. Sci. U.S.A. 106, 5099–5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grizot S., Smith J., Daboussi F., Prieto J., Redondo P., Merino N., Villate M., Thomas S., Lemaire L., Montoya G., Blanco F. J., Pâques F., Duchateau P. (2009) Efficient targeting of a SCID gene by an engineered single chain homing endonuclease. Nucleic Acids Res. 37, 5405–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]