

Background: The active role of SLICK channels and their regulation in neurons are largely unknown.
Results: SLICK transcription is highly dependent upon NFκB activation in peripheral and central neurons.
Conclusion: SLICK is an evolutionarily conserved NFκB-regulated gene.
Significance: SLICK channels may confer neuroprotection during ischemic conditions.
Keywords: hypoxia, molecular evolution, neurobiology, NFκB, potassium channel
Abstract
Although recent studies have shown the sodium-activated potassium channel SLACK (KCNT1) can contribute to neuronal excitability, there remains little information on the physiological role of the closely related SLICK (KCNT2) channel. Activation of SLICK channels may be important during pathological states such as ischemia, in which an increase in intracellular sodium and chloride can perturb membrane potential and ion homeostasis. We have identified two NFκB-binding sites within the promoter region of the human SLICK (KCNT2) and orthologous rat Slick (Kcnt2) genes, suggesting that conditions in which NFκB transcriptional activity is elevated promote expression of this channel. NFκB binding to the rat Slick promoter was confirmed in vivo by ChIP analyses, and NFκB was found differentially bound to the two sites. We verified NFκB transcriptional regulation of SLICK/Slick by mutational analyses and studying gene expression by luciferase assay in P19 cells, where NFκB is constitutively active. For the rat gene, activation of the Slick promoter was found to be additive in single NFκB mutations and synergistic in double mutations. Unexpectedly, for the human gene, NFκB exhibited cooperativity in activating the SLICK promoter. The human SLICK promoter constructs were then tested under hypoxic conditions in PC-12 cells, where NFκB is not active. Only under hypoxic conditions could luciferase activity be detected; the double NFκB mutant construct failed to exhibit activity. Transcriptional regulation of Slick by NFκB was verified in primary neurons. The Slick transcript decreased 24 h after NFκB inhibition. Our data show SLICK expression is predominantly under the control of NFκB. Because neuronal NFκB activation occurs during stressful stimuli such as hypoxia and injury, our findings suggest that SLICK is a neuroprotective gene.
Introduction
Neuronal potassium channels play an essential role in resting membrane potential, action potential firing, synaptic vesicle release, and cell volume regulation (1). SLACK (KCNT1, SLO2.2) is a slowly activating, outwardly rectifying, sodium-activated and chloride-sensitive potassium channel (KNa) found to contribute to neuronal excitability (2). The other member of the KNa channel family, known as SLICK (KCNT2, SLO2.1), has been less studied. The human SLICK gene is ∼400 kb and shares a striking 98% amino sequence identity with the rat protein sequence, exhibiting strong evolutionary conservation. Interestingly, when recorded in heterologous expression systems, Slick channels are more sensitive to intracellular chloride compared with Slack, and Slick activation is very rapid (3). SLICK/Slick channels contain a putative ATP-binding site. However, although rat Slick channel gating was found to be sensitive to intracellular ATP, current studies on human SLICK expressed in Xenopus oocytes did not find similar ATP inhibition (3, 4). Recent findings have shown that in contrast to Slack channels, Slick channels are the only large-conductance potassium channels sensitive to small changes in cell volume (5). Moreover, computer simulations predict that the instantaneous activation properties of Slick could in fact prevent action potential firing (3). These properties suggest that Slick channel expression might be tightly regulated, yet little is known about the channel's pathophysiological role in the nervous system.
A number of transcription factors are responsible for neuronal plasticity in response to stress, such as chronic inflammation and ischemia. NFκB is an inducible transcription factor implicated as a pro-inflammatory and protective mediator in almost all mammalian cell types, with a dominant role for prosurvival in neurons (6). NFκB in mammals is composed of five family members: RelA (p65), RelB, c-Rel, p50 (NFκB1), and p52 (NFκB2) (7). These members can form homo- or heterodimers that initiate or repress transcription. The p65, RelB, and c-Rel proteins contain a C-terminal transactivation domain that is essential for recruitment of the RNA polymerase transcriptional machinery. The p50 and p52 proteins lack this domain and therefore repress transcription if bound, unless bound as a heterodimer to a transactivation domain-containing member. The complexes remain inactive in the cytoplasm, bound by inhibitory family IκB. Once IκB is phosphorylated, NFκB can freely translocate into the nucleus and become active. In in vivo studies using transgenic mice, loss of p50/p65 DNA binding specifically in neurons resulted in prominent cell death after neurotoxic insults in hippocampal slices (8).
NFκB is also found to be a critical component of preconditioning, a biphasic endogenous mechanism for neuroprotection against future insults from ischemia and epilepsy (9). The early phase of preconditioning develops within a few minutes and lasts between 1 and 2 h, whereas the late phase develops between 12 and 24 h and lasts 3–4 days (10). In vivo models of brain tolerance, including ischemic, epileptic, and polyunsaturated fatty acid-induced preconditioning, determined NFκB-dependent neuronal protection in the adult rat hippocampus, where activation of NFκB was an essential step during late preconditioning (9). NFκB p50/p65 heterodimers were identified bound to DNA as early as 1 h after sublethal ischemia.
Slick is highly expressed in cortical and hippocampal areas, regions that constitutively express NFκB (high basal levels of active NFκB) (11). Slick is also moderately expressed peripherally in dorsal root ganglion (DRG)2 neurons (12). In this study, we examined the NFκB-dependent regulation of the SLICK gene.
Experimental Procedures
ChIP
The protocol of Maze et al. (13) was modified to include a nuclear extraction method previous to chromatin shearing. Briefly, Dynabeads (anti-rabbit M-280, catalog no. 112-04, Novex) were blocked in PBS solution containing 0.5% BSA. Beads were then resuspended in blocking solution containing 10 μg of antibody (anti-acetylated Lys-9 histone H3 (H3K9, catalog no. ab4441), anti-NFκB p65 (catalog no. ab7970), and anti-NFκB p105/p50 (catalog no. ab7971), Abcam) and rotated overnight at 4 °C. Beads were washed the next day with blocking solution. Adult Sprague-Dawley rats were decapitated, and fresh punches (3 × 1 mm, six punches per animal) were taken from the hippocampus, caudate putamen, and cerebellum before flash-freezing. Six punches were processed per sample and resuspended in hypotonic lysis buffer with protease inhibitors. Tissue sonication and chromatin shearing were carried out as described previously (13). Part of the sample was used to verify sonication electrophoresing on agarose gel, and part of the sample was used as the input control in quantitative real-time PCR. Shearing of DNA to 400 bp was confirmed by electrophoresing sample on agarose gel electrophoresis. DNA absorbance was measured using a spectrophotometer, and equal amounts of DNA were added to the antibody-bound Dynabeads with overnight rotation at 4 °C. Samples were washed, eluted, and digested with RNase for 1 h at 37 °C, followed by proteinase K for 2 h at 55 °C. DNA was recovered using a Qiagen PCR purification kit (catalog no. 28106).
ChIP/Quantitative Real-Time PCR
Primers were designed based on the two rat NFκB-binding sites, the mammalian conserved site, and the divergent site found only in mice and rats. Gene desert primers were designed by identifying a region in rat chromosome 12 without any genes. SYBR Green PCR Master Mix (catalog no. 4309155, Applied Biosystems) For quantification, a 50-cycle two-step denaturing/annealing was used, with a 15-s absorbance reading on a Bio-Rad iQ5 cycler. Each sample was performed in triplicate. Acetylated H3K9 was used as a positive control for active transcription. The primers used were as follows: NFκB conserved distal site, GCATAAGGGTGCCCTGCAATTTTCC (forward) and CCAGCACCCAGGCTGTGGTCGCC (reverse); NFκB non-conserved proximal site, GGCTGGGCCTTCAACAACTGC (forward) and CGTGATGGCTAGGCACTTTCC (reverse); and gene desert, AGGTCCTTAGAGAGAGCAGGA (forward) and GGAAAAGCAGGTGAGTAGCA (reverse).
Cell Culture
PC-12 cells (a gift from Dr. Richard A. Rabin, The State University of New York at Buffalo) and P19 cells (a gift from Dr. Jerome A. Roth, The State University of New York at Buffalo) were maintained in a 5% CO2 incubator at 37 °C. PC-12 cells were cultured in high-glucose DMEM containing 10% horse serum, 5% fetal bovine serum, and penicillin/streptomycin. P19 cells were cultured in minimum essential medium containing 7.5% newborn calf serum, 2.5% fetal bovine serum, and penicillin/streptomycin.
Dual-Luciferase Assay and Hypoxic Stress
Two kb of sequence upstream of the start site of human SLICK and rat Slick were cloned into the pGL3 luciferase vector. Site-directed mutagenesis of the putative NFκB-binding sites was performed using PfuTurbo DNA polymerase (catalog no. 600250, Agilent). PC-12 and P19 cells lines were plated onto 12-well plates, and constructs were transfected using Lipofectamine 2000 (catalog no. 11668-027, Life Technologies, Inc.) the following day. On day 3, cells were washed and lysed according to the Dual-Luciferase reporter assay system (catalog no. E1960, Promega). For hypoxic stress, transfected PC-12 cells were flushed in a hypoxia chamber (containing a Petri dish with sterile water for adequate humidity) for 10 min with a mixture of 5% CO2 and 95% N2. The chamber was then sealed, and cells were incubated at 37 °C for 1, 6, and 24 h. Luminescence measurements were obtained on BioTek microplate reader. Luciferase and Renilla readings were subtracted from initial blank, divided, and normalized to the pGL3 vector control.
Primary DRG Neuronal Culture
DRG were dissected from day 15 Sprague-Dawley rat embryos. The ganglia were dissociated in trypsin (2.5 mg/ml) for 60 min. Cells were plated on poly-d-lysine (100 μg/ml)- and laminin (3 μg/ml)-coated coverslips, pooled one pup per coverslip, and maintained on serum-free C2 media containing β-NGF (100 ng/ml). Unlike neonatal and adult DRG neurons, embryonic DRG neurons require NGF for survival (14). The day after dissection, cells were treated with 1 μm cytosine β-d-arabinofuranoside for 2 days. The NFκB inhibitor caffeic acid phenethyl ester (CAPE; EMD Millipore) was dissolved in dimethyl sulfoxide (DMSO) and diluted to a final concentration of 2.5 μg/ml in culture growth medium to 0.001% DMSO. On day 5 in culture, CAPE or DMSO was added to neuronal coverslips for 30 min, and cells were washed. For transcriptional analysis, RNA extraction was performed after 24 and 48 h.
RNA Extraction and cDNA Synthesis
An RNeasy micro kit (Qiagen) was used for RNA extraction from neuronal culture. RNA was reverse-transcribed with SuperScript III reverse transcriptase (Life Technologies, Inc.).
cDNA Quantitative Real-time PCR
Transcriptional abundance was measured using a thermocycler and SYBR Green PCR Master Mix. For quantification, a 50-cycle two-step denaturing/annealing was used, with a 15-s absorbance reading on a Bio-Rad iQ5 cycler. Each sample was performed in triplicate. The primers used for DRG neuronal culture were as follows: GAPDH, AACGACCCCTTCATTGAC (forward) and TCCACGACATACTCAGCAC (reverse); and Slick, GGTTTACAGGTCTCGGTGGC (forward) and GAGATGATGAAGGGAACTGC (reverse).
Results
NFκB Transcriptional Consensus Sites in SLICK Are Conserved in Mammals
Promoter analyses for transcription factor binding within the SLICK and rat Slick genes were performed using the web-based program TFSEARCH utilizing 2 kb of the gene sequence upstream of the start codon (Fig. 1). Consensus NFκB-binding sites were identified within the SLICK promoter region. The SLICK promoter region was aligned and compared with other mammalian species, and we determined that NFκB consensus sites are evolutionarily conserved. One putative site was completely conserved across all mammalian species examined, 828 bp upstream of the human SLICK start codon. Another consensus site was identified in human, chimpanzee, rhesus monkey, and dog, 13 bp upstream of the −828 site. In the rat and mouse Slick genes, there was an NFκB consensus site 457 bp upstream of the start codon. Although NFκB-binding sites are conserved across mammalian species, there are variations in the placement of the sites.
FIGURE 1.

Identification of conserved NFκB-binding sites across mammalian species. Shown is the alignment of nucleotide sequences of the SLICK gene in higher and lower order mammalian systems, including human (Homo sapiens), chimpanzee (Pan troglodytes), rhesus monkey (Macaca mulatta), dog (Canis lupus), cow (Bos taurus), guinea pig (Cavia porcellus), rat (Rattus norvegicus), and mouse (Mus musculus). Sequences were aligned using the web-based program ClustalW from the European Bioinformatics Institute. NFκB consensus sites were predicted using the web-based program TFSEARCH from the Computational Biology Research Center. Putative NFκB-binding sites are highlighted in gray, and the start methionine codon is indicated in boldface. Asterisks indicate conservation.
Putative NFκB-binding Sites Verified in Vivo Are Found Differentially Bound to the Slick Promoter
To verify the identified putative NFκB-binding sites of the Slick promoter, ChIP was performed on tissue from the rat hippocampus, caudate putamen, and cerebellum. High-level expression of Slick mRNA and Slick protein was found in the rat hippocampus, with moderate expression in the caudate putamen and undetectable expression in the cerebellum as determined by in situ hybridization and immunohistochemistry (11). We utilized antibodies against the p50 and p65 subunits of NFκB, and acetylated H3K9 was used as a positive control for transcriptional activation. Quantitative PCR Ct values were normalized, dividing IP Ct values by input Ct values to provide ΔCt. These values are relative to 1; therefore, values above 1 represent a higher IP Ct value correlating to a lower IP DNA abundance and therefore less transcription factor binding. Gene desert primers were used as a negative control for nonspecific binding. Gene desert Ct values were undetectable, being above the cutoff threshold (data not shown).3 We compared IP Ct values amplifying the evolutionarily conserved NFκB-binding site, GGGGATTCC (distal site to the start methionine), and the non-conserved NFκB-binding site (proximal site to the start methionine) present only in the rat and mouse genes, GGGGATGCCC. At the distal NFκB-binding site, both the p50 and p65 subunits exhibited a lower mean ΔCt in the hippocampus compared with the caudate putamen and cerebellum, indicating a higher level of NFκB binding at this site within the hippocampus (Fig. 2). However, the data did not achieve statistical significance. At the proximal NFκB-binding site, transcription factor binding was significantly different in the hippocampus versus the caudate putamen and cerebellum (Table 1). Specifically, the p65 subunit was significantly less bound in the proximal NFκB-binding site within the cerebellum compared with the hippocampus. Furthermore, the p50 subunit was significantly less bound in the caudate putamen at the proximal NFκB-binding site compared with every other p50 distal and proximal NFκB-binding site. In other words, both the p50 and p65 subunits exhibited significantly higher binding levels at the proximal NFκB-binding site in the hippocampus compared with the caudate putamen and cerebellum.
FIGURE 2.
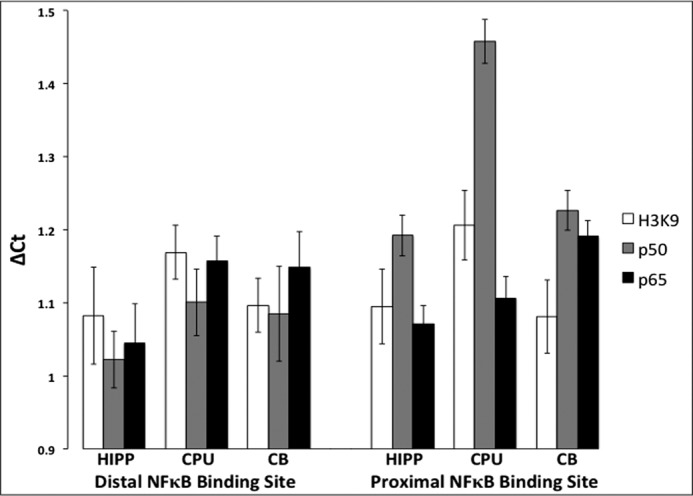
Differential NFκB binding to the Slick gene verified in vivo. Shown are the results from ChIP of NFκB subunits p65 and p50 and acetylated H3K9 bound to the Slick promoter region of rat hippocampal (HIPP), caudate putamen (CPU), and cerebellar (CB) tissue. Primers were designed based on the identified putative mammalian NFκB-binding sites, the evolutionarily conserved distal NFκB-binding site, and the proximal NFκB-binding site identified in only rats and mice. Acetylated H3K9 was used as a positive control for active transcription. Relative ΔCt was calculated by normalizing IP to input. Quantitative real-time PCR was performed in triplicate. Error bars represent S.E. (n = 4–6).
TABLE 1.
Statistical analysis of ChIP ΔCt values by individual t tests
| Significant comparison | p |
|---|---|
| Hippocampal p50 distal NFκB-binding site versus hippocampal p50 proximal NFκB-binding site | p ≤ 0.05 |
| Hippocampal p65 proximal NFκB-binding site versus cerebellar p65 proximal NFκB-binding site | p ≤ 0.05 |
| Hippocampal p50 distal NFκB-binding site versus caudate putamen p50 proximal NFκB-binding site | p ≤ 0.0001 |
| Caudate putamen p50 distal NFκB-binding site versus caudate putamen p50 proximal NFκB-binding site | p ≤ 0.001 |
| Cerebellar p50 distal NFκB-binding site versus caudate putamen p50 proximal NFκB-binding site | p ≤ 0.01 |
| Hippocampal p50 proximal NFκB-binding site versus caudate putamen p50 proximal NFκB-binding site | p ≤ 0.001 |
| Cerebellar p50 proximal NFκB-binding site versus caudate putamen p50 proximal NFκB-binding site | p ≤ 0.01 |
It should also be noted that in the hippocampus, p50 subunit binding to the proximal NFκB-binding site was significantly less compared with the distal NFκB-binding site. This suggests that the proximal NFκB-binding site has lower NFκB-binding affinity. Notwithstanding, these results confirm that NFκB binds to the Slick promoter in vivo and indicates differential NFκB-binding affinity between the two sites. Corroborating the high levels of constitutive NFκB activity and Slick mRNA expression in the adult hippocampus, we found the highest levels of p50 and p65 binding to the Slick promoter within the hippocampus.
NFκB Is Essential for Mammalian SLICK Transcription
To identify whether NFκB can actively recruit the RNA polymerase transcriptional machinery and promote transcription of the SLICK gene, 2 kb of sequence upstream of the start site of the rat and human genes were inserted into the pGL3 promoter-less luciferase vector and designated as the wild-type construct. To observe activation of the luciferase gene, the inserted promoter region must contain regulatory elements that will initiate transcription. One or both NFκB-binding sites were mutated to prevent NFκB binding (Fig. 3A). The constructs were transfected into two separate neuronal model cell lines, PC-12 and P19 cells, for luciferase assays (Fig. 3B). PC-12 cells contain inactive NFκB complexes, whereas P19 cells contain constitutively active p50 and p65 subunits (15, 16). In the PC-12 cell line, activation of luciferase was not observed in the rat or human constructs. In the P19 cell line, luciferase activation increased by ∼17-fold in both the rat and human wild-type constructs compared with the vector control. Nullifying either NFκB-binding site in the rat promoter resulted in an additive decrease in luciferase activity, whereas nullifying both NFκB-binding sites by mutation resulted in a synergistic decrease that did not significantly differ from the empty vector base line. In contrast, nullifying either NFκB-binding site in the human promoter resulted in a complete loss of luciferase activity, not differing from the empty vector (Fig. 3C). However, we found that nullifying both NFκB-binding sites resulted in a small but significant increase in luciferase activity over the base line. This suggests that occupancy of either site by NFκB subunits represses other transcription factors; nonetheless, our data demonstrate that there is strong NFκB-dependent transcription of SLICK across species.
FIGURE 3.

NFκB binding to the SLICK gene promotes transcriptional activation. A, schematic of the mutations made by site-directed mutagenesis to the putative NFκB-binding sites of the rat (r) Slick and human (h) SLICK genes. The promoter regions of the rat Slick (B) and human SLICK (C) genes were cloned into the pGL3 promoter vector. Mutations were made in the rat constructs only in the evolutionarily conserved distal NFκB-binding site (Mut 1), only in the proximal NFκB-binding site (Mut 2), or in both sites (Mut 1–2). Mutations were made in the human constructs only in the distal NFκB-binding site (Mut 1), only in the evolutionarily conserved proximal NFκB-binding site (Mut 2), or in both sites (Mut 1–2). The constructs were transfected into neuronal model cell lines in which NFκB can be inducibly expressed (PC-12 cells) or constitutively expressed (P19 cells). Dual-Luciferase assays were performed using a plate reader to measure luciferase activity, with Renilla activity as an internal control. The pGL3 vector by itself was used as a control to normalize relative expression of each construct. Experiments were performed in triplicate between multiple experiments (n = 2–7). *, p ≤ 0.001; #, p ≤ 0.01 compared with the control by one-way analysis of variance. Error bars represent S.E.
Hypoxic Induction of SLICK Transcription Occurs through NFκB
To further demonstrate the dependence of SLICK transcription on NFκB, we induced NFκB activation in PC-12 cells by hypoxia. Hypoxia is known to cause IκBα degradation, increase NFκB DNA-binding activity, and cause the transactivation of a reporter gene construct containing multiple NFκB DNA-binding sites (17). The human wild-type and double NFκB-binding site mutant SLICK constructs were transfected into PC-12 cells and exposed to hypoxic stress for 1, 6, and 24 h. Luciferase activation was observed in the wild-type construct compared with the vector control at each time point (Fig. 4). The degree of activation was comparable with that seen in P19 cells (Fig. 3), where NFκB is constitutively active. The rat Slick wild-type construct showed a similar increase in luciferase activity when transfected in PC-12 cells after 45 min of hypoxic stress (data not shown).3 The double NFκB-binding site mutant construct exhibited a small but significant increase in activity at 1 h, similar to what we saw in P19 cells, but this disappeared at 6 and 24 h. We assume that another transcription factor that is repressed by NFκB binding during normal conditions loses activity during prolonged hypoxia. Collectively, our results reveal that the SLICK gene can be induced by hypoxia via NFκB activation.
FIGURE 4.

Hypoxic induction of NFκB increases SLICK transcription. The pGL3 promoter human SLICK constructs were transfected into the hypoxic and neuronal model cell line (PC-12) and incubated in a hypoxia chamber for 1, 6, and 24 h at 37 °C. Dual-Luciferase assays were performed using a plate reader to measure luciferase activity, with Renilla activity as an internal control. The pGL3 vector by itself was used as a control to normalize relative expression of each construct. Experiments were performed in triplicate and performed twice. *, p ≤ 0.001 compared with the control by one-way analysis of variance. Error bars represent S.E. Mut 1–2, double NFκB-binding site mutant.
Decreased Slick Expression after NFκB Inhibition in Primary Neurons
Although our data support Slick regulation by NFκB, we further confirmed this by studying the effects of NFκB inhibition on endogenous Slick transcription in native neurons. Using CAPE, a p65 translocation blocker, we assessed Slick expression by quantitative real-time PCR in primary DRG neurons as described previously (18). We elected to use DRG neuronal cultures to study the transcriptional regulation of Slick, as both adult and embryonic DRG neurons express Slick channels (12). This contrasted with embryonic hippocampal neuronal cultures, where we observed little to no Slick expression at this early developmental stage (data not shown).3 Additionally, embryonic DRG neurons were cultured in the presence of NGF, a neurotrophic factor essential for survival of these neurons. NGF-dependent survival was found to be contingent upon NFκB activation (19). Furthermore, cytosine β-d-arabinofuranoside was added to cultures to remove mitotic glial supporting cells, ensuring that the effects seen could be attributed to neuron-specific inhibition of NFκB. We found that when DRG neurons were treated with CAPE for 30 min and RNA was isolated after 24 h, Slick expression was significantly decreased by ∼50% compared with the DMSO control (Fig. 5). Slick transcriptional expression was normalized to the housekeeping gene GAPDH. We also examined Slick transcript levels 48 h after CAPE exposure, and although there was still slightly diminished expression, the data were not statistically significant. This is likely due to the short incubation time and eventual washing out of the inhibitor. Still, our data confirm that endogenous Slick expression, assessed in primary neurons, is dependent upon NFκB activation.
FIGURE 5.

Inhibition of NFκB significantly decreases Slick the transcript in primary neurons. A, a 30-min incubation of neurons with the NFκB inhibitor CAPE significantly reduced Slick transcript levels assayed 24 h later compared with the DMSO control. B, transcript levels returned to near control levels at 48 h compared with the DMSO control. The -fold change (ΔΔCt) was normalized to GAPDH (n = 6 for 24 h, n = 3 for 48 h). *, p ≤ 0.01 by t test. Error bars represent S.E.
Discussion
In this study, we identified NFκB as a positive regulator of SLICK gene expression. Analysis of the promoter region of SLICK identified two NFκB-binding sites, with one site completely conserved across all mammalian species examined. The putative sites were verified in vivo using ChIP assay, and we determined differential binding of p50 and p65 at the proximal NFκB-binding site compared with the conserved distal NFκB-binding site. Mutating the consensus NFκB-binding sites caused a substantial reduction in SLICK transcriptional activity. SLICK transcriptional activation was seen after hypoxic stress and was found to be dependent upon NFκB activation, as mutating the two NFκB-binding sites largely inhibited activation of SLICK. We identified decreased Slick expression after using an NFκB inhibitor in primary neurons. We conclude that the SLICK gene is stringently regulated by NFκB activation.
NFκB subunits p50 and p65 are constitutively active in the glutamatergic neurons of the hippocampus and cortex, and extensive supporting evidence has attributed this high basal expression to be important for synaptic plasticity and morphology (see Ref. 20 for review of supporting evidence). Our ChIP data defined the proximal NFκB-binding site of the Slick gene as a determining factor of differential transcriptional expression because of variable binding of the NFκB p50 and p65 subunits at this site when analyzed in three different brain regions. Using DNA binding preferences by EMSA-Seq, Wong et al. (21) identified the relative binding affinities of nine distinct human NFκB dimers on canonical and non-canonical sequences. In their report, the evolutionarily conserved distal NFκB-binding site was analyzed and found to bind all NFκB dimers with relatively high affinity. The exact sequence of the proximal NFκB-binding site was not assessed in their study; however, a very similar sequence was reported to also have considerable binding affinity for all dimers, but an overall lower binding affinity compared with the distal NFκB-binding site. Our ChIP data support these findings, as p50 and p65 exhibited a lower binding affinity for the proximal NFκB-binding site compared with the distal NFκB-binding site.
The NFκB-binding affinity we observed in the hippocampus from our ChIP correlates with the luciferase expression we observed in the rat Slick promoter. The high levels of constitutive NFκB activity in P19 cells resemble those found in adult hippocampal neurons (22). Therefore, when sufficient levels of both p50 and p65 are available, binding is observed at both NFκB-binding sites, explaining the additive decrease observed after mutation of the distal or proximal NFκB-binding site. Interestingly, we found some differences between the human SLICK and rat Slick promoters. Nullifying either NFκB-binding site in the rat Slick promoter resulted in an additive decrease in luciferase activation and a synergistic decrease in activation when both sites were mutated. This differed from the human SLICK promoter, where single mutations were sufficient to obliterate transcriptional activity. This may be due to the distance between the NFκB-binding sites. Giorgetti et al. (23) found NFκB to additively contribute to polymerase recruitment, which is defined by the strength of the environmental input. The synergistic decrease in luciferase activation of the double NFκB-binding site mutation in the rat Slick promoter supports this idea. However, NFκB cooperativity was observed for the human SLICK promoter, a result that is contrary to the conclusions put forth by Giorgetti et al., who found that NFκB bound independently and without cooperativity. However, it should be noted that although the evidence supports p65 binding as non-cooperative, other NFκB subunits were not examined. NFκB binding predicted by TFSEARCH indicated a high confidence level of c-Rel also potentially binding to the evolutionarily conserved site at −828 bp, along with the other site 13 bp upstream. Immunochemical supershift analyses determined that P19 cells express p50, p65, and c-Rel subunits (22). As c-Rel can activate transcription, it is possible that c-Rel binding may be cooperative at this site. Giorgetti et al. (23) also examined intersite NFκB binding of up to 400 bp for cooperativity. The human NFκB intersite difference is only 13 bp, much smaller than the average intersite distance examined. Cooperativity may arise because of the limited spacing between the two sites. In specific cases, p50/p52 dimers have been found to exhibit cooperativity in a viral enhancer (24). Furthermore, cooperativity of the human NFκB-binding sites would allow for maximal SLICK transcriptional activation after a small increase in NFκB activation, whereas the additive properties in the rat NFκB-binding sites would not. Our data suggest that cooperative NFκB binding in the human SLICK promoter is an evolutionary adaptation that might be important for more complex brains.
With our ChIP data and the fact that Slick channels are highly expressed in adult pyramidal cells of the rat hippocampus and cortex, we can infer that SLICK channels are likely involved in synaptic plasticity (11). However, the two NFκB-binding sites in the human SLICK promoter are predicted to bind to c-Rel as well. The c-Rel protein drives neuroprotective effects during glucose oxygen deprivation, and the neuronal survival factor interleukin-1β can activate c-Rel (25, 26). Therefore, c-Rel activation may be important for inducing SLICK expression during ischemia. Of note, we also identified putative HIF1 (hypoxia-inducible factor 1)-binding sites in the SLICK promoter (data not shown).3 HIF1-binding sites were not present in the rat promoter. Although we determined that SLICK transcriptional initiation occurs primarily through NFκB by hypoxia experiments, we cannot completely rule out an involvement of HIF1-dependent regulation of SLICK.
Slick channels are also expressed in DRG neurons, and the regulation by NFκB may be important during stressful conditions such as nerve injury and neuropathy. Given the regenerative properties of these injured neurons, cytokine and/or neurotrophin signaling through NFκB may allow neurons to adapt to neuropathic conditions and maintain normal firing. Although we saw a significant reduction in the Slick transcript in dissociated DRG neurons after NFκB inhibition, we were not able to detect changes in protein levels after 48–96 h (data not shown).3 This is likely due to the typical longer half-lives of ion channels and also the effect we observed with CAPE inhibition of NFκB. Using the protocol described by Chiechio et al. (18), the significant decrease in the Slick transcript observed at 24 h dissipated after 48 h. Therefore, the level of transcript reduction we saw may not be sufficient to significantly impact protein levels. It should be noted that we tried the potent NFκB-specific peptide inhibitor SN50M (Enzo Life Sciences, Farmingdale, NY), but found substantial neuronal death within 24 h (data not shown),3 as might be expected for a prosurvival transcription factor such as NFκB (19). Still, we cannot conclude that there are no post-transcriptional mechanisms that also govern Slick channel expression.
The small-conductance calcium-activated potassium channels of the SK2 subtype were also shown to have two NFκB-binding sites and to be regulated by NFκB (27). SK2 channels are important for synaptic plasticity and are neuroprotective during cerebral ischemia (28, 29). Indeed, SK2 channel openers were shown to reduce neuronal cell death during ischemia (29). Potassium channels are important for countering the ischemia-induced excitotoxicity in neurons (30, 31). During ischemic excitotoxicity, neurons experience elevations in intracellular calcium, sodium, and chloride, causing changes in neuronal cell volume and eventual death. Potassium channels that are responsive to intracellular ion perturbations would be uniquely suited to confer neuroprotection. This may be one reason why SK2 and SLICK genes are under the control of NFκB. Elevating NFκB signaling could limit ischemia-associated neuronal death by induction of the SK2 and SLICK genes (17). Simply targeting SLICK channels directly during ischemia or neuropathy, channels that are uniquely sensitive to cell volume perturbations, could have neuroprotective effects (5). This remains to be explored.
Author Contributions
D. L. T. and A. B. designed the study and wrote the paper. D. L. T. conducted the luciferase, ChIP, and neuronal transcriptional assays. A. M. G. -K. and D. M. D. assisted with the tissue isolation and design of the ChIP assays. A. B. designed and constructed vectors for luciferase assays. All authors analyzed the results and approved the final version of the manuscript.
Acknowledgments
We thank Dr. Ji Li for the use of hypoxia chambers and gas, Dr. Michael J. Buck for use of the Bioruptor for chromatin shearing, and Monica S. Humby and Aaron Caccamise (Dietz laboratory) for assistance with tissue collection.
This work was supported, in whole or in part, by National Institutes of Health Grant NS078184 (to A. B.). The authors declare that they have no conflicts of interest with the contents of this article.
D. L. Tomasello, A. M. Gancarz-Kausch, D. M. Dietz, and A. Bhattacharjee, unpublished data.
- DRG
- dorsal root ganglion/ganglia
- CAPE
- caffeic acid phenethyl ester
- DMSO
- dimethyl sulfoxide.
References
- 1. Lang F., Föller M., Lang K., Lang P., Ritter M., Vereninov A., Szabo I., Huber S. M., Gulbins E. (2007) Cell volume regulatory ion channels in cell proliferation and cell death. Methods Enzymol. 428, 209–225 [DOI] [PubMed] [Google Scholar]
- 2. Nuwer M. O., Picchione K. E., Bhattacharjee A. (2010) PKA-induced internalization of slack KNa channels produces dorsal root ganglion neuron hyperexcitability. J. Neurosci. 30, 14165–14172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bhattacharjee A., Joiner W. J., Wu M., Yang Y., Sigworth F. J., Kaczmarek L. K. (2003) Slick (Slo2.1), a rapidly-gating sodium-activated potassium channel inhibited by ATP. J. Neurosci. 23, 11681–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garg P., Sanguinetti M. C. (2014) Intracellular ATP does not inhibit Slo2.1 K+ channels. Physiol Rep. 2, e12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tejada M. A., Stople K., Hammami Bomholtz S., Meinild A. K., Poulsen A. N., Klaerke D. A. (2014) Cell volume changes regulate slick (Slo2.1), but not slack (Slo2.2) K+ channels. PLoS ONE 9, e110833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bhakar A. L., Tannis L. L., Zeindler C. (2002) Constitutive nuclear factor-κB activity is required for central neuron survival. J. Neurosci. 22, 8466–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hayden M. S., Ghosh S. (2012) NF-κB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev. 26, 203–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fridmacher V., Kaltschmidt B., Goudeau B., Ndiaye D., Rossi F. M., Pfeiffer J., Kaltschmidt C., Israël A., Mémet S. (2003) Forebrain-specific neuronal inhibition of nuclear factor-κB activity leads to loss of neuroprotection. J. Neurosci. 23, 9403–9408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Blondeau N., Widmann C., Lazdunski M. (2001) Activation of the nuclear factor-κB is a key event in brain tolerance. J. Neurosci. 21, 4668–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Das M., Das D. K. (2008) Molecular mechanism of preconditioning. IUBMB Life 60, 199–203 [DOI] [PubMed] [Google Scholar]
- 11. Bhattacharjee A., von Hehn C. A. A., Mei X., Kaczmarek L. K. (2005) Localization of the Na+-activated K+ channel Slick in the rat central nervous system. J. Comp. Neurol. 484, 80–92 [DOI] [PubMed] [Google Scholar]
- 12. Tamsett T. J., Picchione K. E., Bhattacharjee A. (2009) NAD+ activates KNa channels in dorsal root ganglion neurons. J. Neurosci. 29, 5127–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maze I., Covington H. E., 3rd, Dietz D. M., LaPlant Q., Renthal W., Russo S. J., Mechanic M., Mouzon E., Neve R. L., Haggarty S. J., Ren Y., Sampath S. C., Hurd Y. L., Greengard P., Tarakhovsky A., Schaefer A., Nestler E. J. (2010) Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science 327, 213–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chalazonitis A., Peterson E. R., Crain S. M. (1987) Nerve growth factor regulates the action potential duration of mature sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 84, 289–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Panet H., Barzilai A., Daily D., Melamed E. (2001) Activation of nuclear transcription factor kappa B (NF-κB) is essential for dopamine-induced apoptosis in PC12 cells. J. Neurochem. 77, 391–398 [DOI] [PubMed] [Google Scholar]
- 16. Paradkar P. N., Roth J. A. (2006) Post-translational and transcriptional regulation of DMT1 during P19 embryonic carcinoma cell differentiation by retinoic acid. Biochem. J. 394, 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Koong A. C., Chen E. Y., Giaccia A. (1994) Hypoxia causes the activation of nuclear factor κB through the phosphorylation of IκBα on tyrosine residues. Cancer Res. 54, 1425–1430 [PubMed] [Google Scholar]
- 18. Chiechio S., Copani A., De Petris L., Morales M. E., Nicoletti F., Gereau R. W. (2006) Transcriptional regulation of metabotropic glutamate receptor 2/3 expression by the NF-κB pathway in primary dorsal root ganglia neurons: a possible mechanism for the analgesic effect of l-acetylcarnitine. Mol. Pain 2, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maggirwar S. B., Sarmiere P. D., Dewhurst S., Freeman R. S. (1998) Nerve growth factor-dependent activation of NF-κB contributes to survival of sympathetic neurons. J. Neurosci. 18, 10356–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kaltschmidt B., Kaltschmidt C. (2009) NF-κB in the nervous system. Cold Spring Harb. Perspect. Biol. 1, a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wong D., Teixeira A., Oikonomopoulos S., Humburg P., Lone I. N., Saliba D., Siggers T., Bulyk M., Angelov D., Dimitrov S., Udalova I. A., Ragoussis J. (2011) Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 12, R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Burke M. A., Bothwell M. (2003) p75 neurotrophin receptor mediates neurotrophin activation of NF-κB and induction of iNOS expression in P19 neurons. J. Neurobiol. 55, 191–203 [DOI] [PubMed] [Google Scholar]
- 23. Giorgetti L., Siggers T., Tiana G., Caprara G., Notarbartolo S., Corona T., Pasparakis M., Milani P., Bulyk M. L., Natoli G. (2010) Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol. Cell 37, 418–428 [DOI] [PubMed] [Google Scholar]
- 24. Moorthy A. K., Huang D.-B.., Wang V. Y.-F., Vu D., Ghosh G. (2007) X-ray structure of a NF-κB p50/RelB/DNA complex reveals assembly of multiple dimers on tandem κB sites. J. Mol. Biol. 373, 723–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sarnico I., Lanzillotta A., Boroni F., Benarese M., Alghisi M., Schwaninger M., Inta I., Battistin L., Spano P., Pizzi M. (2009) NF-κB p50/RelA and c-Rel-containing dimers: opposite regulators of neuron vulnerability to ischaemia. J. Neurochem. 108, 475–485 [DOI] [PubMed] [Google Scholar]
- 26. Pizzi M., Goffi F., Boroni F., Benarese M., Perkins S. E., Liou H.-C., Spano P. (2002) Opposing roles for NF-κB/Rel factors p65 and c-Rel in the modulation of neuron survival elicited by glutamate and interleukin-1β. J. Biol. Chem. 277, 20717–20723 [DOI] [PubMed] [Google Scholar]
- 27. Kye M.-J., Spiess J., Blank T. (2007) Transcriptional regulation of intronic calcium-activated potassium channel SK2 promoters by nuclear factor-kappa B and glucocorticoids. Mol. Cell. Biochem. 300, 9–17 [DOI] [PubMed] [Google Scholar]
- 28. Allen D., Bond C. T., Luján R., Ballesteros-Merino C., Lin M. T., Wang K., Klett N., Watanabe M., Shigemoto R., Stackman R. W., Jr., Maylie J., Adelman J. P. (2011) The SK2-long isoform directs synaptic localization and function of SK2-containing channels. Nat. Neurosci. 14, 744–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Allen D., Nakayama S., Kuroiwa M., Nakano T., Palmateer J., Kosaka Y., Ballesteros C., Watanabe M., Bond C. T., Luján R., Maylie J., Adelman J. P., Herson P. S. (2011) SK2 channels are neuroprotective for ischemia-induced neuronal cell death. J. Cereb. Blood Flow Metab. 31, 2302–2312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Deng P., Pang Z.-P., Lei Z., Shikano S., Xiong Q., Harvey B. K., London B., Wang Y., Li M., Xu Z. C. (2011) Up-regulation of A-type potassium currents protects neurons against cerebral ischemia. J. Cereb. Blood Flow Metab. 31, 1823–1835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shah N. H., Schulien A. J., Clemens K., Aizenman T. D., Hageman T. M., Wills Z. P., Aizenman E. (2014) Cyclin E1 regulates Kv2.1 channel phosphorylation and localization in neuronal ischemia. J. Neurosci. 34, 4326–4331 [DOI] [PMC free article] [PubMed] [Google Scholar]