

Background: Steroid receptor coactivator 1 (SRC-1) is a transcriptional coactivator.
Results: SRC-1 promotes human hepatocellular carcinoma (HCC) cell proliferation by coactivating β-catenin to enhance c-Myc expression.
Conclusion: SRC-1 plays an important role in HCC progression by enhancing Wnt/β-catenin signaling.
Significance: SRC-1 is a potential therapeutic molecular target for human HCC.
Keywords: cell proliferation, hepatocellular carcinoma, Myc (c-Myc), transcriptional coactivator, Wnt signaling, steroid receptor coactivator (SRC-1)
Abstract
Steroid receptor coactivator 1 (SRC-1) is a transcriptional coactivator not only for steroid receptors, such as androgen receptor and estrogen receptor, but also for other transcription factors. SRC-1 has been shown to play an important role in the progression of breast cancer and prostate cancer. However, its role in liver cancer progression remains unknown. In this study, we report that SRC-1 was overexpressed in 25 (62.5%) of 40 human hepatocellular carcinoma (HCC) specimens. Down-regulation of SRC-1 decreased HCC cell proliferation and impaired tumor maintenance in HCC xenografts. Knockdown of SRC-1 reduced protein levels of the proliferation marker proliferating cell nuclear antigen (PCNA) and the oncogene c-Myc. Knockout of SRC-1 in mice reduced diethylnitrosamine/CCl4-induced tumor formation in the liver and the expression of c-Myc and PCNA in liver tumors. SRC-1 promoted c-Myc expression, at least in part, by directly interacting with β-catenin to enhance Wnt/β-catenin signaling. Consistent with these results, the expression of SRC-1 was positively correlated with PCNA expression in human HCC specimens, and the expression levels of c-Myc in SRC-1-positive HCC specimens were higher than in SRC-1-negative HCC specimens. In addition, SRC-1 and SRC-3 were co-overexpressed in 47.5% of HCC specimens, and they cooperated to promote HCC cell proliferation. Simultaneous down-regulation of SRC-1 and SRC-3 dramatically inhibited HCC cell proliferation. Our results demonstrate that SRC-1 promotes HCC progression by enhancing Wnt/β-catenin signaling and suggest that SRC-1 is a potential therapeutic molecular target for HCC.
Introduction
Primary liver cancer, particularly hepatocellular carcinoma (HCC),3 is the fifth most common cancer worldwide (1) and the second most common cause of cancer mortality in men, behind lung cancer (2). Among primary liver cancers, HCC represents the major histological subtype, accounting for 70–85% of the total liver cancer burden worldwide (3). HCC grows rapidly and is usually accompanied by vascular invasion, metastasis, recurrence, and poor prognosis. Considering the high incidence and mortality rate of HCC, it is necessary to develop a better understanding of the molecular mechanisms underlying the growth and progression of HCC. However, the molecular mechanism of HCC progression is not well understood.
Steroid receptor coactivator 1 (SRC-1; also known as NCOA1) was first discovered in 1995 as an authentic nuclear receptor coactivator (4). It belongs to the SRC/p160 family, which also includes SRC-2 (also known as NCOA2, TIF2, and GRIP1) and SRC-3 (also known as AIB1, p/CIP, ACTR, RAC3, and NCOA3) (5–7). SRC-1 functions as a transcriptional coactivator for steroid receptors, such as estrogen receptor (8) and androgen receptor (9, 10), as well as other transcriptional factors, such as PEA3 (11), Ets-2 (12), and AP-1 (13).
SRC-1 has been shown to play a role in the progression of prostate cancer and breast cancer. In androgen-dependent prostate tumors, increased SRC-1 expression correlates with lymph node metastasis (9). Ablation of SRC-1 in androgen-dependent LNCaP prostate cancer cells represses the activation of androgen receptor target genes and decreases androgen receptor-dependent cellular proliferation (9). SRC-1 protein is detected in 19–34% of human breast tumors. SRC-1 expression is significantly associated with large, high grade tumors, HER2 positivity, and disease recurrence and resistance to endocrine therapy. SRC-1 expression also serves as an independent predictor of disease-free survival (14–16). SRC-1 has been shown to promote breast cancer cell proliferation by enhancing estrogen receptor-α function (17).
Our previous study demonstrated that SRC-3/AIB1 promotes HCC progression by enhancing cell proliferation and invasion (18). SRC-1 is another member of the SRC/p160 family whose role in liver cancer remains unknown. In this study, we report that SRC-1 was overexpressed in 25 of 40 human HCC specimens, suggesting a potential impact by SRC-1 on HCC progression. Knockdown of SRC-1 decreased HCC cell proliferation and oncogenic potential in vitro by reducing the expression of c-Myc and PCNA. Consistent with these in vitro results, c-Myc expression levels in SRC-1-positive HCC specimens were higher than levels in SRC-1-negative HCC specimens, and expression of SRC-1 was positively correlated with PCNA in human HCC specimens. Taken together, our results show that SRC-1 is an important regulator of HCC growth and progression that has potential as a therapeutic molecular target for HCC.
Experimental Procedures
Patients and Liver Tissue Samples
Forty pairs of HCC specimens and adjacent non-tumorous liver tissues were obtained from the First Affiliated Hospital of Xiamen University (Xiamen, China). Informed consent was obtained from each patient, and the study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institute Research Ethics Committee at Xiamen University.
Cell Lines
Human HCC cell line HepG2, Huh-7, SK-Hep-1, MHCC97H, MHCC97L, and human hepatocyte cell line L-O2 were cultured in DMEM or 1640 with 10% fetal bovine serum (FBS) and penicillin/streptomycin.
Cell Transfection and Luciferase Assay
Cells were transfected with reporter plasmid together with PCR3.1-Rluc as an internal control in the presence of indicated plasmids. Cells were harvested at 48 h post-transfection. Luciferase activity was assayed and normalized to the value of Renilla luciferase activity.
RNA Interference and Establishment of Stable Cell Line
Specific and nonspecific siRNAs were purchased from Invitrogen. siRNA was transfected by Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. To generate stably transfected cells, cells were transfected with the specific shRNA plasmid. The puromycin was used to select stably transfected cells. SRC-1-specific targeting sequences are CCTCAGGGCAGAGAACCATCT and CACGACGAAATAGCCATAC, SRC-3-specific targeting sequence is AGACTCCTTAGGACCGCTT, c-Myc-specific targeting sequence is AGACCTTCATCAAAAACATTT.
Cell Proliferation
An MTT assay was used to detect cell proliferative rate. A total of 3000 cells were seeded into 96-well plates, and MTT was added to each well every day. Cells were incubated for 4 h until the solubilization buffer (10% SDS + 0.01 m HCl) was added. The absorbance was measured at 560 nm by a microplate reader.
Direct Cell Counting
For cell counting, 3 × 104 SRC-1-knockdown cells and control cells were seeded into 12-well plates. Then cells were stained with trypan blue, and live cells were counted every day. The results shown are from triplicate experiments.
Cell Cycle Analysis
For this analysis, 4 × 105 cells were seeded into 6-well plates, synchronized by serum starvation for 24 h, and re-entered into the cell cycle by an exchange of medium with 10% FBS DMEM for 24 h. Both adherent and non-adherent cells were harvested and fixed in 70% ethanol at 4 °C overnight. Cells were incubated with RNase A at 37 °C for 30 min and then stained with propidium iodide. Cell cycle status was measured by flow cytometry.
Measurement of DNA Synthesis
We measured DNA synthesis by using a Click-iT 5-ethynyl-2′-deoxyuridine (EdU) Alexa Fluor Imaging Kit (Invitrogen). Cells were seeded into 6-well cell culture plates with glass coverslips at a density of 2.0 × 105 cells/well and incubated in normal DMEM for 48 h. Culture medium was then replaced with DMEM containing 10 μm EdU (Invitrogen). After a 3-h incubation, the cells on the glass coverslips were fixed with formaldehyde. After permeabilization in PBS containing 0.5% Triton X-100, cells were stained with Alexa 594-azide (Invitrogen). Cells were mounted in mounting medium and imaged with a fluorescence microscope.
Detection of Cell Death Using Flow Cytometry
A total of 4 × 105 cells were seeded into 6-well plates and cultured with either 10% FBS or 1% FBS for 72 h. Both adherent and non-adherent cells were harvested and stained with propidium iodide. Cells were detected and analyzed by flow cytometry.
Reverse Transcription and Real-time PCR
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was obtained from 2 μg of total RNA using a reverse transcription kit (Toyobo). Real-time PCR was performed using universal SYBR Green Master (Roche Applied Science). Relative quantification was achieved by normalization to GAPDH.
Co-immunoprecipitation Assay
For co-immunoprecipitation (co-IP) assays, cells were first lysed with lysis buffer. Cell lysates were immunoprecipitated using the appropriate antibodies or control immunoglobulin G (IgG). After extensive washing, precipitates were analyzed by Western blot analysis.
Western Blot Analysis
Equal amounts of protein lysates were separated by SDS-PAGE and transferred onto PVDF membranes. Filters were probed with the following primary antibodies: anti-SRC-1 (Cell Signaling), anti-SRC-3 (Cell Signaling), anti-PCNA (Cell Signaling), anti-β-actin (Sigma), anti-β-catenin (Santa Cruz Biotechnology), anti-HA (Roche Applied Science), anti-FLAG (Sigma), anti-PARP (Santa Cruz Biotechnology), or anti-c-Myc (Abcam). Blots were then incubated with horseradish peroxidase-conjugated secondary antibody (Pierce) and visualized by chemiluminescence. The band density was quantified by densitometry using Scion imaging software and normalized to β-actin levels.
In Vitro Expression of SRC-1 Protein and GST Pull-down Assays
The coding sequence of SRC-1 was cloned into the pGS5 plasmid at a site downstream of the T7 promoter. Escherichia coli extract-based cell-free in vitro expression of SRC-1 protein was induced using the S30 T7 high yield protein expression system (Promega), following the manufacturer's protocol. For GST pull-down assays, 1 μg of E. coli-produced GST or GST-β-catenin protein was immobilized in glutathione-agarose (Roche Applied Science) for 1 h at room temperature. After several washes, the agarose was resuspended and then incubated with in vitro expressed SRC-1 protein at 4 °C. After extensive washing, bound proteins were eluted, resolved using SDS-PAGE, and visualized by immunoblotting.
Focus Formation
For focus formation assays, 500 cells were cultured in 6-well plates in DMEM with 10% FBS. After 3 weeks, cells were stained with 0.005% crystal violet for 10 min to detect foci, and colonies were counted.
Mice Treated with Lithium Chloride (LiCl)
Male mice were injected with LiCl, through the tail vein, at a dose of 12.9 mg/kg body weight every 12 h for 2 days. Control mice were injected with the same volume of vehicle (0.9% saline solution). Forty-eight hours following LiCl injection, mice were sacrificed, and the livers were harvested for Western blot analysis. All experimental procedures involving animals were performed in accordance with animal protocols approved by the Laboratory Animal Center of Xiamen University.
Tumor Xenograft
Four to six-week-old male nude mice were obtained from the Laboratory Animal Center of Xiamen University. A total of 4 × 106 SRC-1-knockdown cells or control cells were subcutaneously injected into the dorsal flanks of mice. From day 6 after injection, the tumor size was measured along two perpendicular axes every 3 days using a vernier caliper. The volume of the tumor was calculated using the formula, volume = length × width2 × 0.5. All experimental procedures involving animals were performed in accordance with animal protocols approved by the Laboratory Animal Center of Xiamen University.
Diethylnitrosamine (DEN)/CCl4-induced Liver Tumor Formation
SRC-1+/+ and SRC-1−/− mice were maintained on a C57BL/6 background. Fifteen-day-old male SRC-1+/+ and SRC-1−/− mice were intraperitoneally injected with DEN (25 mg/kg). This was followed by biweekly intraperitoneal injections of CCl4 (0.5 ml/kg), starting at 4 weeks of age. After 12 CCl4 injections, when the mice were 27 weeks old, they were sacrificed, and their livers were excised for photographing, quantitation, hematoxylin and eosin staining, and Western blot and immunohistochemical analysis. All experimental procedures involving animals were performed in accordance with animal protocols approved by the Laboratory Animal Center of Xiamen University.
Immunohistochemistry
Slides were soaked in preheated citrate buffer (pH 6.0) and heated in a microwave for 20 min to retrieve antigen. After cooling, slides were washed with PBS, incubated with Ki-67 antibody (1:500) for 1 h, and then incubated with a horseradish peroxidase-conjugated secondary antibody for 1 h. After washing, 3,3′-diaminobenzidine tetrahydrochloride was added to visualize labeled proteins.
Statistical Analysis
Data were collected from several independent experiments, with three replicates performed per experiment. All data are expressed as the mean ± S.E. Statistically significant effects (p < 0.05) were evaluated by a two-tailed Student's test.
Results
SRC-1 Is Frequently Overexpressed in Human HCC Specimens and Is Correlated with PCNA Expression
We previously reported that SRC-3/AIB1 was overexpressed in nearly 70% of human HCC specimens and that it promoted HCC progression by enhancing cell proliferation and invasion (18). As a member of the SRC/p160 family, the role of SRC-1 in HCC progression remains unknown. To evaluate the expression of SRC-1 in HCC, we performed Western blot analysis to detect protein levels of SRC-1 in a set of 40 human HCC specimens. As shown in Fig. 1A, SRC-1 was up-regulated in 25 (62.5%) of the 40 human HCC specimens compared with the surrounding non-tumorous tissues. In addition, mRNA levels of SRC-1 were also up-regulated in HCC specimens compared with non-tumorous tissues (Fig. 1B). Overexpression of SRC-1 in human HCC indicates that SRC-1 may be involved in HCC progression.
FIGURE 1.

SRC-1 is frequently overexpressed in many human HCC specimens and is correlated with PCNA expression. A, Western blot analysis of SRC-1 protein expression in a set of 40 human HCC specimens and surrounding non-tumorous tissue. N, non-tumorous tissue; T, tumor tissue. *, SRC-1-positive (+) HCC specimens. β-Actin was used as a loading control. B, SRC-1 mRNA levels were up-regulated in HCC specimens. SRC-1 mRNA levels in the non-tumorous and tumorous tissues were measured by real-time PCR. Relative quantification was achieved by normalization to GAPDH. **, p < 0.01. C, Western blot analysis of SRC-1 and PCNA protein expression in SRC-1-positive (+) and SRC-1-negative (−) human HCC specimens. β-Actin was used as a loading control. Number 34 specimens were used for quantitative normalization between images. D, quantitative analysis of PCNA protein expression in SRC-1-positive (+) and SRC-1-negative (−) human HCC specimens. **, p < 0.01. Band density in C was quantified by densitometry using Scion Imaging software and normalized to β-actin levels. The relative protein levels were determined by normalization to the number 34 specimen. E, correlation between SRC-1 protein levels and PCNA protein levels in human HCC specimens. Statistical t tests indicated a highly significant positive correlation (r = 0.73, p < 0.01). Band density in C was quantified and normalized to β-actin levels. The relative protein levels were determined by normalization to the number 34 specimen. These experiments were performed at least twice with similar results. Error bars, S.E.
To determine whether SRC-1 enhances HCC cell proliferation, levels of the cell proliferation marker PCNA were analyzed in HCC specimens. Tumors were divided into two groups according to SRC-1 protein expression: SRC-1-positive (+) and SRC-1-negative (−). As shown in Fig. 1, C and D, SRC-1-positive tumors showed significantly higher protein levels of PCNA than SRC-1-negative tumors. In addition, a positive correlation was identified between the protein levels of PCNA and SRC-1 in HCC specimens (Fig. 1E). These results suggest that SRC-1 may enhance HCC cell proliferation.
Down-regulation of SRC-1 Decreases HCC Cell Proliferation
To determine whether SRC-1 expression is up-regulated in the HCC cell lines compared with the hepatocyte cell line, we used Western blot analysis to compare the protein expression of SRC-1 across five human HCC cell lines (HepG2, SK-Hep-1, Huh-7, MHCC97H, and MHCC97L) and the hepatocyte cell line L-O2. As shown in Fig. 2A, SRC-1 was significantly increased in the HCC cell lines compared with L-O2 cells. To determine the role of SRC-1 in cell proliferation, RNA interference was used to knock down SRC-1 expression. Transfection of two SRC-1-specific siRNAs (siSRC-1-1 and siSRC-1-2) decreased the protein level of SRC-1 in HepG2, Huh-7, and SK-Hep-1 cells (Fig. 2B). Significantly, down-regulation of SRC-1 decreased proliferation in HepG2, Huh-7, and SK-Hep-1 cells, as measured by an MTT assay (Fig. 2C).
FIGURE 2.

Down-regulation of SRC-1 decreases HCC cell proliferation. A, protein expression of SRC-1 was up-regulated in the HCC cell lines (HepG2, SK-Hep-1, Huh-7, MHCC97H, and MHCC97L) compared with the hepatocyte cell line L-O2. L-O2 was cultured in 1640, whereas the other cell lines were cultured in DMEM. β-actin was used as a loading control. This experiment was performed twice with similar results. B, transfection of two SRC-1-specific siRNAs decreased protein levels of SRC-1 in HepG2, Huh-7, and SK-Hep-1 cells. Cells were transfected with siRNAs for 72 h and then harvested for Western blot analysis. This experiment was performed twice with similar results. C, knockdown of SRC-1 decreased proliferation in HepG2, Huh-7, and SK-Hep-1 cells, as measured by an MTT assay. Cells were transfected with siRNAs for 48 h and then seeded into 96-well plates for the MTT assay. This experiment was performed twice with similar results. D, proliferation was reduced in stable SRC-1-knockdown HepG2 cells, as measured by the MTT assay. This experiment was performed twice with similar results. E, proliferation was reduced in stable SRC-1-knockdown HepG2 cells, as measured by direct cell counting. This experiment was performed twice with similar results. F, knockdown of SRC-1 inhibited HCC cell colony formation. *, p < 0.05; **, p < 0.01. This experiment was performed twice with similar results. Error bars, S.E.
To establish stable SRC-1-knockdown cell lines for further study, HepG2 cells were transfected with pSuper-SRC-1-shRNA (shSRC-1) or control (shCtrl) constructs. Puromycin was used to select stably transfected cells. Two SRC-1-knockdown stable cell lines (shSRC-1-14 and shSRC-1-15) were established, and the down-regulation of SRC-1 was confirmed in these cell lines by Western blot analysis (Fig. 2D, inset). Stable knockdown of SRC-1 significantly decreased cell proliferation, as measured by MTT assay and direct cell counting (Fig. 2, D and E), and colony formation, as measured by a colony formation assay (Fig. 2F). These results indicate that SRC-1 promotes HCC cell growth.
Down-regulation of SRC-1 Decreases the Expression of c-Myc
Because down-regulation of SRC-1 decreased HCC cell proliferation, the cell cycle was analyzed to determine whether down-regulation of SRC-1 inhibits cell cycle progression. Flow cytometric analysis showed that the percentage of SRC-1-knockdown cells in G1 phase was increased compared with control cells. This increase was associated with a reduction in the number of cells at S and G2/M phase (Fig. 3A). To detect proliferative cells, EdU staining was performed, and EdU-incorporated cells were calculated. Consistent with a reduced number of SRC-1-knockdown cells in S phase, as measured by flow cytometric analysis, down-regulation of SRC-1 decreased the proportion of EdU-incorporated cells (Fig. 3B). In addition, the percentage of cells in sub-G1 phase, which indicates apoptotic cells, was higher in SRC-1-knockdown cell lines (Fig. 3A), indicating that down-regulation of SRC-1 significantly increases cell apoptosis under normal growth conditions.
FIGURE 3.

Down-regulation of SRC-1 decreases c-Myc expression. A, cell cycle analyses of stable SRC-1-knockdown cells and control cells were performed by flow cytometry. A total of 4 × 105 cells were seeded into 6-well plates, synchronized by serum starvation for 24 h, and re-entered into the cell cycle by an exchange of 10% FBS DMEM for 24 h. Cell were harvested, and cell cycle status was measured by flow cytometry. This experiment was performed three times with similar results. B, down-regulation of SRC-1 decreased DNA synthesis. DNA synthesis analyses of stable SRC-1-knockdown cells and control cells were performed by EdU staining. This experiment was performed twice with similar results. C, c-Myc mRNA levels were significantly decreased in stable SRC-1-knockdown cells. c-Myc mRNA levels were measured by real-time PCR. Relative quantification was achieved by normalization to GAPDH. This experiment was performed twice with similar results. D, c-Myc and PCNA protein levels were significantly decreased in stable SRC-1-knockdown cells, as measured by Western blot analysis. This experiment was performed twice with similar results. E, SRC-1 positively regulated c-Myc protein levels. Either SRC-1-specific siRNAs or SRC-1 expression constructs were transfected into HepG2 cells for 48 h. Cells were then harvested for Western blot analysis. This experiment was performed three times with similar results. F, knockdown of c-Myc decreased proliferation in HepG2 cells, as measured by MTT assay. This experiment was performed twice with similar results. G, knockdown of c-Myc decreased DNA synthesis. DNA synthesis analyses of c-Myc-knockdown cells and control cells were performed by EdU staining. This experiment was performed twice with similar results. H, transfection of c-Myc into SRC-1-knockdown cells restored cell proliferation. Control constructs were transfected into wild-type control cells, and control constructs and c-Myc expression constructs were transfected into SRC-1-knockdown cells for 48 h, respectively. A total of 3000 cells were then seeded into 96-well plates for cell proliferation analyses using an MTT assay. This experiment was performed twice with similar results. I, c-Myc mRNA levels in SRC-1-positive (+) human HCC specimens were significantly higher than those in SRC-1-negative (−) HCC specimens. c-Myc mRNA levels were measured by real-time PCR. Relative quantification was achieved by normalization to GAPDH. *, p < 0.05; **, p < 0.01. Error bars, S.E.
It has been reported that SRC-1 can regulate the expression of c-Myc in breast cancer cells (12, 19). To determine whether c-Myc expression is affected by SRC-1 in HCC cells, the mRNA levels of c-Myc in stable SRC-1-knockdown and control cells was examined. As shown in Fig. 3C, SRC-1-knockdown cells exhibited a significant decrease in mRNA levels of c-Myc compared with control cells. Consistent with these results, protein levels of c-Myc were also significantly decreased in SRC-1-knockdown cells compared with control cells (Fig. 3D). In addition, SRC-1-knockdown cells showed a significant reduction in the levels of PCNA protein (Fig. 3D). Consistent with data obtained previously from stable SRC-1-knockdown cells, the transient knockdown of SRC-1 significantly decreased protein levels of c-Myc (Fig. 3E, left). By contrast, overexpression of SRC-1 significantly increased protein levels of c-Myc (Fig. 3E, right). To confirm the role of c-Myc in HepG2 cell proliferation, c-Myc-specific siRNA was used to knock down c-Myc expression in HepG2 cells, and cell proliferation was measured. As shown in Fig. 3, F and G, down-regulation of c-Myc did indeed significantly decrease proliferation in HepG2 cells, as measured by an MTT assay and EdU staining.
To determine whether SRC-1 affects cell proliferation through regulation of c-Myc, SRC-1-knockdown cells were transfected with c-Myc expression constructs to restore c-Myc expression, and then cell proliferation was examined. The results showed that transfection of c-Myc expression constructs rescued cell proliferation in SRC-1-knockdown cells (Fig. 3H), indicating that SRC-1 affects cell proliferation, at least in part, through the regulation of c-Myc.
In agreement with the results obtained from the in vitro experiments, c-Myc expression was significantly higher in SRC-1-positive human HCC tissue than in SRC-1-negative HCC tissue (Fig. 3I), indicating that SRC-1 up-regulates c-Myc to promote human HCC growth.
SRC-1 Cooperates with β-Catenin to Enhance the Expression of c-Myc
It has been reported that SRC-2, which belongs to the SRC/p160 family, can interact with β-catenin to promote its transactivation activity (20, 21). Because c-Myc can be directly regulated by the Wnt/β-catenin signaling pathway, we hypothesized that SRC-1 might cooperate with β-catenin to enhance the transactivation of c-Myc. We therefore transfected SRC-1 and/or β-catenin expression constructs, together with a c-Myc promoter reporter, into cells in the absence or presence of LiCl, a reagent that can enhance the Wnt/β-catenin signaling pathway. In the absence of LiCl, c-Myc promoter activity was increased 4- and 11-fold by SRC-1 and β-catenin transfections, respectively, whereas it was increased 57-fold when co-transfected with both SRC-1 and β-catenin (Fig. 4A). In the presence of LiCl, the effects of SRC-1 and/or β-catenin on the promoter activity of c-Myc were further enhanced (Fig. 4A). These results suggest that SRC-1 cooperates with β-catenin to enhance the promoter activity of c-Myc.
FIGURE 4.

SRC-1 cooperates with β-catenin to enhance c-Myc expression. A, SRC-1 cooperated with β-catenin to enhance the promoter activity of c-Myc. 293T cells were transfected with c-Myc promoter reporter and SRC-1 and/or β-catenin expression plasmids. Cells were then treated with 50 mm LiCl or vehicle for 24 h. Luciferase activity was measured 48 h after transfection. **, p < 0.01. This experiment was performed three times with similar results. B, overexpression of SRC-1 and β-catenin significantly increased TOPFlash reporter activity. 293T cells were transfected with TOPFlash reporter and SRC-1 and/or β-catenin expression plasmids. Cells were then treated with 50 mm LiCl or vehicle for 24 h. Luciferase activity was measured 48 h after transfection. This experiment was performed three times with similar results. C, down-regulation of SRC-1 decreased c-Myc protein levels normally induced by β-catenin. This experiment was performed twice with similar results. D, the expression of c-Myc, normally induced by LiCl treatment, was significantly decreased in SRC-1−/− mice. SRC-1+/+ and SRC-1−/− mice were treated with LiCl for 48 h, and then c-Myc protein levels in the liver were detected. This experiment was performed three times with similar results. E, co-IP analysis of the interaction between transfected SRC-1 and β-catenin. 293T cells were transfected with FLAG-SRC-1 and HA-β-catenin for 48 h. Cell lysates were immunoprecipitated using the appropriate antibodies (FLAG or HA) or control IgG. This experiment was performed twice with similar results. F, co-IP analysis of the interaction between endogenous SRC-1 and β-catenin in HepG2 cells. Cell lysates of HepG2 cells were immunoprecipitated using the appropriate antibodies (β-catenin or SRC-1) or control IgG. This experiment was performed twice with similar results. G, GST pull-down analysis of the in vitro interaction between SRC-1 and β-catenin. E. coli-produced GST or GST-β-catenin protein was incubated with SRC-1 protein that was produced by an E. coli extract-based cell-free protein synthesis system for GST pull-down assays. This experiment was performed twice with similar results. H, SRC-1 interacted with the Arm3–10 domains of β-catenin. Shown is GST pull-down analysis of the interaction between SRC-1 and the different domains of β-catenin. Arrows, GST fusion proteins. This experiment was performed twice with similar results. I, β-catenin interacted with the bHLH-S/T and HAT domains of SRC-1. Shown is co-IP analysis of the interaction between β-catenin and the different domains of SRC-1. Arrows, immunoprecipitated FLAG-tagged proteins. This experiment was performed twice with similar results. Error bars, S.E.
To determine whether SRC-1 specifically enhances β-catenin transactivation activity, we examined the effects of SRC-1 on the activity of the TOPFlash reporter, a well established Wnt/β-catenin reporter. Overexpression of SRC-1 and β-catenin synergistically increased TOPFlash reporter activity in the absence or presence of LiCl (Fig. 4B), indicating that SRC-1 is involved in the Wnt/β-catenin signaling pathway.
To test whether SRC-1 regulates the expression of c-Myc, at least in part, through β-catenin, HepG2 cells were transfected with β-catenin expression constructs together with siCtrl or siSRC-1. As shown in Fig. 4C, overexpression of β-catenin significantly enhanced c-Myc protein levels. However, this enhancement was dramatically suppressed when SRC-1 expression was knocked down (Fig. 4C).
To detect whether SRC-1 is involved in Wnt/β-catenin signaling in normal hepatocytes in vivo, SRC-1+/+ and SRC-1−/− mice were treated with LiCl, which has been shown to activate the Wnt/β-catenin signaling pathway in the liver (22). As shown in Fig. 4D, LiCl treatment induced levels of c-Myc in the livers of SRC-1+/+ mice significantly higher than those in SRC-1−/− mice, indicating that SRC-1 is required for optimal activation of the Wnt/β-catenin signaling pathway for inducing c-Myc expression not only in HCC cells but also in normal hepatocytes.
To detect whether SRC-1 and β-catenin interact with each other, FLAG-tagged SRC-1 and HA-tagged β-catenin were co-transfected into 293T cells, and co-IP was performed. Anti-HA antibodies, but not control IgG, immunoprecipitated SRC-1 from cell lysates (Fig. 4E, top). Reciprocally, anti-FLAG antibodies, but not control IgG, immunoprecipitated β-catenin from the cell lysates (Fig. 4E, bottom). In addition, the interaction between endogenous SRC-1 and β-catenin in HepG2 cells was detected by co-IP assays using anti-SRC-1 and anti-β-catenin antibodies (Fig. 4F). To determine whether SRC-1 can directly bind to β-catenin, E. coli-produced GST-β-catenin protein was incubated with SRC-1 protein produced by an E. coli extract-based cell-free protein synthesis system developed for GST pull-down assays. The results showed that the GST-β-catenin protein, but not GST, was able to pull down SRC-1 (Fig. 4G), indicating that SRC-1 directly binds to β-catenin. This is in agreement with a previous report (23). Furthermore, GST pull-down assays showed that SRC-1 protein interacted with the Arm3–10 domains of β-catenin (Fig. 4H), and co-IP assays showed that β-catenin protein interacted with the bHLH-S/T and HAT domains of SRC-1 (Fig. 4I).
Down-regulation of SRC-1 Impairs Xenograft Tumor Maintenance
To investigate the role of SRC-1 in HCC progression in vivo, control cells and cells from two stable SRC-1-knockdown cell lines were injected into nude mice, and the growth of xenograft tumor was measured every 3 days. SRC-1-knockdown tumors grew much slower than control tumors (Fig. 5A). Strikingly, SRC-1-knockdown tumors began to shrink on day 12 and were completely regressed on day 21 (Fig. 5A), indicating that SRC-1 plays an important role in xenograft tumor maintenance.
FIGURE 5.

Down-regulation of SRC-1 impairs xenograft tumor maintenance. A, knockdown of SRC-1 inhibited HCC cell tumorigenesis in vivo. Stable SRC-1-knockdown HepG2 cells and control cells were subcutaneously injected into the backs of nude mice. Beginning 6 days after cell injection, tumor volume was measured every 3 days. n = 5; *, p < 0.05; **, p < 0.01. This experiment was performed twice with similar results. B, on day 15 after injection, SRC-1-knockdown tumors showed a significant decrease in tumor volume compared with control tumors. An image of a tumor is shown. This experiment was performed twice with similar results. C, Ki-67 and H&E staining of tissue sections from SRC-1-knockdown and control tumors. A representative image is shown. D, the expression of the intranuclear proteins poly(ADP-ribose) polymerase and PCNA is lost in SRC-1-knockdown tumors. This experiment was performed three times with similar results. E, down-regulation of SRC-1 significantly increased serum starvation-induced cell death. A total of 4 × 105 cells were seeded into 6-well plates and cultured with either 10% FBS or 1% FBS for 72 h. Both adherent and non-adherent cells were harvested for cell death detection. This experiment was performed three times with similar results. Error bars, S.E.
To identify the reason why SRC-1-knockdown tumors regressed, control cells and stable SRC-1-knockdown cells were injected into nude mice, and the mice were sacrificed on day 15. As shown in Fig. 5B, tumor size in the SRC-1-knockdown group was smaller than in the SRC-1 control group. Consistent with previous data showing SRC-1-knockdown tumors grew much slower than control tumors, many Ki-67-positive cells were detected in the control tumors, whereas few Ki-67-positive cells were detected in the SRC-1-knockdown tumors (Fig. 5C). H&E staining showed that control tumor cells exhibited normal cellular morphologies, but the SRC-1-knockdown tumor cells exhibited characteristics of necrosis. The nuclei of SRC-1-knockdown tumor cells were ruptured, and inflammatory cell infiltration was observed in SRC-1-knockdown tumors (Fig. 5C). Consistent with these results, expression of the intranuclear proteins poly(ADP-ribose) polymerase and PCNA was lost in SRC-1-knockdown tumors, as measured by Western blot analysis (Fig. 5D).
SRC-1-knockdown tumors exhibited extensive cell death, suggesting that a lack of nutrients and growth factors in vivo may enhance death of SRC-1-knockdown cells. Therefore, we speculated that SRC-1 may prevent cell death induced by nutrient and growth factor deficiency. To mimic nutrient and growth factor deficiency conditions in vitro, we induced serum starvation by culturing SRC-1-knockdown and control cells for 72 h in medium supplemented with 1% FBS. As shown in the left panel of Fig. 5E, serum-starved SRC-1-knockdown cells expressed significantly higher levels of cleaved poly(ADP-ribose) polymerase than serum-starved control cells, indicating that SRC-1-knockdown cells experience significantly higher levels of cell death than serum-starved control cells. Similar results were obtained by measuring cell death with flow cytometry (Fig. 5E, right). Collectively, these results indicate that SRC-1 plays an important role in tumor maintenance, at least in part, by preventing cell death induced by nutrient/growth factor deficiency.
SRC-1−/− Mice Are Resistant to DEN/CCl4-induced Liver Tumor Formation
To determine whether loss of SRC-1 reduces the development and growth of HCC in mice, we used an in vivo DEN/CCl4-induced liver tumor formation model for study (24). After 25 weeks of DEN/CCl4 treatment, SRC-1+/+ mice developed extensive liver tumors, whereas SRC-1−/− mice developed fewer and smaller liver tumors (Fig. 6, A–C). Protein levels of c-Myc and PCNA were significantly higher in liver tumors from SRC-1+/+ mice than in tumors from SRC-1−/− mice (Fig. 6D), whereas the expression of β-catenin was comparable in liver tumors from both types of mice (Fig. 6D). Consistently, SRC-1+/+ tumors exhibited a significantly higher proliferation rate compared with SRC-1−/− tumors, as determined by Ki-67 staining (Fig. 6, E and F). These in vivo results provide strong genetic evidential support for an important role for SRC-1 in liver tumor formation.
FIGURE 6.

SRC-1−/− mice were resistant to DEN/CCl4-induced liver tumor formation. A, images of livers with surface tumors from SRC-1+/+ and SRC-1−/− mice after 25 weeks of DEN/CCl4 treatment. B, quantitation of the numbers of surface liver tumors from SRC-1+/+ and SRC-1−/− mice (*, p < 0.05; n = 5). C, quantitation of the diameter of the largest surface liver tumors from SRC-1+/+ and SRC-1−/− mice (*, p < 0.05; n = 5). D, c-Myc and PCNA protein levels were significantly decreased in liver tumors from SRC-1−/− mice compared with SRC-1+/+ mice. Liver tissue lysates from five SRC-1+/+ or SRC-1−/− mice were mixed together for Western blot analysis of SRC-1, c-Myc, PCNA, and β-catenin. β-Actin was used as a loading control. N, non-tumorous tissue; T, tumor tissue. This experiment was performed three times with similar results. E, Ki-67 staining of tissue sections from non-tumorous liver tissues and liver tumors from SRC-1+/+ and SRC-1−/− mice. A representative image is shown. F, quantitation of Ki-67-positive tumor cells (**, p < 0.01; NS, not significant; n = 4 tumors/group). Error bars, S.E.
SRC-1 Cooperates with SRC-3 to Promote HCC Cell Proliferation
We previously reported that SRC-3 was frequently overexpressed in human HCC specimens and that it promoted HCC progression by enhancing cell proliferation and invasiveness (18, 25), indicating that SRC-3 is an oncogene in liver cancer. Because both SRC-1 and SRC-3 are overexpressed in human HCC and promote HCC progression, there was an interest in examining the expression profiles of SRC-1 and SRC-3 in same sets of HCC specimens. As summarized in Table 1, simultaneous overexpression of both SRC-1 and SRC-3 (SRC-1(+)/SRC-3(+) group) was observed in 19 (47.5%) of the 40 HCC specimens. Overexpression of SRC-1 or SRC-3 alone, in the SRC-1(+)/SRC-3(−) group or SRC-1(−)/SRC-3(+) group, was observed in 6 (15%) or 4 (10%) of the 40 HCC specimens, respectively. A total of 11 (27.5%) of the 40 HCC specimens did not exhibit overexpression of either SRC-1 or SRC-3 in the SRC-1(−)/SRC-3(−) group. To determine whether there was a correlation between cell growth and the expression profiles of SRC-1 and SRC-3, we examined the expression of PCNA in the four different groups of HCC specimens. As shown in Fig. 7A, compared with the SRC-1(−)/SRC-3(−) group, PCNA expression was significantly increased in the SRC-1(+)/SRC-3(−), SRC-1(−)/SRC-3(+), and SRC-1(+)/SRC-3(+) HCC specimens, with SRC-1(+)/SRC-3(+) HCC specimens exhibiting the highest PCNA expression. These results indicate that SRC-1 and SRC-3 cooperate to promote HCC cell proliferation. This notion was corroborated by the finding that simultaneous knockdown of SRC-1 and SRC-3 significantly decreased proliferation in HepG2 cells compared with control cells and SRC-1 or SRC-3 single knockdown cells (Fig. 7B). Knockdown of SRC-1 and SRC-3 in HepG2 cells was confirmed by Western blot analysis (Fig. 7C). The c-Myc protein was decreased by knockdown of SRC-1 or SRC-3 and further decreased by simultaneous knockdown of SRC-1 and SRC-3 (Fig. 7C), indicating that both SRC-1 and SRC-3 regulate c-Myc expression.
TABLE 1.
Expression profiles of SRC-1 and SRC-3 in human HCC specimens
| Expression profile | HCC specimen number | Percentage |
|---|---|---|
| % | ||
| SRC-1(+)/SRC-3(+) | 1, 6, 12, 13, 15, 21, 24, 26, 29, 30, 32, 34, 37, 38, 39, 41, 45, 46, 49 | 47.5 |
| SRC-1(+)/SRC-3(−) | 7, 19, 40, 54, 55, 59 | 15.0 |
| SRC-1(−)/SRC-3(+) | 9, 11, 22, 58 | 10.0 |
| SRC-1(−)/SRC-3(−) | 3, 4, 5, 28, 42, 43, 44, 48, 50, 56, 57 | 27.5 |
FIGURE 7.

SRC-1 cooperates with SRC-3 to promote HCC cell proliferation. A, PCNA protein levels were increased in SRC-1(+)/SRC-3(−), SRC-1(−)/SRC-3(+), and SRC-1(+)/SRC-3(+) HCC specimens compared with SRC-1(−)/SRC-3(−) HCC specimens. B, simultaneous down-regulation of SRC-1 and SRC-3 led to a more significant reduction in cell proliferation than down-regulation of SRC-1 or SRC-3 alone. HepG2 cells were transfected with siRNAs specific for SRC-1 and/or SRC-3 for 48 h and seeded into 96-well plates for MTT assays. This experiment was performed three times with similar results. C, c-Myc protein levels were further decreased after simultaneous knockdown of SRC-1 and SRC-3. This experiment was performed three times with similar results. D, bufalin treatment for 24 h dramatically reduced the protein levels of SRC-1, SRC-3, and c-Myc in HepG2 and SK-Hep-1 cells. HepG2 and SK-Hep-1 cells were treated with 10 or 20 nm bufalin for 24 h, and cells were lysed for Western blot analysis. This experiment was performed twice with similar results. E, bufalin treatment significantly inhibited proliferation in HepG2 and SK-Hep-1 cells. A total of 3000 cells were seeded into 96-well plates and treated with 10 or 20 nm bufalin for the indicated time before MTT was added. This experiment was performed twice with similar results. *, p < 0.05; **, p < 0.01. Error bars, S.E.
Recently, Wang et al. (26) reported that the cardiac glycoside bufalin is a potent small molecule inhibitor of SRC-1 and SRC-3. Bufalin strongly reduced protein levels of SRC-1 and SRC-3 and blocked breast and lung cancer cell growth (26). To determine whether bufalin reduces protein levels of SRC-1 and SRC-3 in HCC cells and inhibits the proliferation of HCC cells, we treated the HCC cell lines HepG2 and SK-Hep-1 with 10 and 20 nm bufalin for the indicated time, respectively. As shown in Fig. 7D, bufalin treatment for 24 h dramatically reduced protein levels of SRC-1, SRC-3, and c-Myc in both HepG2 and SK-Hep-1 cells. Bufalin treatment also significantly inhibited proliferation in HepG2 and SK-Hep-1 cells (Fig. 7E). These results indicate that bufalin is able to inhibit HCC cell proliferation, at least in part, by reducing the protein levels of SRC-1 and SRC-3.
The Expression of SRC-1 Positively Correlates with PCNA and c-Myc in Human HCC Specimens from a Gene Expression Omnibus (GEO) Profile Data Set
To determine whether the positive correlation between the expression of SRC-1 and PCNA/c-Myc can be verified in a larger cohort of human HCC specimens, we analyzed the expression of SRC-1, PCNA, and c-Myc in 135 human HCC specimens from a publicly available GEO profile data set (GSE20017) (27). As shown in Fig. 8, the expression of SRC-1 was positively correlated with PCNA (r = 0.51, p < 0.001) and c-Myc (r = 0.24, p = 0.005). These results support our notion that SRC-1 plays an important role in HCC progression.
FIGURE 8.
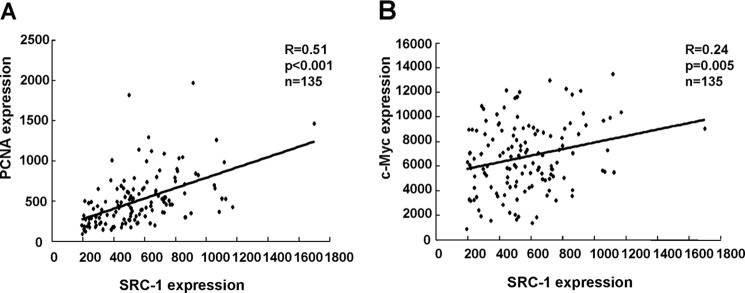
The expression of SRC-1 positively correlates with PCNA and c-Myc in human HCC specimens from a GEO profile data set. A, positive correlation between SRC-1 and PCNA (r = 0.51, p < 0.001, n = 135). Shown are expression profiles of 135 human HCC specimens from a GEO data set (GSE20017). The PCNA probe ID used was ILMN_1694177, and the SRC-1 probe ID used was ILMN_2335198. B, positive correlation between SRC-1 and c-Myc (r = 0.24, p = 0.005, n = 135). Expression profiles of 135 human HCC specimens from a GEO data set (GSE20017), the c-Myc probe ID used was ILMN_2110908, and the SRC-1 probe ID used was ILMN_2335198.
Discussion
It has been reported that SRC-1 is frequently overexpressed and plays a crucial promoting role in several human cancers, including breast cancer and prostate cancer. However, the role of SRC-1 in liver cancer progression remains unknown. In this study, we found that SRC-1 was overexpressed in 62.5% of human HCC specimens and that it was positively correlated with the expression of the cell proliferation marker PCNA, suggesting that SRC-1 plays an important role in HCC growth. Furthermore, down-regulation of SRC-1 decreased proliferation and oncogenic potential in HCC cells. Knock-out of SRC-1 in mice reduced DEN/CCl4-induced liver tumor formation and the expression of c-Myc and PCNA in liver tumors. These results demonstrate that SRC-1 indeed plays an important role in HCC development. The oncogenic protein c-Myc is frequently overexpressed in human liver cancers, where it plays a critical role in regulating cellular growth and survival. Overexpression of c-Myc in hepatic cells has been found to lead to HCC formation in mice (28). In this study, protein and mRNA levels of c-Myc were decreased in SRC-1-knockdown cells but increased in SRC-1-overexpressing cells, indicating that SRC-1 positively regulates c-Myc expression. Transfection of c-Myc expression constructs into SRC-1-knockdown cells restored cell proliferation, suggesting that SRC-1 promotes HCC cell proliferation via positive regulation of c-Myc expression. It has been reported that c-Myc is required for tumor maintenance in vivo (29, 30). Therefore, the phenomena that down-regulation of SRC-1 dramatically impaired HCC xenograft tumor maintenance, and knock-out of SRC-1 in mice significantly reduced DEN/CCl4-induced liver tumor formation, may be due to down-regulation of c-Myc expression.
Given that SRC-1 and β-catenin had a synergistic effect on the induction of c-Myc promoter activity and that c-Myc was identified as a direct target of the Wnt/β-catenin signaling pathway, we hypothesized that SRC-1 may act as a coactivator, cooperating with β-catenin to enhance c-Myc expression. Co-overexpression of SRC-1 and β-catenin significantly increased Wnt/β-catenin reporter activity, indicating that SRC-1 is involved in the Wnt/β-catenin signaling pathway. Co-IP and GST pull-down analysis demonstrated that SRC-1 directly interacted with β-catenin. These results indicate that SRC-1 serves as a coactivator, cooperating with β-catenin to enhance c-Myc expression.
The SRC/p160 coactivator family consists of only three members: SRC-1, SRC-2, and SRC-3 (7). SRC-1, -2, and -3 proteins share 50–55% similarity. Due to the high structural and functional similarities shared by the SRC/p160 coactivators, all SRC/p160 family members coactivate many nuclear receptors, including androgen receptor and estrogen receptor, as well as other transcription factors, in cell-based reporter assays (31). However, SRC-1, -2, and -3 also play distinct roles in the context of different biological processes. For example, SRC/p160 family individual knock-out mice display differentially altered expression profiles for a large number of genes, indicating that SRC-1, -2, and -3 also have many non-overlapping functions in the liver (32). We reported in this and a previous study that SRC-1 and SRC-3 were overexpressed in human HCC specimens and that they promoted HCC progression by enhancing cell proliferation (18, 25), indicating that SRC-1 and SRC-3 are oncogenes in liver cancer. Surprisingly, it has been reported that a loss-of-function mutation in SRC-2 in a mouse model predisposed the mice to DEN-induced liver tumorigenesis (33), indicating that SRC-2 is a tumor suppressor in liver cancer.
Given that both SRC-1 and SRC-3 play important roles in HCC progression and that they are both overexpressed in 47.5% of HCC specimens, the simultaneous down-regulation of SRC-1 and SRC-3 may be an effective strategy for treating liver cancer. Indeed, double knockdown of SRC-1 and SRC-3 significantly decreased proliferation of HCC cells compared with control cells and single knockdown cells. It was reported that miR-206 targets the mRNAs encoding SRC-1 and SRC-3 but not SRC-2, resulting in a decrease in the expression of SRC-1 and SRC-3 (34). These results suggest that administration of miR-206 may also be a therapeutic approach for treating liver cancer. In addition, it was reported that a natural polyphenol, gossypol, and a cardiac glycoside, bufalin, significantly reduced the protein levels of SRC-1 and SRC-3, but not SRC-2 (26, 35). These results suggest that gossypol and bufalin are also potential new drugs for the treatment of HCC that can simultaneously target the oncogenes SRC-1 and SRC-3 without affecting the tumor suppressor SRC-2. In conclusion, the current and our previous studies demonstrate that SRC-1 and SRC-3 are overexpressed in HCC and that they promote HCC progression by enhancing cell proliferation, suggesting that down-regulation of SRC-1 as well as SRC-3 may be an effective therapeutic strategy for treating HCC.
Author Contributions
Z. T., M. L., and C. Y. conceived and coordinated the study and wrote the paper. Z. T., M. L., W. W., P. M., L. Y., K. L., and W. R. designed, performed, and analyzed the experiments shown in Figs. 1–7. W. L. collected the human HCC specimens and analyzed the experiments shown in Fig. 1. H. Z. analyzed the data shown in Fig. 8. J. X. provided the SRC-1 knockout mice, analyzed the experiments shown in Fig. 6, and revised the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgment
We thank Zhimeng Yao (Cancer Research Center, Shantou University Medical College) for GEO data analysis.
This work was supported by National Basic Research Program of China (973 Program) Grant 2015CB553800 (to C. Y.); Natural Science Foundation of China Grants 81372176 (to C. Y.) and 31301128 (to P. M.); Fundamental Research Funds for the Central Universities Grant 2012121038 (to C. Y.); and Xiamen Science and Technology Plan Grant 3502Z20124049 (to W. L.). This work was also supported in part by National Institutes of Health Grant R01 CA112403 (to J. X.). The authors declare that they have no conflicts of interest with the contents of this article.
- HCC
- human hepatocellular carcinoma
- SRC
- steroid receptor coactivator
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- EdU
- 5-ethynyl-2′-deoxyuridine
- IP
- immunoprecipitation
- DEN
- diethylnitrosamine
- PCNA
- proliferating cell nuclear antigen
- GEO
- Gene Expression Omnibus.
References
- 1. Farazi P. A., DePinho R. A. (2006) Hepatocellular carcinoma pathogenesis: from genes to environment. Nat. Rev. Cancer 6, 674–687 [DOI] [PubMed] [Google Scholar]
- 2. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global cancer statistics. CA Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 3. Perz J. F., Armstrong G. L., Farrington L. A., Hutin Y. J., Bell B. P. (2006) The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 45, 529–538 [DOI] [PubMed] [Google Scholar]
- 4. Oñate S. A., Tsai S. Y., Tsai M. J., O'Malley B. W. (1995) Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science 270, 1354–1357 [DOI] [PubMed] [Google Scholar]
- 5. Hong H., Kohli K., Trivedi A., Johnson D. L., Stallcup M. R. (1996) GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc. Natl. Acad. Sci. U.S.A. 93, 4948–4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Torchia J., Rose D. W., Inostroza J., Kamei Y., Westin S., Glass C. K., Rosenfeld M. G. (1997) The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 387, 677–684 [DOI] [PubMed] [Google Scholar]
- 7. Walsh C. A., Qin L., Tien J. C., Young L. S., Xu J. (2012) The function of steroid receptor coactivator-1 in normal tissues and cancer. Int. J. Biol. Sci. 8, 470–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tai H., Kubota N., Kato S. (2000) Involvement of nuclear receptor coactivator SRC-1 in estrogen-dependent cell growth of MCF-7 cells. Biochem. Biophys. Res. Commun. 267, 311–316 [DOI] [PubMed] [Google Scholar]
- 9. Agoulnik I. U., Vaid A., Bingman W. E., 3rd, Erdeme H., Frolov A., Smith C. L., Ayala G., Ittmann M. M., Weigel N. L. (2005) Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 65, 7959–7967 [DOI] [PubMed] [Google Scholar]
- 10. Gregory C. W., He B., Johnson R. T., Ford O. H., Mohler J. L., French F. S., Wilson E. M. (2001) A mechanism for androgen receptor-mediated prostate cancer recurrence after androgen deprivation therapy. Cancer Res. 61, 4315–4319 [PubMed] [Google Scholar]
- 11. Qin L., Liu Z., Chen H., Xu J. (2009) The steroid receptor coactivator-1 regulates twist expression and promotes breast cancer metastasis. Cancer Res. 69, 3819–3827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Al-azawi D., Ilroy M. M., Kelly G., Redmond A. M., Bane F. T., Cocchiglia S., Hill A. D., Young L. S. (2008) Ets-2 and p160 proteins collaborate to regulate c-Myc in endocrine resistant breast cancer. Oncogene 27, 3021–3031 [DOI] [PubMed] [Google Scholar]
- 13. Qin L., Chen X., Wu Y., Feng Z., He T., Wang L., Liao L., Xu J. (2011) Steroid receptor coactivator-1 upregulates integrin α(5) expression to promote breast cancer cell adhesion and migration. Cancer Res. 71, 1742–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Myers E., Hill A. D., Kelly G., McDermott E. W., O'Higgins N. J., Buggy Y., Young L. S. (2005) Associations and interactions between Ets-1 and Ets-2 and coregulatory proteins, SRC-1, AIB1, and NCoR in breast cancer. Clin. Cancer Res. 11, 2111–2122 [DOI] [PubMed] [Google Scholar]
- 15. Hudelist G., Czerwenka K., Kubista E., Marton E., Pischinger K., Singer C. F. (2003) Expression of sex steroid receptors and their co-factors in normal and malignant breast tissue: AIB1 is a carcinoma-specific co-activator. Breast Cancer Res. Treat. 78, 193–204 [DOI] [PubMed] [Google Scholar]
- 16. Fleming F. J., Hill A. D., McDermott E. W., O'Higgins N. J., Young L. S. (2004) Differential recruitment of coregulator proteins steroid receptor coactivator-1 and silencing mediator for retinoid and thyroid receptors to the estrogen receptor-estrogen response element by β-estradiol and 4-hydroxytamoxifen in human breast cancer. J. Clin. Endocrinol. Metab. 89, 375–383 [DOI] [PubMed] [Google Scholar]
- 17. Karmakar S., Foster E. A., Smith C. L. (2009) Unique roles of p160 coactivators for regulation of breast cancer cell proliferation and estrogen receptor-α transcriptional activity. Endocrinology 150, 1588–1596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu Y., Chen Q., Li W., Su X., Chen T., Liu Y., Zhao Y., Yu C. (2010) Overexpression of transcriptional coactivator AIB1 promotes hepatocellular carcinoma progression by enhancing cell proliferation and invasiveness. Oncogene 29, 3386–3397 [DOI] [PubMed] [Google Scholar]
- 19. McBryan J., Theissen S. M., Byrne C., Hughes E., Cocchiglia S., Sande S., O'Hara J., Tibbitts P., Hill A. D., Young L. S. (2012) Metastatic progression with resistance to aromatase inhibitors is driven by the steroid receptor coactivator SRC-1. Cancer Res. 72, 548–559 [DOI] [PubMed] [Google Scholar]
- 20. Li H., Kim J. H., Koh S. S., Stallcup M. R. (2004) Synergistic effects of coactivators GRIP1 and β-catenin on gene activation: cross-talk between androgen receptor and Wnt signaling pathways. J. Biol. Chem. 279, 4212–4220 [DOI] [PubMed] [Google Scholar]
- 21. Song L. N., Gelmann E. P. (2005) Interaction of β-catenin and TIF2/GRIP1 in transcriptional activation by the androgen receptor. J. Biol. Chem. 280, 37853–37867 [DOI] [PubMed] [Google Scholar]
- 22. Du Q., Park K. S., Guo Z., He P., Nagashima M., Shao L., Sahai R., Geller D. A., Hussain S. P. (2006) Regulation of human nitric oxide synthase 2 expression by Wnt β-catenin signaling. Cancer Res. 66, 7024–7031 [DOI] [PubMed] [Google Scholar]
- 23. Fonte C., Grenier J., Trousson A., Chauchereau A., Lahuna O., Baulieu E. E., Schumacher M., Massaad C. (2005) Involvement of β-catenin and unusual behavior of CBP and p300 in glucocorticosteroid signaling in Schwann cells. Proc. Natl. Acad. Sci. U.S.A. 102, 14260–14265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Uriarte I., Latasa M. U., Carotti S., Fernandez-Barrena M. G., Garcia-Irigoyen O., Elizalde M., Urtasun R., Vespasiani-Gentilucci U., Morini S., de Mingo A., Mari M., Corrales F. J., Prieto J., Berasain C., Avila M. A. (2015) Ileal FGF15 contributes to fibrosis-associated hepatocellular carcinoma development. Int. J. Cancer 136, 2469–2475 [DOI] [PubMed] [Google Scholar]
- 25. Liu Y., Tong Z., Li T., Chen Q., Zhuo L., Li W., Wu R. C., Yu C. (2012) Hepatitis B virus X protein stabilizes amplified in breast cancer 1 protein and cooperates with it to promote human hepatocellular carcinoma cell invasiveness. Hepatology 56, 1015–1024 [DOI] [PubMed] [Google Scholar]
- 26. Wang Y., Lonard D. M., Yu Y., Chow D. C., Palzkill T. G., Wang J., Qi R., Matzuk A. J., Song X., Madoux F., Hodder P., Chase P., Griffin P. R., Zhou S., Liao L., Xu J., O'Malley B. W. (2014) Bufalin is a potent small-molecule inhibitor of the steroid receptor coactivators SRC-3 and SRC-1. Cancer Res. 74, 1506–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mínguez B., Hoshida Y., Villanueva A., Toffanin S., Cabellos L., Thung S., Mandeli J., Sia D., April C., Fan J. B., Lachenmayer A., Savic R., Roayaie S., Mazzaferro V., Bruix J., Schwartz M., Friedman S. L., Llovet J. M. (2011) Gene-expression signature of vascular invasion in hepatocellular carcinoma. J. Hepatol. 55, 1325–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lin C. P., Liu C. R., Lee C. N., Chan T. S., Liu H. E. (2010) Targeting c-Myc as a novel approach for hepatocellular carcinoma. World J. Hepatol. 2, 16–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shachaf C. M., Gentles A. J., Elchuri S., Sahoo D., Soen Y., Sharpe O., Perez O. D., Chang M., Mitchel D., Robinson W. H., Dill D., Nolan G. P., Plevritis S. K., Felsher D. W. (2008) Genomic and proteomic analysis reveals a threshold level of MYC required for tumor maintenance. Cancer Res. 68, 5132–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Soucek L., Whitfield J., Martins C. P., Finch A. J., Murphy D. J., Sodir N. M., Karnezis A. N., Swigart L. B., Nasi S., Evan G. I. (2008) Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu J., Wu R. C., O'Malley B. W. (2009) Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nature Rev. Cancer 9, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jeong J. W., Kwak I., Lee K. Y., White L. D., Wang X. P., Brunicardi F. C., O'Malley B. W., DeMayo F. J. (2006) The genomic analysis of the impact of steroid receptor coactivators ablation on hepatic metabolism. Mol. Endocrinol. 20, 1138–1152 [DOI] [PubMed] [Google Scholar]
- 33. O'Donnell K. A., Keng V. W., York B., Reineke E. L., Seo D., Fan D., Silverstein K. A., Schrum C. T., Xie W. R., Mularoni L., Wheelan S. J., Torbenson M. S., O'Malley B. W., Largaespada D. A., Boeke J. D. (2012) A Sleeping Beauty mutagenesis screen reveals a tumor suppressor role for Ncoa2/Src-2 in liver cancer. Proc. Natl. Acad. Sci. U.S.A. 109, E1377–E1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Adams B. D., Cowee D. M., White B. A. (2009) The role of miR-206 in the epidermal growth factor (EGF) induced repression of estrogen receptor-α (ERα) signaling and a luminal phenotype in MCF-7 breast cancer cells. Mol. Endocrinol. 23, 1215–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang Y., Lonard D. M., Yu Y., Chow D. C., Palzkill T. G., O'Malley B. W. (2011) Small molecule inhibition of the steroid receptor coactivators, SRC-3 and SRC-1. Mol. Endocrinol. 25, 2041–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]