

Background: It was reported that amyloid precursor-like protein 2 (APLP2) increases PCSK9-mediated low-density lipoprotein receptor (LDLR) degradation, and sortilin facilitates PCSK9 secretion.
Results: APLP2 or sortilin deficiency/overexpression in cells/mice did not affect LDLR degradation by PCSK9. However, APLP2 binds sortilin, and PCSK9 enhances their degradation.
Conclusion: APLP2/sortilin are not required for PCSK9 activity on LDLR, but their interaction may modulate APLP2 functions.
Significance: APLP2 and sortilin do not affect LDLR levels.
Keywords: cardiovascular disease, dyslipidemia, lipoprotein metabolism, low-density lipoprotein (LDL), proprotein convertase subtilisin/kexin type 9 (PCSK9), APLP2, Sortilin (SORT1), low-density lipoprotein receptor (LDLR) degradation
Abstract
Amyloid precursor-like protein 2 (APLP2) and sortilin were reported to individually bind the proprotein convertase subtilisin/kexin type 9 (PCSK9) and regulate its activity on the low-density lipoprotein receptor (LDLR). The data presented herein demonstrate that mRNA knockdowns of APLP2, sortilin, or both in the human hepatocyte cell lines HepG2 and Huh7 do not affect the ability of extracellular PCSK9 to enhance the degradation of the LDLR. Furthermore, mice deficient in APLP2 or sortilin do not exhibit significant changes in liver LDLR or plasma total cholesterol levels. Moreover, cellular overexpression of one or both proteins does not alter PCSK9 secretion, or its activity on the LDLR. We conclude that PCSK9 enhances the degradation of the LDLR independently of either APLP2 or sortilin both ex vivo and in mice. Interestingly, when co-expressed with PCSK9, both APLP2 and sortilin were targeted for lysosomal degradation. Using chemiluminescence proximity and co-immunoprecipitation assays, as well as biosynthetic analysis, we discovered that sortilin binds and stabilizes APLP2, and hence could regulate its intracellular functions on other targets.
Introduction
The proprotein convertase subtilisin/kexin 9 (PCSK9)5 is the ninth member of a family of serine proteinases usually implicated in the cleavage of protein precursors (1, 2). Independent of its catalytic activity (3, 4), PCSK9 binds the low-density lipoprotein receptor (LDLR), mainly at the cell surface of hepatocytes, and favors its transit into the endosomal/lysosomal pathway (5). PCSK9 is thus an important post-translational regulator of the LDLR (6–8). Gain-of-function mutations in the PCSK9 gene are responsible for lower LDLR levels and accumulation of plasma LDL-cholesterol (LDLc), and thus result in autosomal dominant hypercholesterolemia (9), whereas loss-of-function mutations lead to hypocholesterolemia (10). Because circulating PCSK9 mostly originates from liver hepatocytes (11, 12), this has led a number of pharmaceutical companies to develop neutralizing monoclonal antibodies (mAb) that target plasma PCSK9 and thereby prevent its binding to the LDLR and the subsequent PCSK9-LDLR complex internalization and lysosomal degradation (13, 14). These mAb were injected every 2 weeks to patients that led to a sustained 60–70% reduction of circulating LDLc for more than 1 year (13–15). These data underlined the key role of extracellular PCSK9 in the human liver. Phase III clinical trials are now being evaluated worldwide in >100,000 subjects that suffer from hypercholesterolemia.
Despite its critical role as a regulator of the LDLR and attractiveness as a pharmacological target, the mechanism(s) by which extracellular PCSK9 is sorted into clathrin-coated endosomes and subsequently to lysosomes for degradation (5) remain largely unknown. Previous studies showed that PCSK9 can target the LDLR for endosomes/lysosomes degradation either intracellularly from the trans-Golgi network (16) or extracellularly from the cell surface (17), with the latter seemingly the predominant route in liver (13–15). The catalytic domain of PCSK9 binds to the EGFA-like repeat of the LDLR (18, 19). However, the ability of PCSK9 to escort the LDLR to endosomes/lysosomes and enhance the degradation of the PCSK9-LDLR complex requires the presence of its C-terminal Cys-His-rich domain (20–22).
An additional factor that has been demonstrated to be implicated in the PCSK9-mediated extracellular degradation of the LDLR in hepatocytes is the autosomal recessive hypercholesterolemia (ARH) adaptor protein (23–25). ARH binds the FDNPVY motif in the cytosolic tail (CT) of the LDLR, the β2-adaptin subunit of AP-2, and the clathrin heavy chain, thereby recruiting the receptor into clathrin-coated pits (26, 27). Mutations in, or deletion of, ARH render the LDLR at the cell surface of hepatocytes insensitive to extracellular PCSK9, emphasizing the importance of ARH in the mechanism of extracellular PCSK9-induced LDLR degradation in liver (23).
However, a truncated LDLR mutant lacking its CT (K811X; ΔCT) is still degraded in CHO cells treated exogenously with the PCSK9 gain-of-function mutant D374Y (PCSK9-D374Y) (28). This was confirmed in HEK293 cells treated with exogenous wild-type (WT) PCSK9 (29). Moreover, the substitution of the transmembrane domain of LDLR-ΔCT by that of the very low density lipoprotein receptor (VLDLR) or angiotensin converting enzyme 2 did not hamper degradation, revealing that the wild type (WT) CT or transmembrane domains are not critical for the internalization and subsequent degradation of the PCSK9-LDLR complex (29).
Altogether, these findings suggest the existence of an additional transmembrane protein containing an ARH binding motif, (F/Y)XNPXY, that would bind ARH and interact with the luminal PCSK9-LDLR complex, allowing the internalization of LDLR-ΔCT mutants. Although we recently showed that the LDLR-related protein 1 (LRP1) is a PCSK9 target in cell lines, we eliminated the possibility that it is an essential factor for LDLR regulation, even though the LDLR can compete with LRP1 for PCSK9 activity (29).
In a study analyzing a blocking antibody of PCSK9 that efficiently targeted PCSK9 for degradation independently of a direct interaction with the LDLR (30), the amyloid precursor-like protein 2 (APLP2), which contains an YENPTY motif, was identified as a PCSK9 partner binding its Cys-His-rich domain. This suggested that PCSK9 can divert LDLR to lysosomes from the luminal side of the membrane via its Cys-His-rich domain interaction with APLP2 (31). Recent evidence also showed that the LDLR forms a complex with APLP2 at the cell surface (32). This finding was not unprecedented, as APLP2 is involved in the transport of transmembrane proteins such as MHC class I molecules to lysosomes (33, 34).
Another recent study reported that sortilin can bind PCSK9 in vitro at neutral or slightly acidic pH, and enhance its secretion (35). Sortilin, like APLP2, is a type-I membrane-bound protein that acts as a sorting receptor regulating the traffic of proteins from the Golgi/cell surface to lysosomes (36). Whether sortilin regulates the ability of PCSK9 to enhance the degradation of the LDLR by either the intracellular or extracellular pathways is unknown.
In the present study, we sought to better characterize the role of APLP2 and sortilin in PCSK9 trafficking and activity in cell lines and in Aplp2−/− and Sort1−/− mice. Our data revealed that, in hepatocyte cell lines and in the liver, the ability of extracellular and intracellular PCSK9 to enhance the degradation of the LDLR, ex vivo and in vivo, does not require the presence of APLP2 or sortilin. In addition, no change in liver LDLR levels were observed in Aplp2- or sortilin-deficient mice. However, using a proximity assay based on a split luciferase (37), we showed that APLP2 and sortilin can interact with each other in HEK293 cells.
Experimental Procedures
Cell Culture, Plasmids, siRNA, Antibodies and Reagents
Human embryonic kidney (HEK293) cells and HepG2 cells were purchased from American Type Culture Collection. Hepatoma Huh7 cells were a gift of François Jean (University of British Columbia). HepG2 cells stably expressing human PCSK9 were generated in the laboratory and were found to secrete 25 ng of PCSK9/ml. All cells were maintained at 37 °C under 5% CO2, and grown in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (Invitrogen). The bicistronic pIRES2-EGFP vector (Clontech) allowed the expression of human PCSK9 without or with a C-terminal V5 tag (1), human ER-localized PCSK9-KDEL with a C-terminal V5 tag (5), and human APLP2 with C-terminal V5 and His6 tags. The pcDNA3 vector (Invitrogen) was used to express an unrelated protein to this study, mouse 7B2 (38), as a negative control and human sortilin carrying a C-terminal Myc tag. The pCMV6-XL5 vector (Origene) was used to express human sortilin (no tag).
Human untagged or V5-tagged PCSK9 was detected with a rabbit polyclonal (5) or monoclonal V5 (mAb-V5; Invitrogen) antibody, whereas human V5-tagged PCSK9-KDEL was detected with a mAb-V5 antibody. Sortilin was detected using a mAb-Myc (Santa Cruz Biotechnology), rabbit polyclonal antibody (Abcam), or mouse mAb (BD Biosciences). Mouse APLP2 was detected using a rabbit polyclonal antibody kindly provided by Dr. G. Thinakaran (University of Chicago), whereas human APLP2 was detected with either mAb-V5 or a rabbit polyclonal antibody (Novus Biologicals). Human LDLR was detected using a goat polyclonal antibody (R/D Systems). Finally, β-actin and GAPDH antibodies were from Sigma and Abcam, respectively.
Purified PCSK9 was a generous gift from Dr. Rex Parker (Bristol-Myers Squibb; 1.7 μg/μl). Conditioned media were produced by incubating HEK293 cells stably transfected with the pIRES2-EGFP vector empty (−PCSK9) or expressing PCSK9-V5 (+PCSK9) overnight in serum-free DMEM. Media were then assessed by Western blotting, and the “+PCSK9” medium was found to contain ∼1 μg/ml of PCSK9 by ELISA (39, 40). The media were stored at −80 °C until use.
Co-expression of PCSK9 with Sortilin and/or APLP2 in HEK293, Huh7, and HepG2 Cells
Cells were transfected with vectors expressing human PCSK9, APLP2, and sortilin or in combinations using either jetPRIME (Polyplus) or FuGENE HD (Promega) transfection reagents for 48 h, according to the manufacturer's recommendations. An empty pIRES2-EGFP or pcDNA3–7B2 (38) vectors were used as controls. 24 h before harvest, cells were placed in serum-free DMEM.
siRNA of Sortilin and APLP2 in HepG2 and Huh7 Cells
Human APLP2-specific and control siRNAs were purchased from Origene (Fig. 1) or GE Healthcare BioSciences (Dharmacon; siGENOME SMARTpool; Fig. 3). Human sortilin siRNAs were purchased from the latter company.
FIGURE 1.
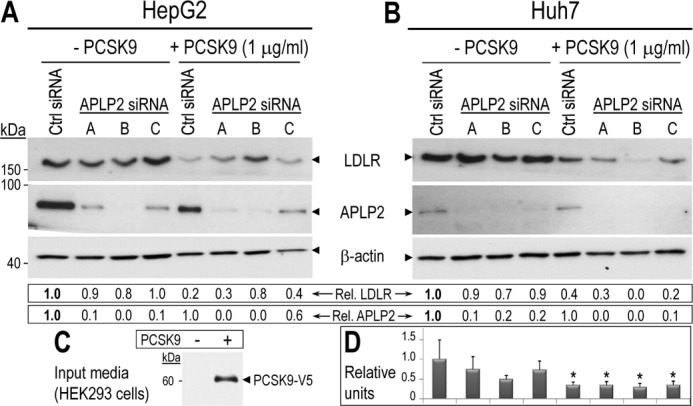
Exogenous PCSK9 can induce degradation of the LDLR in the absence of APLP2. HepG2 (A) and Huh7 (B) cells were transfected with a control non-target siRNA (Ctrl) or 3 different siRNAs targeting APLP2. Cells were incubated overnight with serum-free conditioned media lacking or containing 1 μg/ml of PCSK9-V5. HepG2 and Huh7 cell lysates were then subjected to Western blotting using LDLR, APLP2, and β-actin antibodies. LDLR and APLP2 signals were normalized to that of β-actin. C, the input HEK293 conditioned medium was analyzed using mAb-V5 to detect PCSK9-V5. D, duplicate samples of Huh7 cells matching those in panel B were analyzed by FACS to assess the cell surface LDLR levels. Values were normalized to that of the first lane (control non-target siRNA in the absence of PCSK9). Error bars represent S.E. *, p < 0.05 (Student's t test). The data shown here are representative of two to three independent experiments.
FIGURE 3.
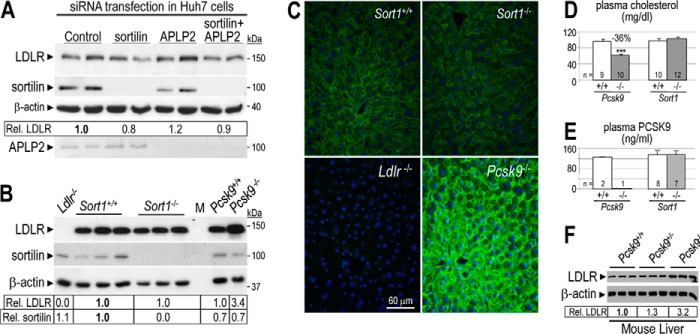
Sortilin depletion does not affect LDLR degradation by PCSK9 and has no cholesterol phenotype in mice. A, Huh7 cells were transfected with control non-target, APLP2-, and/or sortilin-specific siRNAs. After 48 h, cells were incubated in serum-free media for 24 h. Cell lysates were then subjected to Western blotting using LDLR, sortilin, APLP2, and β-actin antibodies. B, total liver extracts of Sort+/+ (n = 3), Sort1−/− (n = 3), Ldlr−/− (n = 1), Pcsk9+/+ (n = 1), and Pcsk9−/− (n = 1) mice were analyzed by Western blot analysis. Accurate quantification of LDLR and sortilin signals and their normalization to that of β-actin was obtained by LI-COR analysis of a duplicate gel. Relative LDLR/β-actin signals were normalized to that of control (A and B in bold). C, immunohistochemistry of surface LDLR (green) in the liver of Sort+/+, Sort1−/−, Ldlr−/−, and Pcsk9−/− mice. Surface LDLR levels in the liver of Sort1−/− and Sort+/+ mice were similar. Bar = 60 μm. Plasma (D) total cholesterol and (E) PCSK9 measured by ELISA, in control Pcsk9+/+ and Pcsk9−/− mice, and in Sort+/+ and Sort1−/− mice. Error bars represent S.E. ***, p < 3 × 10−5 (Student's t test). F, Western blot analysis of total liver extracts of Pcsk9+/+ (n = 3), Pcsk9+/− (n = 3), and Pcsk9−/− (n = 3) mice. Accurate quantification of LDLR and its normalization to that of β-actin was obtained by BioRad Image Lab 5.2 analysis of the same gel. Relative LDLR/β-actin signals were normalized to that of Pcsk9+/+ (in bold). These data are representative of at least two independent experiments.
For APLP2 depletion in Fig. 1, cells were transfected with control or APLP2-specific siRNA using jetPRIME (Polyplus) transfection reagent. For sortilin and/or APLP2 depletion in Fig. 3, cells were transfected using DharmaFECT 4 transfection reagent (GE Healthcare BioSciences) according to the manufacturer's instruction.
Western Blot Analyses
Cells were washed three times in PBS and lysed on ice in 1× RIPA buffer (50 mm Tris-HCl, pH 8.0, 1% (v/v) Nonidet P-40, 0.5% sodium deoxycholate, 150 mm NaCl, and 0.1% (v/v) SDS) supplemented with 1× complete protease inhibitor mixture (Roche Applied Science). Proteins were separated by 8% SDS-PAGE and transferred overnight to 0.45 μm PVDF membranes (PerkinElmer Life Sciences). The membranes were blocked for 1 h at room temperature in TBS-T (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat dry milk. Membranes were incubated with primary antibodies overnight at 4 °C in a 5% milk/TBS-T solution at the following dilutions: LDLR (1:1,000), PCSK9 (1:3,000), APLP2 (1:1,000), sortilin (1:5,000), mAb-Myc (1:1,000), mAb-V5 (1:5,000), and β-actin (1:2,500). Appropriate horseradish peroxidase-conjugated secondary antibodies were used at 1:10,000 in 5% milk/TBS-T and revealed by chemiluminescence using the Clarity Western ECL Substrate (Bio-Rad). Quantification in all cases was performed relative to β-actin using the NIH ImageJ software.
Biosynthetic Analyses and Immunoprecipitations
HEK293 cells were transiently transfected as described above. At 48 h post-transfection, the cells were washed in Cys/Met-free RPMI 1640 medium containing 0.2% BSA (Sigma) and pulse-labeled for 4 h with 250 μCi/ml of [35S]Met/Cys (PerkinElmer Life Sciences) in the presence or absence of 5 mm NH4Cl (Sigma). After the pulse, cells were lysed as reported previously (41) and cells lysates were immunoprecipitated with mAb-V5, mAb-Myc, or a rabbit polyclonal PCSK9 antibody (5). The immunoprecipitated proteins were resolved by SDS-PAGE on 8% Tricine gels, dried, and autoradiographed, as described (41).
Fluorescence-associated Cell Sorting (FACS)
Twenty-four hours post-transfection, cells were incubated overnight with serum-free DMEM and washed three times with calcium/magnesium-free Dulbecco's PBS containing 0.5% BSA and 1 g/liter of glucose (Solution A). Cells were then incubated for 5 min at 37 °C with 500 μl of 1× Versene solution (Invitrogen) and layered onto 5 ml of Solution A. Following centrifugation for 5 min at 1,000 rpm, cells were re-suspended in 1 ml of Solution A containing 1:100 of monoclonal human LDLR antibody (mAb-C7, Santa Cruz Biotechnology) and incubated for 30 min. After one wash with 5 ml of Solution A, cells were centrifuged, and incubated for 30 min in 1 ml of PBS containing 1:250 of Alexa Fluor 647 donkey anti-mouse (Molecular Probes). Cells were then washed in PBS, re-suspended in 300 μl of PBS containing 0.2% of propidium iodide, and analyzed by FACS for both propidium iodide (viable cells are propidium iodide-negative) and Alexa Fluor 647 using the CyAn flow cytometer (Beckman Coulter).
Isolation and Culture of Primary Hepatocytes
Hepatocytes were isolated from 8 to 10-week-old male livers using a two-step collagenase perfusion method as previously described (42). Following anesthesia by 2% isoflurane inhalation, the liver was perfused in situ via the inferior vena cava for 6 min at 37 °C with calcium-free HEPES buffer I (142 mm NaCl, 6.7 mm KCl, 10 mm HEPES, pH 7.6), and for 8 min with calcium-supplemented HEPES buffer II (4.7 mm CaCl2, 66.7 mm NaCl, 6.7 mm KCl, 100 mm HEPES, pH 7.4) containing 0.5 mg/ml of collagenase Type V (Sigma). The perfusion rates were set to 8 and 6 ml/min, respectively. In 3.5-cm Petri dishes coated with fibronectin (0.5 mg/ml, Sigma), 5 × 105 cells were seeded in Williams' medium E supplemented with 10% fetal bovine serum (GIBCO BRL). After 2 h, the medium was replaced with hepatozyme medium (GIBCO BRL) 12 h prior to treatment.
Mice and Genotyping
Aplp2 mice (43) were obtained from The Jackson Laboratory (stock number 004142) and genotyped as recommended. Liver samples from Aplp2+/+ and Aplp2−/− mice and analyzed in Fig. 2A were obtained from Dr. Zheng (Baylor College of Medicine). Sort1 mice were a kind gift from V. Wallace (University of Ottawa) and originally generated by C. R. Morales (44). The tail DNA of these mice was genotyped using the following forward 5′-GGGGTGAGAGGAACAGGAGGCA and reverse 5′-CCCCAGTGCTGTCTCCAACCCA (WT) or 5′-TCGGGGTAGCGGCTGAAGCA (EGFP; KO) primers. PCSK9 knock-out and transgenic mice were described previously (11). Mice were housed in a 12-h light/dark cycle and fed a standard diet (2018 Teklad global 18% protein rodent diet; Harlan Laboratories). All procedures were approved by the IRCM bioethics committee for animal care.
FIGURE 2.
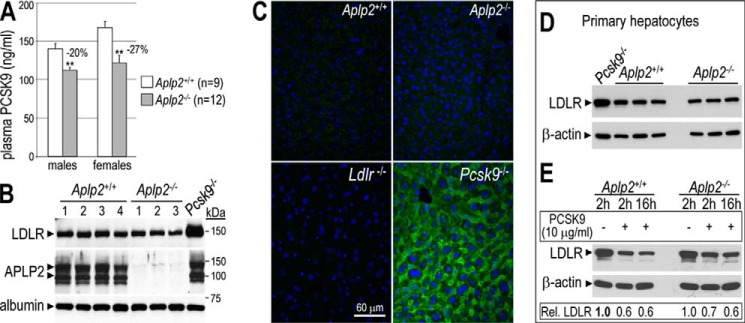
APLP2-deficient mice exhibit reduced levels of circulating PCSK9 but similar levels of total and cell surface LDLR in the liver. A, plasma levels of PCSK9 were measured by ELISA in Aplp2+/+ (WT) and Aplp2−/− mice. Error bars represent S.E. **, p < 0.01 (Student's t test). B, liver extracts of Aplp2+/+ (n = 4), Aplp2−/− (n = 3), and Pcsk9−/− (n = 1) mice were analyzed by Western blot analysis. The latter showed a 2.5-fold higher level of LDLR. Note that liver APLP2 levels were not affected in Pcsk9−/− mice (last lane). C, immunohistochemistry of surface LDLR (green) in the liver of Aplp2+/+, Aplp2−/−, Ldlr−/−, and Pcsk9−/− mice. Bar = 60 μm. D, LDLR levels were analyzed in primary hepatocytes isolated from Pcsk9−/− (n = 1), Aplp2+/+ and Aplp2−/− (n = 3) mice. E, Aplp2+/+ and Aplp2−/− primary hepatocytes were incubated without or with purified human PCSK9 (10 μg/ml) for 2 and/or 16 h. LDLR relative intensities to β-actin obtained by Western blotting were normalized to that of the Aplp2+/+ signal in the absence of PCSK9 (first lane in bold). These data are representative of at least two independent experiments.
Immunohistochemistry of LDLR in the Liver
Cryosections were incubated with a mouse LDLR antibody (1:150; R&D Systems number F2255) at 4 °C overnight and with Alexa Fluor 488-labeled anti-goat IgGs (1:150; Invitrogen) for 1 h at room temperature as previously described (12). Nuclei were counterstained with Hoechst dye (Sigma). Images were acquired using a Retiga EXi camera (Q-Imaging) mounted on a Leica DMRB microscope.
Quantitation of Plasma PCSK9 by ELISA
Mouse plasma were collected in EDTA-coated microtainer tubes (BD Biosciences) and 1 μl was used to measure PCSK9 using the CircuLex mouse/rat PCSK9 ELISA kit (number CY-8078; Boca Scientific).
Split-luciferase Assay
In this Gaussia princeps luciferase protein complementation assay, protein pairs assessed for their interaction are N or C terminally fused to one of the luciferase halves (Gluc1, Gluc2). When the two inactive complementary halves are brought in close proximity by interacting protein pairs, a significant luciferase activity is restored. The complete ORFs clones from the Human ORFeome resource consortium for human APLP2, Sec24A, Sec24B, Sec24C, Sec23B, Sec23IP, and sortilin were transferred by recombinational cloning (LR clonase reaction, Invitrogen) into Gateway-compatible destination vectors. Because of the presence of a signal peptide in secretory proteins, the luciferase moiety was fused at the C terminus of sortilin and APLP2 (in pC-Gluc1 or pC-Gluc2 vectors). DNA constructs were verified by sequencing.
HEK293 cells were seeded at a concentration of 3 × 104 cells/well in 96-well plates (Greiner Bio-One, number 655 083). After 24 h, cells were transfected using jetPEI (Polyplus) with 100 ng of each recombinant vector. At 24 h post-transfection, after a washing step in PBS, cells were harvested with 30 μl of Renilla lysis buffer (Promega, E2820) for 30 min, and luciferase enzymatic activity was measured in triplicates using a Berthold Centro XS LB960 luminometer by injecting 100 μl of Renilla luciferase assay reagent (Promega, E2820) into cell lysates and counting luminescence for 10 s. Results were expressed as a fold-change normalized over the sum of controls (normalized luminescence ratio) (37, 45).
APLP2 and sortilin interaction was validated by co-expression and immunoprecipitation. Cell extracts were incubated with mAb-Myc and protein A/G-agarose beads (Santa Cruz Biotechnology) at 4 °C overnight. The beads were washed with cell extraction buffer (20 mm HEPES, pH 7.4, 20 mm CaCl2 150 mm NaCl, and 0.5% Triton X-100). Laemmli buffer was added to the washed beads, boiled for 5 min, and subjected to SDS-PAGE followed by Western blot analysis.
In Situ Hybridization
Frozen whole bodies of postnatal mice (p10) and adult mouse livers were cut into 12-μm thick sections that were mounted onto glass slides and fixed in fresh 4% formaldehyde. RNA sense and antisense riboprobes were synthesized in vitro from recombinant PCRII vectors (Invitrogen) using the T7 and SP6 RNA polymerase promoters in the presence of [35S]UTP (>1,000 Ci/mmol; number NEG039H, PerkinElmer LAS Canada, Inc.), as previously reported (1). The RNA probes for mouse PCSK9, sortilin, and APLP2 covered amino acids 352–648, 468–825, and 384–722 in the protein sequence, respectively. Briefly, and as described previously (46), tissue sections were hybridized overnight with 50–80,000 cpm/μl of 35S-labeled cRNA probe, washed at 65 °C, and treated with 20 μg/ml of RNase A at 37 °C. After washing, the slides were dehydrated, exposed for 4 days, then dipped in Kodak NTB nuclear track emulsion, and finally exposed for 12 days at 4 °C.
Results
PCSK9 Can Induce Degradation of the LDLR in the Absence of APLP2
A recent report suggested that the extracellular PCSK9-induced post-endocytic sorting of the LDLR to endosomes/lysosomes is dependent on APLP2 (31). To assess whether APLP2 is an essential factor in the ability of PCSK9 to induce degradation of the LDLR, hepatocyte-derived HepG2 and Huh7 cells were transfected with control or APLP2 siRNAs and incubated overnight in a conditioned medium from HEK293 cells lacking or containing ∼1 μg/ml of PCSK9-V5 (Fig. 1). Cells were then lysed and the LDLR and APLP2 levels analyzed by Western blotting. The APLP2 knockdown efficiency ranged from 40 to 95% in HepG2 (Fig. 1A) and 80 to 95% in Huh7 cells (Fig. 1B). Importantly, under significantly reduced levels of APLP2, exogenous PCSK9 (input media; Fig. 1C) still induced degradation of up to 70% of the LDLR in HepG2 (Fig. 1A) and ≥90% in Huh7 cells (Fig. 1B). In addition, FACS analysis of the latter cells revealed a significant 60–70% decrease in cell surface levels of LDLR (Fig. 1D).
The original study by DeVay et al. (31) also used treatments with 50 μg/ml of purified PCSK9 for 2 h. Because our findings may be affected by the presence of additional factors in the HEK293 cell-conditioned media, or by the overnight incubation with PCSK9, we repeated the experiment described in Fig. 1 with pure PCSK9. Following transfection of APLP2 siRNA-B, HepG2, and Huh7 cells were incubated for 2 h or overnight with 50 μg/ml of pure PCSK9 (39). Our data showed that this high concentration of pure PCSK9 in the medium similarly enhanced the degradation of the LDLR independently of APLP2 (not shown). In conclusion, a substantial APLP2 deficiency did not block the ability of extracellular PCSK9 to induce the degradation of the LDLR in HepG2 and Huh7 cells.
APLP2-deficient Mice Exhibit Slightly Reduced Levels of Circulating PCSK9 but Similar Levels of Plasma Cholesterol, as Well as Total and Cell Surface LDLR in the Liver
To corroborate our results in vivo, we first analyzed the circulating levels of PCSK9 in Aplp2+/+ (WT) and Aplp2−/− mice, and found them slightly but significantly reduced by 20 and 27% in Aplp2−/− males and females, respectively (Fig. 2A). However, similar plasma total cholesterol levels were observed in both genotypes (116 versus 123 mg/dl in WT mice). In agreement with this observation, the lack of APLP2 did not affect the levels of liver total LDLR (Fig. 2B), nor those at the cell surface of hepatocytes (Fig. 2C). In contrast, livers lacking PCSK9 had 2.5- and 60-fold higher levels of total and surface LDLR, respectively (Fig. 2, B and C) (11, 12). Primary hepatocytes isolated from three WT and three Aplp2−/− littermates also exhibited the same levels of total LDLR (Fig. 2D). Furthermore, the LDLR levels were similarly reduced (30–40%) by exogenous PCSK9 (2- or 16-h incubations) in primary hepatocytes isolated from both genotypes (Fig. 2E). Finally, WT or Aplp2−/− primary hepatocytes secreted similar levels of endogenous PCSK9 (98.1 ± 4.2 versus 74.3 ± 20.3 ng/ml for WT mice; n = 3 and p = 0.12), suggesting that lower circulating levels of PCSK9 in Aplp2−/− male and female mice (Fig. 2A) are not due to a secretion defect of PCSK9 by hepatocytes. We also observed that the protein levels of APLP2 in the liver were not sensitive to the absence or overexpression of PCSK9 in either Pcsk9−/− or Tg(PCSK9) mice (11) (not shown).
Sortilin Depletion Does Not Affect LDLR Degradation by PCSK9 and Has No Cholesterol Phenotype in Mice
As this study was progressing, it was reported that sortilin facilitates PCSK9 secretion from mouse primary hepatocytes and that Sort1−/− mice displayed ∼30% lower levels of circulating PCSK9, whereas mice acutely overexpressing sortilin in the liver via adenoviral infections with recombinant AAV-sortilin exhibited increased levels of plasma PCSK9 (35). We thus assessed the role of sortilin in PCSK9-mediated LDLR degradation. Huh7 cells were transfected with either control siRNA or siRNAs specific to sortilin or APLP2, or both, and LDLR levels were estimated in cell lysates by Western blotting (Fig. 3A). Knockdown of sortilin, APLP2, or both revealed that lack of sortilin, alone or in combination with APLP2, had no significant impact on LDLR levels, suggesting that PCSK9-triggered LDLR degradation was not sensitive to lower levels of these transmembrane proteins in Huh7 cells. A similar lack of effect was observed in sortilin knock-out (KO; Sort1−/−) mice (44) as compared with Sort1+/+ (WT) mice (Fig. 3B). Thus, in the liver, neither total LDLR levels examined by Western blotting (Fig. 3B) nor surface levels examined by immunohistochemistry (Fig. 3C) were affected. In addition, plasma total cholesterol (Fig. 3D) and PCSK9 (Fig. 3E) levels were similar in WT and Sort1−/− littermates. Additionally, LDLR, PCSK9, and SREBP-2 mRNA expressions in WT and Sort1−/− livers were comparable (not shown).
Heterozygous Pcsk9+/− mice exhibit a ∼70% decrease in circulating PCSK9 levels as compared with WT mice (29.7 ± 11.0 versus 94.2 ± 11.0 ng/ml, respectively; n = 6–7 mice; p = 5 × 10−6). This higher than 50% decrease is likely due to increased LDLR levels on Pcsk9+/− hepatocytes (∼30% increase; Fig. 3F), thereby contributing to a further clearance of circulating PCSK9. Thus, a ∼70% decrease in circulating PCSK9 only resulted in a 1.3-fold increased liver LDLR levels (Fig. 3F). In contrast, liver extracts from Pcsk9−/− mice showed a 3.2–3.4-fold increase in LDLR levels (Fig. 3, B and F). Thus, our data suggest that the 2.2-fold increased levels of LDLR reported in Sort1−/− mice could not be primarily attributed to the loss of ∼30% of circulating PCSK9 (35).
Sortilin and APLP2 Are Novel Cellular Targets of PCSK9
Because the extracellular pathway of PCSK9 is more dominant in the liver (13–15), we next assessed whether APLP2 and sortilin could affect the PCSK9-triggered LDLR degradation via the intracellular pathway. We thus co-expressed PCSK9 and sortilin-Myc (or untagged sortilin; not shown) or APLP2-V5 in HEK293 (Fig. 4A). Unexpectedly, the intracellular levels of sortilin and APLP2 were dramatically decreased in HEK293 cells co-expressing PCSK9 (−90 and −40%, respectively; Fig. 4A), whereas neither protein was degraded in the presence of exogenous PCSK9 (not shown). This suggested that both sortilin and APLP2 are new targets of intracellular PCSK9. Note that the absence of any apparent reduction in intracellular PCSK9 levels is likely due to the fact that, at a given time, most of the protein is in the endoplasmic reticulum (ER), as evidenced by its sensitivity to endoglycosidase H digestion (1, 7).
FIGURE 4.
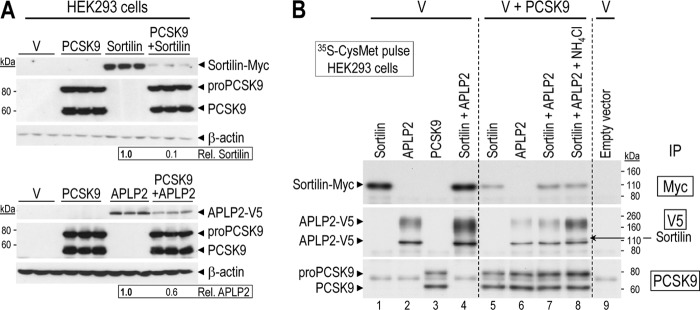
Sortilin and APLP2 are novel cellular targets of PCSK9. A, overexpressed PCSK9 induces sortilin and APLP2 degradation in HEK293 cells. Triplicate Western blot analyses revealing that both sortilin-Myc and APLP2-V5 expression levels in HEK293 cells were reduced by 90 and 40%, respectively, upon transfection with a PCSK9 plasmid, as compared with a control empty pIRES vector (V). Quantification of sortilin and APLP2 band intensities were normalized against those of β-actin. B, HEK293 cells transfected with a cDNA coding for an empty vector control (pIRES; V) or individually with human sortilin or APLP2 tagged at the C terminus with a Myc or V5 epitope, respectively, or together in the absence or presence of a cDNA coding for untagged PCSK9. The following day the cells were washed and then pulsed for 4 h with [35S]Met + Cys in the presence or absence of 5 mm NH4Cl. The cells were then extracted and their lysates immunoprecipitated (IP) with a mAb-V5 or mAb-Myc or a polyclonal antibody for PCSK9. The precipitates were separated on an 8% SDS-PAGE. The dried gel was then autoradiographed. Notice the co-precipitation of sortilin and APLP2 in the presence of NH4Cl. These data are representative of at least three independent experiments.
To better understand the dynamics of their PCSK9-induced disappearance, we analyzed the biosynthesis of APLP2 and sortilin (Fig. 4B) in the absence or presence of PCSK9. APLP2-V5 and sortilin-Myc were expressed in HEK293 cells that were pulse-labeled for 4 h with [35S]Met + Cys. Cell lysates were immunoprecipitated with corresponding antibodies and the precipitated proteins were separated by SDS-PAGE followed by autoradiography.
APLP2 seems to be first synthesized as an immature, likely ER-localized protein with an apparent molecular mass of ∼100 kDa, and to be subsequently shifted to a ∼220 kDa broad protein band, likely corresponding to chondroitin sulfate-modified forms (47, 48). Because the ∼100 kDa form was strongly increased in the presence of brefeldin A (not shown), which collapses the cis-medial Golgi cisternae with the ER (49), this suggests that the majority of nascent APLP2 undergoes maturation and subsequent degradation. The latter may occur in acidic endosomes/lysosomes, because preincubation with 5 mm NH4Cl (to alkalinize the cells) increased the levels of the ∼220 kDa wider band, with no effect on the ∼100 kDa one that is likely in the ER (neutral pH compartment; not shown).
Newly synthesized sortilin in HEK293 cells migrates with an apparent molecular mass of ∼105 kDa, just above APLP2 (Fig. 4B). Interestingly, co-expression of sortilin with APLP2 resulted in higher levels of the latter, suggesting that sortilin binding stabilizes APLP2 and partially rescues it from degradation. Conversely, APLP2 had no effect on the levels of sortilin (Fig. 4B, lanes 1 and 4).
Co-expression of PCSK9 with sortilin or APLP2 (Fig. 4B, lanes 5–8) resulted in a very significant reduction in the levels of either protein (compare lanes 1 and 5, and 2 and 6), as expected from Western blot analysis (Fig. 4A). The levels of APLP2 were mildly rescued by the co-expression of sortilin, but substantially rescued upon incubation of cells with 5 mm NH4Cl (compare lanes 6–8), resulting in a visible co-immunoprecipitated sortilin signal (see lane 8). These data suggest that PCSK9 can target APLP2 to endosomes/lysosomes for degradation.
In contrast, the PCSK9-induced degradation of sortilin is neither rescued by the presence of APLP2 nor 5 mm NH4Cl (compare lanes 1, 5, 7, and 8). This might be due to the fact that sortilin is more sensitive to PCSK9 than APLP2 (Fig. 4A). However, upon incubation of cells with both 5 mm NH4Cl and 10 μg/ml of the cell permeable Cys-protease inhibitor E64d, we did observe a partial rescue of sortilin (not shown), suggesting that here also PCSK9 induces the degradation of sortilin in the acidic compartments of endosomes/lysosomes.
To better understand the mechanism of PCSK9-induced degradation of sortilin and APLP2, we co-expressed sortilin (no tag), APLP2-V5, or soluble sAPLP2-V5 (lacking the transmembrane-cytosolic tail) with either PCSK9-V5 or ER-localized PCSK9-V5-KDEL (5) in HEK293 cells (Fig. 5). Western blot analysis showed that WT PCSK9 and ER-retained PCSK9-KDEL were able to degrade sortilin, APLP2, and sAPLP2. This suggested that all of these proteins bind to PCSK9 possibly already in the ER, and are then targeted for degradation. However, because PCSK9-KDEL is also able to degrade APLP2, sAPLP2, and sortilin (Fig. 5), this suggests that ER retention of the complex of PCSK9 with these proteins likely leads to their degradation within the ERAD system (50). Thus, it is probable that PCSK9 could enhance the degradation of APLP2 and sortilin by both the ERAD and the endosomes/lysosome hydrolases.
FIGURE 5.

Sortilin, APLP2, and soluble APLP2 are degraded by both PCSK9 and ER-localized PCSK9-KDEL isoforms. HEK293 cells were transfected with indicated DNA amounts of vectors encoding a control protein 7B2, sortilin (no tag), APLP2-V5, soluble APLP2-V5 (sAPLP2-V5), PCSK9-V5, or PCSK9-V5-KDEL, as indicated. After 48 h, lysates and media were analyzed by Western blotting for the indicated proteins. The data show that overexpressed PCSK9 or PCSK9-KDEL induces degradation of sortilin (A), APLP2 (B), and sAPLP2 (C) in HEK293 cells. Quantification of sortilin and APLP2 band intensities were normalized against those of β-actin or GAPDH. These data are representative of two independent experiments.
Co-expression of Sortilin, APLP2, or Both with PCSK9 Has No Major Effect on LDLR Degradation
Sortilin and APLP2 were targeted for degradation only by high levels of intracellular PCSK9. To verify whether this process modulates the activity of PCSK9 on the LDLR, Huh7 cells were transfected with vectors allowing the expression of either APLP2 and/or sortilin without or with that of PCSK9 (Fig. 6). As in HEK293 cells (Figs. 4 and 5) APLP2 and sortilin levels were also significantly decreased when these proteins were co-expressed with PCSK9 in Huh7 cells. Although the amount of LDLR estimated by Western blotting tended to decrease upon co-expression of sortilin and/or APLP2, the 62–67% enhancement of LDLR degradation by PCSK9 did not seem to be vary (Fig. 6). Thus neither APLP2 nor sortilin modify the activity of PCSK9 on the LDLR.
FIGURE 6.
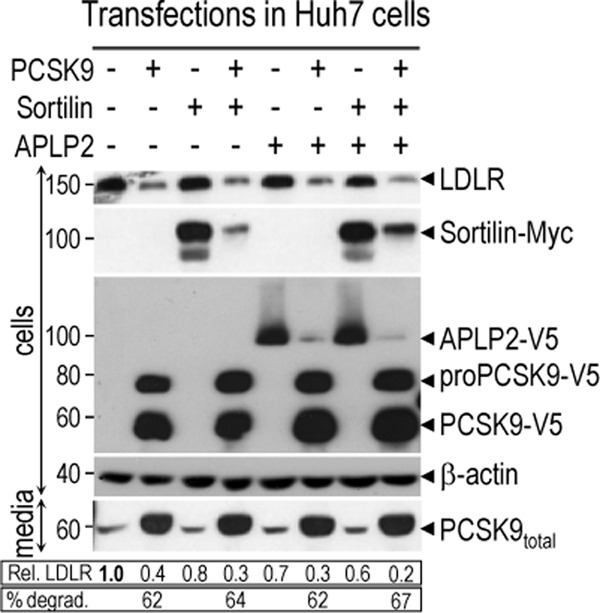
Co-expression of sortilin, APLP2, or both with PCSK9 has no major effect on LDLR degradation. Huh7 cells were transfected with a total of 3 μg using 1 μg of each vector encoding for either a control protein 7B2 (−), sortilin (+), APLP2 (+), or PCSK9 (+), as indicated. After 48 h, lysates were analyzed by Western blotting for expression of the LDLR, sortilin-Myc, APLP2-V5, intracellular pro- and mature-PCSK9-V5, and β-actin. Media were analyzed for secreted endogenous and overexpressed PCSK9-V5 using a rabbit polyclonal human PCSK9 antibody. Quantification of LDLR expression was normalized against that of β-actin. These data are representative of at least 3 different experiments showing similar results.
Sortilin Binds APLP2
A recent study showed by a genetic approach that the cytosolic adaptor protein Sec24A implicated in the ER-Golgi trafficking of COPII vesicles was critical for the secretion of PCSK9 from the ER into the medium (51). This facilitation is thought to be mediated by a membrane-bound protein that binds PCSK9 in the ER-lumen and Sec24A in the cytosol via its CT.
Because both APLP2 and sortilin were targeted for degradation by intracellular PCSK9 (Fig. 4A), and sortilin-stabilized APLP2 (Fig. 4B), a G. princeps luciferase-based complementation assay (37, 45) was used to assess a possible interaction of their CT with Sec24A (Figs. 7, A and B). Positive and negative interaction controls included the co-expression of Sec23 and Sec24 family members known to dimerize or not in the cytosol (52). As expected, the strongest interaction signal was observed between Sec24B and Sec23B. The lack of detection of interaction with Sec24B-Gluc2 C-term fusion/Sec23B-Gluc1 N-term protein pair suggested that the C-terminal region of Sec24B could be involved in forming a complex with Sec23B and Sec23IP. From this assay, we found that Sec24A did not bind APLP2 or sortilin, whereas Sec23B bound weakly to sortilin and APLP2, with the latter interaction best seen when Sec23B has the Gluc-1 domain at its N terminus (Fig. 7B). This ruled out sortilin or APLP2 as candidates for the putative ER-localized protein whose CT binds Sec24A (51). However, the assay unraveled an unsuspected strong interaction between sortilin and APLP2 (Fig. 7B, upper right corner). The binding domains are not defined, but the interaction was strong enough to result in close proximity of the two halves of the G. princeps luciferase, resulting in a strong luminescence signal. This interaction was validated by co-expressing in HEK293 cells sortilin-Myc and APLP2-V5 (Fig. 7C). Lysates of control cells (empty vector) or cells expressing sortilin-Myc and/or APLP2-V5 were immunoprecipitated with a Myc mAb. Western blot analyses of the precipitates revealed that sortilin co-immunoprecipitated with APLP2. The relative binding of sortilin to APLP2 compared with the input was estimated to be ∼10%. These data demonstrate that in HEK293 cells sortilin and APLP2 can bind to each other.
FIGURE 7.
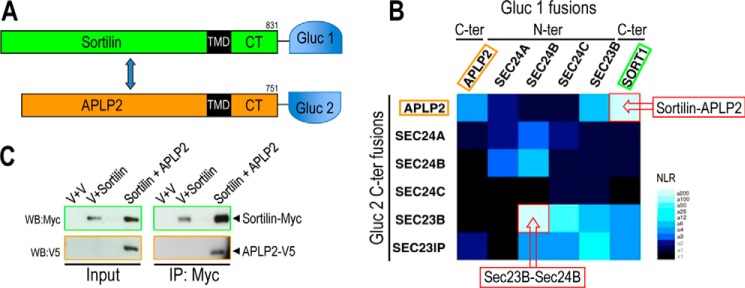
Sortilin binds APLP2. A, schematic diagram of sortilin and APLP2 fused to the G. princeps luciferase half-domains, Gluc-1 and Gluc-2, respectively. B, heat map generated by G. princeps luciferase complementation assay showing the interaction profile of 36 protein pairs. Normalized luminescence ratio ranging from strong to null interactions is displayed on a light blue to black scale. C, validation of sortilin-Myc and APLP2-V5 interaction was confirmed by co-expression in HEK293 cells and immunoprecipitation with mAb-Myc followed by Western blotting (WB) using a mAb-Myc or mAb-V5.
Discussion
Although targeting circulating PCSK9 is a very promising treatment for hypercholesterolemia, the mechanism(s) behind the ability of PCSK9 to enhance the degradation of the LDLR in endosomes/lysosomes is not yet elucidated. Because PCSK9 can still degrade a truncated form of the LDLR lacking its CT (28, 29), we hypothesized the existence of another bridging transmembrane partner able to bind, via its extracellular domain, the PCSK9-LDLR complex, and to interact via its CT with ARH and/or other cytosolic trafficking partners.
Recently, two proteins were suggested to enhance the PCSK9-mediated degradation of the LDLR. APLP2 was reported to act as a bridge protein that facilitates trafficking of the PCSK9-LDLR complex to endosomes/lysosomes (31), and sortilin was shown to bind PCSK9 in the trans-Golgi network and possibly facilitates its secretion (35). Herein, we attempted to elucidate the mechanisms underlying the role of APLP2 and sortilin in PCSK9 trafficking.
In this study, LDLR levels were not significantly affected upon APLP2 depletion by siRNA in HepG2 and Huh7 cells. In both cell lines, the ability of 1 μg/ml of exogenous PCSK9 to promote the degradation of the LDLR was unchanged (overnight in serum-free HEK293 conditioned media; Fig. 1). Importantly, the same lack of effect was observed by adding 50 μg/ml of pure recombinant PCSK9 for 2 h, the conditions used by DeVay et al. (31), or overnight (not shown).
Similar to APLP2, depletion of either sortilin alone or in combination with APLP2 in Huh7 cells had no significant effect on LDLR levels (Fig. 3A). In addition, LDLR levels and PCSK9 activity/secretion remained unaffected by overexpression of APLP2, sortilin, or both proteins (Fig. 6).
We then tested whether APLP2 or sortilin, which had no effect on the PCSK9 activity in cell lines, may in vivo regulate liver LDLR levels. Our data showed that the LDLR levels in the livers and primary hepatocytes of Aplp2−/− mice, and those of circulating total cholesterol were unchanged (Fig. 2, B–D). However, Aplp2−/− mice exhibited a 20–27% decrease in plasma PCSK9 levels, suggesting that APLP2 can partially regulate the levels of circulating PCSK9 (Fig. 2A). The observed reduction in plasma PCSK9 in Aplp2−/− mice could be due to a direct effect of APLP2 on PCSK9 secretion/degradation by hepatocytes, independent of the LDLR. An indirect effect through increased LDLR uptake of PCSK9 is unlikely as the LDLR levels remained unchanged in Aplp2−/− mice (Fig. 2, B–D). Therefore, we conclude that APLP2 does not affect the ability of PCSK9 to enhance the degradation of the LDLR in cells and in vivo.
In Sort1−/− mice the levels of LDLR, circulating PCSK9 and total cholesterol were not affected (Fig. 3, B–D). Our results on Sort1−/− mice differ from those of Gustafsen et al. (35) who reported that Sort1−/− mice exhibit a ∼30% reduction in the levels of circulating PCSK9 and ∼2.2-fold increase in liver LDLR protein. This is surprising because heterozygote Pcsk9+/− mice exhibit a ∼70% reduction in circulating PCSK9, but only a 1.3-fold increase in liver LDLR levels (Fig. 3F). Could this discrepancy be due to the use of different knock-out strategies? In our Sort1−/− mice, the KO strategy consisted of replacing segments of exon 2-intron 3 with a neomycin-resist-and EGFP cassette by homologous recombination (44). Western blot analysis using an antibody directed against amino acids 300–422 of human sortilin encoded by exons 8 to 11 (98% identical protein sequence between human and mouse orthologues) showed a ∼105-kDa mature sortilin only in wild type, but not KO liver (Fig. 3B), confirming the complete loss of protein expression in our KO livers. In the mice used by Gustafsen et al. (35) the KO strategy was to disrupt the reading frame of the Sort1 gene by deleting 41 codons in exon 14 (53). This also resulted in the loss of sortilin protein expression, as evidenced by the same antibody used in this study, suggesting that the residual protein and/or mRNA are unstable. We thus have no explanation for the difference between this study and that of Gustafsen et al. (35) as both Sort1−/− mouse models have comparable genetic backgrounds (C57BL/6) and the mice used were fed similar chow diets.
Even though we concluded that APLP2 or sortilin did not significantly affect PCSK9 activity on the LDLR, we were surprised by the fact that APLP2 and sortilin were targeted for degradation in both the ER and acidic compartments by PCSK9 in HEK293 and Huh7 cells (Figs. 4–6). This likely involves the intracellular PCSK9 pathway, because incubation of these cells with exogenous PCSK9 had no effect on either protein (not shown). However, this did not happen in the liver of transgenic mice overexpressing PCSK9 (not shown). This phenomenon was also observed for LRP1 that was efficiently targeted for degradation by PCSK9 in various cell lines, but not in mouse liver (29). One explanation would be that the high ability of the liver to secrete PCSK9 may hamper the intracellular PCSK9-mediated degradation of sortilin or APLP2. Alternatively, the ER exit and secretion of PCSK9 in hepatocytes may be very fast and efficient as compared with cell lines where PCSK9 may spend more time intracellularly. Another explanation is that intrinsic high levels of PCSK9 may result in an adaptive response in transgenic mice, unlike an acute overexpression in cells.
The above data and the fact that sortilin can stabilize APLP2 (Fig. 4B) suggest that both proteins together with PCSK9 meet early in the secretory pathway and not at the cell surface. This is further evidenced by the ability of ER-retained PCSK9-KDEL to enhance the degradation of APLP2, sAPLP2, and sortilin likely in the ERAD pathway (Fig. 5).
Proteins are shuttled from the ER to the Golgi apparatus by COPII vesicles coated with the cytosolic Sec23/Sec24 adaptors heterodimers that play a key role in cargo recruitment to the transport vesicle (54). In mouse, Sec24A deficiency led to ∼60% lower levels of circulating PCSK9 and up-regulated levels of the LDLR protein in the liver (51). Sec24A was thus suggested to promote PCSK9 exit from the ER. To assess whether APLP2 and/or sortilin were involved in this process, a split G. princeps luciferase assay (37, 45) was used. Surprisingly, although APLP2 and sortilin did not bind Sec24, they interacted strongly with each other, as also confirmed by co-immunoprecipitation (Fig. 7).
It was shown that sortilin and the amyloid precursor protein (APP) bind to each other via luminal (head-to-head) and cytosolic (tail-to-tail) interaction domains (55). Their cytosolic tails interact via the FLVHRY motif of sortilin with the YNPTYKFFE motif in APP, which overlaps with the ARH binding consensus YENPTY (26, 27). Because the APP motives in its luminal and CT domains implicated in sortilin binding are either identical or similar in APLP2 (not shown), this suggests that sortilin may also bind APLP2 via these motives and regulate its trafficking. Interestingly, sortilin was shown to target APP for lysosomal degradation (55), whereas our data revealed that sortilin stabilizes APLP2 (Fig. 4B), suggesting a functional difference between the interaction of sortilin with APP or APLP2.
In situ hybridization in adult mouse liver or 10-day-old whole body revealed that the colon, small intestine, retina, olfactory tract, and cerebellum express high levels of PCSK9, APLP2, and sortilin (Fig. 8, left panel), whereas in the mouse (Fig. 8, right panel) and human (56) liver, sortilin expression is rather poor. These results were also confirmed by quantitative PCR analyses of various tissues (not shown). Quantitative PCR analysis of the mouse mRNA levels of liver PCSK9, LDLR, APLP2, sortilin, and HMG-CoA reductase were analyzed. The data revealed that although PCSK9, LDLR, and HMG-CoA reductase mRNA levels are reduced by about 2–3-fold in the liver of mice fed a high cholesterol versus chow diet, the levels of APLP2 or sortilin are not significantly affected by a high cholesterol diet (not shown). This suggests that different for PCSK9, LDLR, and HMG-CoA reductase (57), the transcription of Aplp2 or Sort1 is not regulated by SREBP2.
FIGURE 8.
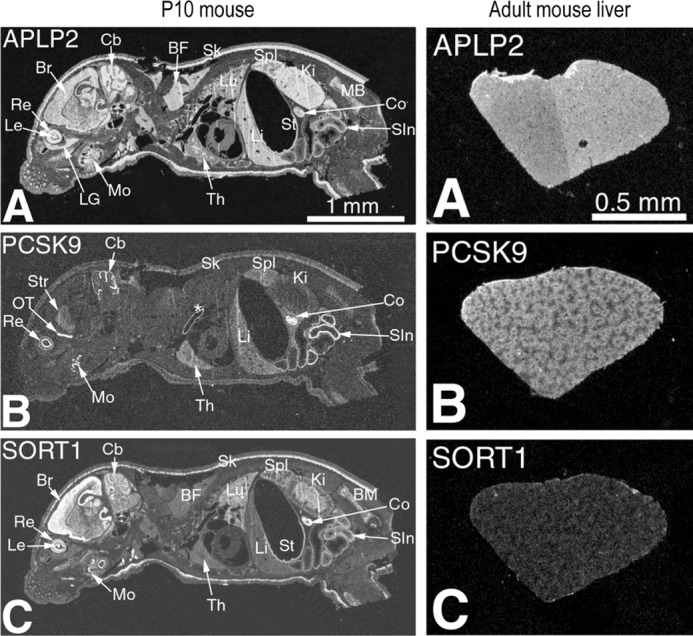
Comparative in situ hybridization histochemistry of APLP2, PCSK9, and sortilin mRNA expression. Comparative in situ hybridization localization of APLP2, PCSK9, and sortilin mRNA in cryostat sagittal sections of whole body postnatal mouse at post-partum day 10 (P10) mice (left panel, magnification ×2.4) and in the liver of adult mice (right panel, magnification ×4). A, x-ray film autoradiography showing widespread APLP2 expression pattern with high mRNA concentration in the eye retina and lens, brain, cerebellum, brown fat, liver, spleen, skin, small intestine, colon, and kidney. B, tissue-specific PCSK9 expression pattern with moderate to high level mRNA concentration in the eye retina inner nuclear layer, brain olfactory tract and cerebellum, spleen, liver, small intestine, and colon. C, tissue-specific sortilin expression pattern with moderate to high level mRNA concentration in the eye retina and lens, brain, cerebellum, skin, kidney medulla, colon, and small intestine. Abbreviations: BF, brown fat; BM, bone marrow; Br, brain; Cb, cerebellum; Co, colon; Ki, kidney; LG, lacrimal gland; Le, eye lens; Li, liver; Lu, lung; Mo, molars; OT, olfactory tract; Re, retina; SIn, small intestine; Sk, skin; Spl, spleen; St, stomach; Str, brain striatum; Th, thymus. *, indicates nonspecific staining of the aorta.
Because our data suggest that APLP2 and sortilin can interact, and that sortilin enhances the stability of APLP2 (Fig. 4B), it will be important in the future to test the effect of sortilin on the functions of APLP2 in specific tissues, especially in the brain, small intestine, and colon where the expression of both transcripts is quite high (Fig. 8). For example, all three members of the APP gene family (APP, APLP1, and APLP2) behave similarly in that they each contribute to the regulation of cell surface NMDA receptor homeostasis in neurons (58). Thus, it is possible that sortilin would differentially regulate the activity of APP and APLP2 on NMDA receptors in the brain (Fig. 8).
The main conclusion of this work is that, in the liver or derived cell lines, neither APLP2 nor sortilin appear to be critical for PCSK9 activity. To date, attempts to identify a protein with a cytosolic (F/Y)XNPXY motif that is critical for the extracellular PCSK9 function on the LDLR have not been successful. An alternative hypothesis postulates that, upon extracellular PCSK9 binding to the cell surface LDLR, the endocytosed membrane-bound PCSK9-LDLR complex is exposed to sheddase(s) that release the LDLR from the membrane (59), and accelerate its degradation. Whether the C-terminal domain of PCSK9, which is essential for LDLR degradation, acts through reinforcing the PCSK9-LDLR interaction (60, 61), rather than through binding a third partner, remains to be proven (20–22).
Author Contributions
C. B. and M. C. performed most of the experiments, analyzed the data, and wrote the paper. R. E. generated the knockout mice and prepared primary hepatocytes for analysis. S. P. provided the analysis of Aplp2 KO mice by Western blotting. S. B. performed the biosynthetic analysis. M. C. A. prepared all the cells and cell lines for analysis. A. R. performed the immunocytochemistry, plasma and cholesterol analyses. J. H. prepared all recombinant vectors and their mutants. J. M. performed all in situ hybridization analyses. A. C. made all the quantitative PCR analyses. J. G. helped in the analyses of primary hepatocytes. G. M. and S. S. read and corrected the manuscript and provided Aplp2 KO mice. Y. J. made all the proximity assays with the cytosolic tail of APLP2, sortilin, and the Sec proteins. A. P. provided the genetic analysis of the mice and made a substantial contribution to the manuscript writing and figure editing. N. G. S. directed the whole process, analyzed the data, and wrote the manuscript.
Acknowledgments
We are grateful to all the members of the Seidah laboratory for helpful discussions and to Brigitte Mary for efficacious editorial assistance. We also thank Dr. Wang of Baylor College of Medicine for collecting some samples from Aplp2−/− mice.
This work was supported in part by Canadian Institutes of Health Research Grants MOP-102741 and CTP-82946, Canada Chair Grant 216684, and Leducq Foundation Grant 13CVD03. The authors declare that they have no conflicts of interest with the contents of this article.
- PCSK9
- proprotein convertase subtilisin/kexin type 9
- APLP2
- amyloid precursor-like protein 2
- LDLR
- low-density lipoprotein receptor
- ARH
- autosomal recessive hypercholesterolemia
- CT
- cytosolic tail
- ER
- endoplasmic reticulum
- APP
- amyloid precursor protein
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
References
- 1. Seidah N. G., Benjannet S., Wickham L., Marcinkiewicz J., Jasmin S. B., Stifani S., Basak A., Prat A., Chretien M. (2003) The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc. Natl. Acad. Sci. U.S.A. 100, 928–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seidah N. G., Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 3. McNutt M. C., Lagace T. A., Horton J. D. (2007) Catalytic activity is not required for secreted PCSK9 to reduce low density lipoprotein receptors in HepG2 cells. J. Biol. Chem. 282, 20799–20803 [DOI] [PubMed] [Google Scholar]
- 4. Poirier S., Mayer G., Benjannet S., Bergeron E., Marcinkiewicz J., Nassoury N., Mayer H., Nimpf J., Prat A., Seidah N. G. (2008) The proprotein convertase PCSK9 induces the degradation of low density lipoprotein receptor (LDLR) and its closest family members VLDLR and ApoER2. J. Biol. Chem. 283, 2363–2372 [DOI] [PubMed] [Google Scholar]
- 5. Nassoury N., Blasiole D. A., Tebon Oler A., Benjannet S., Hamelin J., Poupon V., McPherson P. S., Attie A. D., Prat A., Seidah N. G. (2007) The cellular trafficking of the secretory proprotein convertase PCSK9 and its dependence on the LDLR. Traffic 8, 718–732 [DOI] [PubMed] [Google Scholar]
- 6. Maxwell K. N., Breslow J. L. (2004) Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc. Natl. Acad. Sci. U.S.A. 101, 7100–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Benjannet S., Rhainds D., Essalmani R., Mayne J., Wickham L., Jin W., Asselin M. C., Hamelin J., Varret M., Allard D., Trillard M., Abifadel M., Tebon A., Attie A. D., Rader D. J., Boileau C., Brissette L., Chrétien M., Prat A., Seidah N. G. (2004) NARC-1/PCSK9 and its natural mutants: zymogen cleavage and effects on the low density lipoprotein (LDL) receptor and LDL cholesterol. J. Biol. Chem. 279, 48865–48875 [DOI] [PubMed] [Google Scholar]
- 8. Park S. W., Moon Y. A., Horton J. D. (2004) Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J. Biol. Chem. 279, 50630–50638 [DOI] [PubMed] [Google Scholar]
- 9. Abifadel M., Varret M., Rabès J. P., Allard D., Ouguerram K., Devillers M., Cruaud C., Benjannet S., Wickham L., Erlich D., Derré A., Villéger L., Farnier M., Beucler I., Bruckert E., Chambaz J., Chanu B., Lecerf J. M., Luc G., Moulin P., Weissenbach J., Prat A., Krempf M., Junien C., Seidah N. G., Boileau C. (2003) Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 34, 154–156 [DOI] [PubMed] [Google Scholar]
- 10. Cohen J., Pertsemlidis A., Kotowski I. K., Graham R., Garcia C. K., Hobbs H. H. (2005) Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat. Genet. 37, 161–165 [DOI] [PubMed] [Google Scholar]
- 11. Zaid A., Roubtsova A., Essalmani R., Marcinkiewicz J., Chamberland A., Hamelin J., Tremblay M., Jacques H., Jin W., Davignon J., Seidah N. G., Prat A. (2008) Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 48, 646–654 [DOI] [PubMed] [Google Scholar]
- 12. Roubtsova A., Munkonda M. N., Awan Z., Marcinkiewicz J., Chamberland A., Lazure C., Cianflone K., Seidah N. G., Prat A. (2011) Circulating proprotein convertase subtilisin/kexin 9 (PCSK9) regulates VLDLR protein and triglyceride accumulation in visceral adipose tissue. Arterioscler. Thromb. Vasc. Biol. 31, 785–791 [DOI] [PubMed] [Google Scholar]
- 13. Seidah N. G. (2013) Proprotein convertase subtilisin kexin 9 (PCSK9) inhibitors in the treatment of hypercholesterolemia and other pathologies. Curr. Pharm. Des 19, 3161–3172 [DOI] [PubMed] [Google Scholar]
- 14. Seidah N. G., Awan Z., Chrétien M., Mbikay M. (2014) PCSK9: a key modulator of cardiovascular health. Circ. Res. 114, 1022–1036 [DOI] [PubMed] [Google Scholar]
- 15. Stein E. A., Raal F. (2014) Reduction of low-density lipoprotein cholesterol by monoclonal antibody inhibition of PCSK9. Annu. Rev. Med. 65, 417–431 [DOI] [PubMed] [Google Scholar]
- 16. Poirier S., Mayer G., Poupon V., McPherson P. S., Desjardins R., Ly K., Asselin M. C., Day R., Duclos F. J., Witmer M., Parker R., Prat A., Seidah N. G. (2009) Dissection of the endogenous cellular pathways of PCSK9-induced LDLR degradation: evidence for an intracellular route. J. Biol. Chem. 284, 28856–28864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holla Ø. L., Cameron J., Berge K. E., Ranheim T., Leren T. P. (2007) Degradation of the LDL receptors by PCSK9 is not mediated by a secreted protein acted upon by PCSK9 extracellularly. BMC Cell Biol. 8, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fisher T. S., Lo Surdo P., Pandit S., Mattu M., Santoro J. C., Wisniewski D., Cummings R. T., Calzetta A., Cubbon R. M., Fischer P. A., Tarachandani A., De Francesco R., Wright S. D., Sparrow C. P., Carfi A., Sitlani A. (2007) PCSK9-dependent LDL receptor regulation: effects of pH and LDL. J. Biol. Chem. 282, 20502–20512 [DOI] [PubMed] [Google Scholar]
- 19. McNutt M. C., Kwon H. J., Chen C., Chen J. R., Horton J. D., Lagace T. A. (2009) Antagonism of secreted PCSK9 increases low-density lipoprotein receptor expression in HEPG2 cells. J. Biol. Chem. 284, 10561–10570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang D. W., Garuti R., Tang W. J., Cohen J. C., Hobbs H. H. (2008) Structural requirements for PCSK9-mediated degradation of the low-density lipoprotein receptor. Proc. Natl. Acad. Sci. U.S.A. 105, 13045–13050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cunningham D., Danley D. E., Geoghegan K. F., Griffor M. C., Hawkins J. L., Subashi T. A., Varghese A. H., Ammirati M. J., Culp J. S., Hoth L. R., Mansour M. N., McGrath K. M., Seddon A. P., Shenolikar S., Stutzman-Engwall K. J., Warren L. C., Xia D., Qiu X. (2007) Structural and biophysical studies of PCSK9 and its mutants linked to familial hypercholesterolemia. Nat. Struct. Mol. Biol. 14, 413–419 [DOI] [PubMed] [Google Scholar]
- 22. Saavedra Y. G., Day R., Seidah N. G. (2012) The M2 module of the Cys-His-rich domain (CHRD) of PCSK9 is needed for the extracellular low density lipoprotein receptor (LDLR) degradation pathway. J. Biol. Chem. 287, 43492–43501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lagace T. A., Curtis D. E., Garuti R., McNutt M. C., Park S. W., Prather H. B., Anderson N. N., Ho Y. K., Hammer R. E., Horton J. D. (2006) Secreted PCSK9 decreases the number of LDL receptors in hepatocytes and in livers of parabiotic mice. J. Clin. Invest. 116, 2995–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fasano T., Sun X. M., Patel D. D., Soutar A. K. (2009) Degradation of LDLR protein mediated by “gain of function” PCSK9 mutants in normal and ARH cells. Atherosclerosis 203, 166–171 [DOI] [PubMed] [Google Scholar]
- 25. Wang Y., Huang Y., Hobbs H. H., Cohen J. C. (2012) Molecular characterization of proprotein convertase subtilisin/kexin type 9-mediated degradation of the LDLR. J. Lipid Res. 53, 1932–1943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia C. K., Wilund K., Arca M., Zuliani G., Fellin R., Maioli M., Calandra S., Bertolini S., Cossu F., Grishin N., Barnes R., Cohen J. C., Hobbs H. H. (2001) Autosomal recessive hypercholesterolemia caused by mutations in a putative LDL receptor adaptor protein. Science 292, 1394–1398 [DOI] [PubMed] [Google Scholar]
- 27. He G., Gupta S., Yi M., Michaely P., Hobbs H. H., Cohen J. C. (2002) ARH is a modular adaptor protein that interacts with the LDL receptor, clathrin, and AP-2. J. Biol. Chem. 277, 44044–44049 [DOI] [PubMed] [Google Scholar]
- 28. Ström T. B., Holla Ø. L., Tveten K., Cameron J., Berge K. E., Leren T. P. (2010) Disrupted recycling of the low density lipoprotein receptor by PCSK9 is not mediated by residues of the cytoplasmic domain. Mol. Genet. Metab. 101, 76–80 [DOI] [PubMed] [Google Scholar]
- 29. Canuel M., Sun X., Asselin M.-C., Paramithiotis E., Prat A, Seidah N. G. (2013) Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chaparro-Riggers J., Liang H., DeVay R. M., Bai L., Sutton J. E., Chen W., Geng T., Lindquist K., Casas M. G., Boustany L. M., Brown C. L., Chabot J., Gomes B., Garzone P., Rossi A., Strop P., Shelton D., Pons J., Rajpal A. (2012) Increasing serum half-life and extending cholesterol lowering in vivo by engineering antibody with pH-sensitive binding to PCSK9. J. Biol. Chem. 287, 11090–11097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. DeVay R. M., Shelton D. L., Liang H. (2013) Characterization of proprotein convertase subtilisin/kexin type 9 (PCSK9) trafficking reveals a novel lysosomal targeting mechanism via amyloid precursor-like protein 2 (APLP2). J. Biol. Chem. 288, 10805–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. DeVay R. M., Yamamoto L., Shelton D. L., Liang H. (2015) Common proprotein convertase subtilisin/kexin type 9 (PCSK9) epitopes mediate multiple routes for internalization and function. PLoS One 10, e0125127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tuli A., Sharma M., Capek H. L., Naslavsky N., Caplan S., Solheim J. C. (2009) Mechanism for amyloid precursor-like protein 2 enhancement of major histocompatibility complex class I molecule degradation. J. Biol. Chem. 284, 34296–34307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tuli A., Sharma M., McIlhaney M. M., Talmadge J. E., Naslavsky N., Caplan S., Solheim J. C. (2008) Amyloid precursor-like protein 2 increases the endocytosis, instability, and turnover of the H2-K(d) MHC class I molecule. J. Immunol. 181, 1978–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gustafsen C., Kjolby M., Nyegaard M., Mattheisen M., Lundhede J., Buttenschøn H., Mors O., Bentzon J. F., Madsen P., Nykjaer A., Glerup S. (2014) The hypercholesterolemia-risk gene SORT1 facilitates PCSK9 secretion. Cell Metab 19, 310–318 [DOI] [PubMed] [Google Scholar]
- 36. Nielsen M. S., Madsen P., Christensen E. I., Nykjaer A., Gliemann J., Kasper D., Pohlmann R., Petersen C. M. (2001) The sortilin cytoplasmic tail conveys Golgi-endosome transport and binds the VHS domain of the GGA2 sorting protein. EMBO J. 20, 2180–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cassonnet P., Rolloy C., Neveu G., Vidalain P. O., Chantier T., Pellet J., Jones L., Muller M., Demeret C., Gaud G., Vuillier F., Lotteau V., Tangy F., Favre M., Jacob Y. (2011) Benchmarking a luciferase complementation assay for detecting protein complexes. Nat. Methods 8, 990–992 [DOI] [PubMed] [Google Scholar]
- 38. Mbikay M., Grant S. G., Sirois F., Tadros H., Skowronski J., Lazure C., Seidah N. G., Hanahan D., Chrétien M. (1989) cDNA sequence of neuroendocrine protein 7B2 expressed in beta cell tumors of transgenic mice. Int. J. Pept. Protein Res. 33, 39–45 [DOI] [PubMed] [Google Scholar]
- 39. Benjannet S., Saavedra Y. G., Hamelin J., Asselin M. C., Essalmani R., Pasquato A., Lemaire P., Duke G., Miao B., Duclos F., Parker R., Mayer G., Seidah N. G. (2010) Effects of the prosegment and pH on the activity of PCSK9: evidence for additional processing events. J. Biol. Chem. 285, 40965–40978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Benjannet S., Hamelin J., Chrétien M., Seidah N. G. (2012) Loss- and gain-of-function PCSK9 variants: clevage specificity, dominant negative effects, and low density lipoprotein receptor (LDLR) degradation. J. Biol. Chem. 287, 33745–33755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Benjannet S., Elagoz A., Wickham L., Mamarbachi M., Munzer J. S., Basak A., Lazure C., Cromlish J. A., Sisodia S., Checler F., Chrétien M., Seidah N. G. (2001) Post-translational processing of β-secretase (β-amyloid- converting enzyme) and its ectodomain shedding: the pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-β production. J. Biol. Chem. 276, 10879–10887 [DOI] [PubMed] [Google Scholar]
- 42. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. von Koch C. S., Zheng H., Chen H., Trumbauer M., Thinakaran G., van der Ploeg L. H., Price D. L., Sisodia S. S. (1997) Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 18, 661–669 [DOI] [PubMed] [Google Scholar]
- 44. Zeng J., Racicott J., Morales C. R. (2009) The inactivation of the sortilin gene leads to a partial disruption of prosaposin trafficking to the lysosomes. Exp. Cell Res. 315, 3112–3124 [DOI] [PubMed] [Google Scholar]
- 45. Neveu G., Cassonnet P., Vidalain P. O., Rolloy C., Mendoza J., Jones L., Tangy F., Muller M., Demeret C., Tafforeau L., Lotteau V., Rabourdin-Combe C., Travé G., Dricot A., Hill D. E., Vidal M., Favre M., Jacob Y. (2012) Comparative analysis of virus-host interactomes with a mammalian high-throughput protein complementation assay based on Gaussia princeps luciferase. Methods 58, 349–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Essalmani R., Hamelin J., Marcinkiewicz J., Chamberland A., Mbikay M., Chrétien M., Seidah N. G., Prat A. (2006) Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol. Cell. Biol. 26, 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thinakaran G., Sisodia S. S. (1994) Amyloid precursor-like protein 2 (APLP2) is modified by the addition of chondroitin sulfate glycosaminoglycan at a single site. J. Biol. Chem. 269, 22099–22104 [PubMed] [Google Scholar]
- 48. Thinakaran G., Slunt H. H., Sisodia S. S. (1995) Novel regulation of chondroitin sulfate glycosaminoglycan modification of amyloid precursor protein and its homologue, APLP2. J. Biol. Chem. 270, 16522–16525 [DOI] [PubMed] [Google Scholar]
- 49. Klausner R. D., Donaldson J. G., Lippincott-Schwartz J. (1992) Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116, 1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hampton R. Y. (2002) ER-associated degradation in protein quality control and cellular regulation. Curr. Opin. Cell Biol. 14, 476–482 [DOI] [PubMed] [Google Scholar]
- 51. Chen X. W., Wang H., Bajaj K., Zhang P., Meng Z. X., Ma D., Bai Y., Liu H. H., Adams E., Baines A., Yu G., Sartor M. A., Zhang B., Yi Z., Lin J., Young S. G., Schekman R., Ginsburg D. (2013) SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. Elife 2, e00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Russell C., Stagg S. M. (2010) New insights into the structural mechanisms of the COPII coat. Traffic 11, 303–310 [DOI] [PubMed] [Google Scholar]
- 53. Jansen P., Giehl K., Nyengaard J. R., Teng K., Lioubinski O., Sjoegaard S. S., Breiderhoff T., Gotthardt M., Lin F., Eilers A., Petersen C. M., Lewin G. R., Hempstead B. L., Willnow T. E., Nykjaer A. (2007) Roles for the pro-neurotrophin receptor sortilin in neuronal development, aging and brain injury. Nat. Neurosci. 10, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 54. Lee M. C., Miller E. A., Goldberg J., Orci L., Schekman R. (2004) Bi-directional protein transport between the ER and Golgi. Annu. Rev. Cell Dev. Biol. 20, 87–123 [DOI] [PubMed] [Google Scholar]
- 55. Yang M., Virassamy B., Vijayaraj S. L., Lim Y., Saadipour K., Wang Y. J., Han Y. C., Zhong J. H., Morales C. R., Zhou X. F. (2013) The intracellular domain of sortilin interacts with amyloid precursor protein and regulates its lysosomal and lipid raft trafficking. PLoS One 8, e63049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Petersen C. M., Nielsen M. S., Nykjaer A., Jacobsen L., Tommerup N., Rasmussen H. H., Roigaard H., Gliemann J., Madsen P., Moestrup S. K. (1997) Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. J. Biol. Chem. 272, 3599–3605 [DOI] [PubMed] [Google Scholar]
- 57. Dubuc G., Chamberland A., Wassef H., Davignon J., Seidah N. G., Bernier L., Prat A. (2004) Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler. Thromb. Vasc. Biol. 24, 1454–1459 [DOI] [PubMed] [Google Scholar]
- 58. Cousins S. L., Dai W., Stephenson F. A. (2015) APLP1 and APLP2, members of the app family of proteins, behave similarly to APP in that they associate with NMDA receptors and enhance NMDA receptor surface expression. J. Neurochem. 133, 879–885 [DOI] [PubMed] [Google Scholar]
- 59. Leren T. P. (2014) Sorting an LDL receptor with bound PCSK9 to intracellular degradation. Atherosclerosis 237, 76–81 [DOI] [PubMed] [Google Scholar]
- 60. Lo Surdo P., Bottomley M. J., Calzetta A., Settembre E. C., Cirillo A., Pandit S., Ni Y. G., Hubbard B., Sitlani A., Carfí A. (2011) Mechanistic implications for LDL receptor degradation from the PCSK9/LDLR structure at neutral pH. EMBO Rep. 12, 1300–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Abifadel M., Guerin M., Benjannet S., Rabès J. P., Le Goff W., Julia Z., Hamelin J., Carreau V., Varret M., Bruckert E., Tosolini L., Meilhac O., Couvert P., Bonnefont-Rousselot D., Chapman J., Carrié A., Michel J. B., Prat A., Seidah N. G., Boileau C. (2012) Identification and characterization of new gain-of-function mutations in the PCSK9 gene responsible for autosomal dominant hypercholesterolemia. Atherosclerosis 223, 394–400 [DOI] [PubMed] [Google Scholar]