

Background: YchF is a universally conserved hydrophobic amino acid-substituted ATPase whose catalytic mechanism is not understood.
Results: Biochemical, rapid kinetics, mutagenesis, and molecular dynamics data identify His-114 as critical for ATP hydrolysis.
Conclusion: The His-114-containing flexible loop exists in nucleotide-dependent conformations and is involved in catalysis.
Significance: These results expand the current understanding of enzymatic strategies employed by nucleotide hydrolases.
Keywords: ATPase, conformational change, molecular dynamics, pre-steady-state kinetics, ribosome, HAS-ATPase, YchF, catalytic mechanism, molecular switch, nucleotide binding
Abstract
GTPases perform a wide range of functions, ranging from protein synthesis to cell signaling. Of all known GTPases, only eight are conserved across all three domains of life. YchF is one of these eight universally conserved GTPases; however, its cellular function and enzymatic properties are poorly understood. YchF differs from the classical GTPases in that it has a higher affinity for ATP than for GTP and is a functional ATPase. As a hydrophobic amino acid-substituted ATPase, YchF does not possess the canonical catalytic Gln required for nucleotide hydrolysis. To elucidate the catalytic mechanism of ATP hydrolysis by YchF, we have taken a two-pronged approach combining classical biochemical and in silico techniques. The use of molecular dynamics simulations allowed us to complement our biochemical findings with information about the structural dynamics of YchF. We have thereby identified the highly conserved His-114 as critical for the ATPase activity of YchF from Escherichia coli. His-114 is located in a flexible loop of the G-domain, which undergoes nucleotide-dependent conformational changes. The use of a catalytic His is also observed in the hydrophobic amino acid-substituted GTPase RbgA and is an identifier of the translational GTPase family.
Introduction
GTPases are essential to all living organisms; are present in all kingdoms of life; and perform critical cell functions, including protein synthesis, intracellular signaling, cell cycle regulation, cytoskeletal rearrangement, and differentiation (1, 2). Interestingly, only eight GTPases (elongation factor (EF)2 Tu, EF-G, IF2, FtsY, Ffh, YihA, HflX, and YchF) are universally conserved among the domains of life (3). Of these eight GTPases, the functional mechanisms and cellular roles of three (YihA, HflX, and YchF) are only poorly understood.
The mechanisms employed by these GTPases to catalyze GTP hydrolysis can differ significantly, but typically rely on a conserved catalytic residue found immediately downstream of the conserved G3 motif (DXXG), a sequence motif present in all GTPases that is responsible for magnesium/γ-phosphate coordination (1, 4). In the model GTPase Ras, a conserved glutamine residue that aligns a catalytic water molecule for an in-line attack on the γ-phosphate of GTP is found in this position (5, 6). This catalytic Gln is replaced with His in the translation factor superfamily (including EF-Tu, EF-G, and IF2), Thr in Rap, and Arg in Ffh (7).
In the hydrophobic amino acid-substituted (HAS) class of GTPases, the catalytic Gln is substituted with a hydrophobic amino acid (e.g. Ile or Leu) (8) requiring an alternative mode of catalysis (Fig. 1 and Table 1). Members of this class include HflX, EH, Era, EngA, EngB, MnmE, NogI, FeoB, Rsr1, Rb25, and YchF, as well as the circularly permuted GTPases YqeH and RbgA (YlqF) (8). It is of great interest to understand the mechanistic details of how these proteins catalyze nucleotide hydrolysis, how the activity is regulated, and the consequences on cellular functions. Despite recent work focusing on the HAS-GTPase family, the catalytic mechanisms for most of the known HAS-GTPases remain poorly understood. Examining other members of this GTPase family will shed light on the catalytic strategies employed.
FIGURE 1.
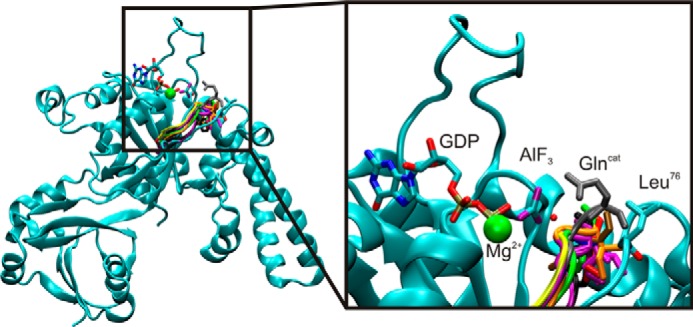
YchF is a member of the HAS-GTPase family. Shown is an alignment of the G3 motif of Ras (gray), YqeH (black), MnmE (yellow), HflX (purple), EHD2 (green), FeoB (pink), Era (red), YsxC (brown), and YlqF (orange) with YchF (cyan). Also shown is GDP·AlF3 and the attacking water molecule (red sphere in line with AlF3) from Ras·RasGap (PDB ID 1WQ1). Alignment was based on the structure of the conserved G1 motif (P-loop). Glncat, catalytic Gln.
TABLE 1.
Structural alignment of the G3 motif of nine HAS-GTPases and Ras
Bold denotes hydrophobic residue substitutions in HAS GTPases.
| Protein | PDB ID | G3 motif (DXXG) |
|---|---|---|
| YchF | DIAGL | |
| FeoB | 3LX5 | DLPGL |
| RbgA (YlqF) | 1PUJ | DTPGI |
| MnmE | 2GJ8 | DTAGL |
| Era | 3IEU | DTPGI |
| YqeH | 3H2Y | DTPGI |
| HflX | 2QTH | DTVGF |
| EH | 2QPT | DTPGI |
| YsxC (EngB) | 1PUI | DVPGY |
| Ras | 1WQ1 | DTAGQ |
Although categorized as a HAS-GTPase due to its conserved Leu immediately following the G3 motif, YchF also contains an altered specificity motif. In YchF, the canonical (N/T)KXD sequence (G4 motif), which normally provides specificity for guanine nucleotides, is changed to NXXE (9). This results in an altered nucleotide affinity, as YchF binds and hydrolyzes ATP more efficiently than GTP (9–11). We therefore refer to YchF as a HAS-ATPase, although initially identified as a GTPase (12–18). To date, YchF has been implicated in numerous cell functions, including protein synthesis in Escherichia coli, iron utilization in Brucella melitensis and Vibrio vulnificus, protein degradation and virulence in Streptococcus pneumoniae and V. vulnificus, the oxidative stress response and infection defense response in rice, and centrosome regulation through interaction of its human homolog with the breast and ovarian cancer-specific tumor suppressor BRCA1 (9, 11, 19–28). Due to its initial characterization as a GTPase, only one x-ray crystallography structure of YchF bound to an adenine nucleotide is available, that of the human homolog OLA1 in complex with AMPPCP (10). Unfortunately, these x-ray crystallography structures do not provide structural information for critical regions of the protein, including both switch regions and a flexible loop located in the G-domain. Furthermore, mutational analyses have revealed that two residues, Asn-13 and Lys-78 (E. coli numbering), play important roles in the potassium dependence of ATP hydrolysis, but amino acid residues essential for ATP hydrolysis have yet to be identified (12). Thus, the current structural and biochemical information on YchF is insufficient to devise a reliable mechanism for ATP hydrolysis by YchF.
Experimental Procedures
All chemicals were obtained from VWR International, Sigma-Aldrich, or Invitrogen unless indicated otherwise. DH5α cells were purchased from New England Biolabs, and BL21(DE3) competent cells were purchased from Novagen. Restriction enzymes were obtained from Fermentas, radiochemicals were from PerkinElmer Life Sciences, and nucleotides and fluorescent nucleotide analogs were purchased from Invitrogen. All buffers were filtered through Whatman 0.45-μm nitrocellulose membranes.
Sequence Alignments
All YchF protein sequences were obtained from the UniProt and Entrez Gene databases. Multiple-sequence alignments (data not shown) were performed using ClustalW2 provided by the European Bioinformatics Institute (EBI).
Cloning and Site-directed Mutagenesis
Amino acid substitutions were introduced into pET28a containing the full-length E. coli sequence coding for N-terminal His6-tagged YchF (11) using the QuikChange method. Ala, Lys, Arg, or Phe was introduced at position 114 to substitute for the native His. Forward primers (H114A, 5′-GACAACATCATTGCCGTTTCGGGCAA-3′; H114K, 5′-GACAACATCATTAAAGTTTCCGGAAAAGTTAACCCGGC-3′; H114F, 5′-GACAACATCATTTTCGTTTCCGGAAAAGTTAACCCGGC-3′; H114R, 5′-GACAACATCATTCGCGTTTCCGGAAAAGTTAACCCGGC-3′; H102Q, 5′-CCGTGAAACCGAAGCGATTGGTCAGGTTGTTCGCTGC-3′; and L76Q, 5′-GATATTGCCGGCCAGGTAAAAGGC-3′, where mutations are indicated in boldface, restriction sites are in italics, and newly introduced restriction sites are underlined) and reverse primers (reverse complements of forward primers) were obtained from Invitrogen. All mutations were confirmed by sequencing (GENEWIZ). The resulting plasmids were each transformed into the E. coli BL21(DE3) strain for YchF overexpression.
Protein Expression and Purification
Overexpression and protein purifications were carried out as described previously (11).
CD Spectroscopy
CD spectroscopy was performed using a Jasco J-815 CD spectrometer with a cuvette of 1-mm path length. WT YchF and variants were dialyzed overnight in 50 mm potassium phosphate buffer (pH 7.5) to reduce the sodium ion concentration to <15 mm. The dialyzed samples were diluted with phosphate buffer to a final concentration of 1.5 μm YchF and then equilibrated to 20 °C before measuring. All samples were subjected to 10 successive scans (320 to 190 nm) with a digital integration time of 1 s, a bandwidth of 1 nm, and a scanning speed of 50 nm/min. The obtained spectra were averaged and corrected for the phosphate buffer spectrum.
Fluorescence Measurements
Fluorescence measurements were obtained using a QuantaMaster fluorescence spectrophotometer (Photon Technology International, London, Ontario, Canada) with a 3 × 3 mm quartz cuvette (Starna Cells, Inc., Atascadero, CA). YchF from E. coli (E. coli YchF) contains a single Trp residue located in its C-terminal domain whose fluorescence decreases upon nucleotide binding (11). YchF (1 μm) in Buffer A (50 mm Tris-Cl (pH 7.5), 70 mm NH4Cl, 30 mm KCl, and 7 mm MgCl2) was titrated with the respective adenine nucleotides and excited at 280 nm with a slit width of 1 nm. Fluorescence emission was monitored from 295 to 400 nm at a 1-nm step size with an emission slit width of 5 nm. Following the addition of nucleotide, the solution was equilibrated for 1 min prior to excitation. Equilibration was not performed when ATP was added due to the intrinsic ATPase activity of YchF. The fluorescence of Buffer A with nucleotides was subtracted from all nucleotide-binding experiments, and fluorescence intensities were corrected for dilution. The equilibrium dissociation constant (KD) was determined by plotting the change in fluorescence at 337 nm as a function of the nucleotide concentration. Each data set was fit with a hyperbolic function (Equation 1) using TableCurve software (Jandel Scientific Software) and Prism (GraphPad Software).
Here, F represents the fluorescence at 337 nm, [nt] is the nucleotide concentration, F0 is the initial fluorescence, and ΔFmax is the amplitude of the signal change. Final KD values and their S.D. values were calculated from at least three independent experiments.
ATP Hydrolysis Assays
The multiple-turnover ATP hydrolysis (ATPase) activities of WT YchF and YchF variants were measured at 20 °C in Buffer A (unless indicated otherwise) by following the liberation of 32Pi from [γ-32P]ATP (200 dpm/pmol). To convert any ADP present, the ATP-containing solutions were charged by incubation with 0.25 μg/μl pyruvate kinase and 3 mm phosphoenolpyruvate for 15 min at room temperature and then for 15 min at 37 °C.
The intrinsic ATPase activities of WT YchF and variants were determined as described (11) using reactions containing 5 μm YchF and 125 μm [γ-32P]ATP. Thin-layer chromatography was used to separate 32Pi from [γ-32P]ATP, followed by visualization using a Typhoon Trio scanner (GE Healthcare). The relative amount of 32Pi formed was determined using ImageJ (29). Ribosome-stimulated ATPase activity was determined under these conditions in the presence of 6 μm ribosomes (70S) purified from E. coli MRE600 cells (11).
The pH-dependent ATPase assays were performed similarly to the intrinsic ATPase assays with Buffer A used at pH 7–9. Experiments at pH <7 were performed in MES-containing buffer. Results were corrected for the background hydrolysis of ATP at 20 °C, and ATPase assays were performed at least in triplicate to determine S.D. values. Michaelis-Menten analyses of the ribosome-dependent ATPase activities of YchF(H114A) and YchF(H114R) were performed similarly to the method described previously (11) at 37 °C using purified 70S ribosomes and the respective YchF variant (1 μm).
Pre-steady-State Kinetics
All rapid kinetics measurements were performed using a KinTek SF-2004 stopped-flow apparatus at 20 °C. N-ethylanthraniloyl (mant)-nucleotides were excited via FRET from the single Trp (λex = 280 nm) present in E. coli YchF and measured after passing through LG-400-F cutoff filters (Newport Corp., Irvine, CA).
For dissociation experiments, 20 μm mant-nucleotide and 2 μm YchF were preincubated in Buffer A for 30 min at 37 °C. Pyruvate kinase (0.25 μg/μl final concentration) and phosphoenolpyruvate (3 mm final concentration) were added to the mixture when mant-ATP was used. To observe dissociation, 25 μl of YchF·mant-ATP/mant-ADP (1 μm after mixing) was rapidly mixed with 25 μl of ATP/ADP (100 μm after mixing). Due to the excess of unlabeled nucleotide present, only the dissociation of the mant-labeled nucleotide contributed to the observed fluorescence change, and rebinding of mant-nucleotide was negligible. Each time course was fit with a one- or two-exponential function (Equations 2 and 3, respectively),
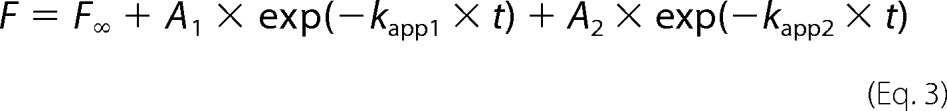 |
where F is the mant fluorescence at time t, F∞ is the final fluorescence, and A is the respective amplitude of the observed fluorescence change. The characteristic apparent rate constants (kapp, kapp1, and kapp2) correspond to the respective nucleotide dissociation constants (koff, koff1, and koff2). Calculations were performed using TableCurve and Prism.
For association experiments, 25 μl of 2 μm YchF in Buffer A was rapidly mixed with 25 μl of increasing concentrations of mant-nucleotide (1–10 μm mant-ADP or 4–40 μm mant-ATP). Similar to the nucleotide dissociation experiments, phosphoenolpyruvate and pyruvate kinase were added to solutions containing mant-ATP, and the mixtures were preincubated for 30 min at 37 °C. Each time course was fitted to a function containing one (mant-ADP, Equation 2), two (mant-ATP, Equation 3), or three (mant-ATP, Equation 4) exponentials.
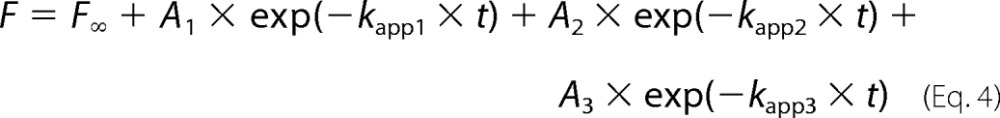 |
The corresponding apparent rate constants for association were kapp1, kapp2, and kapp3. The bimolecular association rate constant (k1) was determined from the slope of the linear concentration dependence of kapp1.
Molecular Dynamics (MD) Simulations
The initial model for E. coli YchF was obtained by constructing a homology model using the SWISS-MODEL server (30) and the crystal structure of Haemophilus influenzae YchF as a template (Protein Data Bank (PDB) ID 1JAL) (31). The conformation of Switch I (residues 28–41, which are missing in the 1JAL structure) was modeled based on the structure of Switch I in Thermus thermophilus YchF bound to GDP (PDB ID 2DBY). To model the adenine nucleotide bound to E. coli YchF, we used the conformation of AMPPCP bound to the human homolog of YchF, OLA1 (PDB ID 2OHF). Transformation of the AMPPCP to ATP and ADP was done manually. To position the magnesium ion associated with the bound nucleotide, we aligned the nucleotide with crystal structures of EF-Tu bound to either GDP or GMPPNP (PDB ID 1EFC and 1EFT, respectively). Hydrogen atoms were added to all models using psfgen in the NAMD software package, and His side chains were protonated at the ϵ-nitrogen only (32). Initial models were minimized and then placed in a water box extending at least 10 Å from the protein in all directions. Water molecules present in the template crystal structures were included in this box, and all other waters were added at random using the Solvate package in NAMD (32). Relaxation of the solvated system was achieved by minimizing the positions of water molecules, followed by minimization of the protein/ligand atoms in two iterative rounds (10,000 steps each). Sodium ions were then added in random positions by the Autoionize package in VMD to neutralize the total charge of the system, followed by a final minimization of all components of the system. YchF·ATP·Mg2+ and YchF·ADP·Mg2+ had total charges of −17 and −16; therefore, 17 and 16 sodium ions were added, respectively. The system was considered to be minimized when no change in energy was observed for at least 1000 steps (33). Minimizations, subsequent equilibrations, and equilibrium MD simulations were performed with periodic boundary conditions using the NAMD software package (32). Minimized models were initially equilibrated at 300 and 350 K for 150 ps at constant pressure (1 atm). Production-phase simulations were started using velocities from the 300 K equilibration and coordinates from the 350 K equilibration. Simulations were performed over 50 ns at 300 K with a step size of 0.5 fs using the CHARMM22 parameters for proteins and CHARMM27 parameters for nucleic acids as implemented in the NAMD package (32, 34). MD simulations were performed in an NPT ensemble in which the pressure was maintained at 1 atm with a Nosé-Hoover Langevin piston, and the temperature was controlled using Langevin dynamics. All simulations were performed in the NAMD software package, and visualization was carried out in VMD (32, 33).
Snapshots of each MD simulation were saved every 0.5 ps, and trajectories were fitted with the software Carma to remove any rotations of the protein complex or translation of the center of mass (35). The root mean square deviation (r.m.s.d.) and root mean square fluctuation (r.m.s.f.) calculations were performed using scripts written in-house and invoked with the VMD software package (33). To assess whether the nucleotide present had an effect on the conformation of the His-114-containing loop, we measured the distance between the α-carbons of Ser-16 in the P-loop (as a reference point) and His-114. The distribution of distances during the different simulations was analyzed using a histogram plot for each simulation with bin sizes of 0.4 Å. Distance measurements were taken every 0.5 ps during the equilibrium MD simulations.
Results
pH Dependence of ATPase Activity
The mechanistic details of how ATP is hydrolyzed by the HAS-ATPase YchF are not understood. To identify amino acid residues that are involved in the catalytic mechanism, we determined the pH dependence of the intrinsic ATPase activity of E. coli YchF (Fig. 2). Such an analysis can provide information regarding the number of ionizable groups involved and their respective pKa values. The latter can indicate the identity of the involved ionizable group. Our data reveal a YchF activity profile consistent with a single ionizable group (Fig. 2B). The intrinsic ATPase activity was maximal at pH ≥7.5 (0.20 ± 0.02 min−1) and negligible at pH <6. From the corresponding log plot of the ATPase activities as a function of pH, a pKa of ∼6.7 for the single ionizable group could be obtained (Fig. 2B). A pKa of 6.7 suggests the participation of a His residue (pKa of 6.04) during catalysis (36).
FIGURE 2.
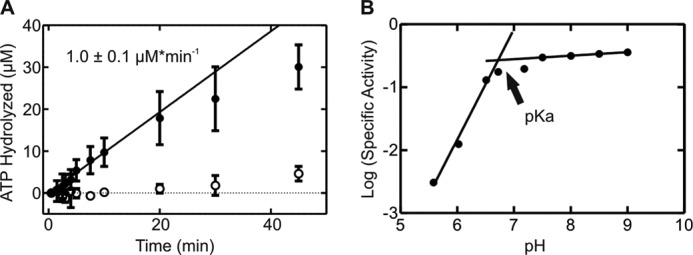
pH dependence of the intrinsic ATPase activity of YchF. A, time dependence of ATP hydrolysis in the presence (●) or absence (○) of YchF. A linear function was fit to the initial phase of each reaction (first 10 min) to determine the rate of ATP hydrolysis (μm min−1). B, pH dependence of the ATPase activity of YchF (●).
The two common mechanisms of catalysis in GTPases involve aligning a water molecule for nucleophilic attack of the γ-phosphate (Gln-61 in Ras and His-85 in EF-Tu) and the stabilization of a developing negative charge in the transition state (Arg-178 in Giα1 and Arg-174 in Gtα) (5, 37, 38). Of the four histidines in YchF (positions 102, 114, 145, and 308), only positions 102 and 114 are located within the G-domain and therefore close enough to the γ-phosphate of the bound nucleotide to participate in hydrolysis (Fig. 3). Because the catalytic amino acid in YchF is likely to be conserved, we investigated the conservation of each amino acid in E. coli YchF to assist in the search for a catalytic residue. To this end, we aligned 116 bacterial YchF sequences to determine the conservation of the histidines present in E. coli YchF. Histidines at positions 102 and 114 are found in 76 and 98% of the bacterial YchF sequences used in the alignment. Position 102 shows significant variability, containing Asn (8%), Met (1%), Gln (13%), or Tyr (2%) instead of His. On the other hand, His-114 is 98% conserved, and only Lys (1%) or Arg (1%) is also found at this position. However, His-114 and His-102 are >10 Å away from a position that would be compatible with a catalytic role such as aligning a nucleophilic water or stabilizing a developing negative charge in the transition state.
FIGURE 3.
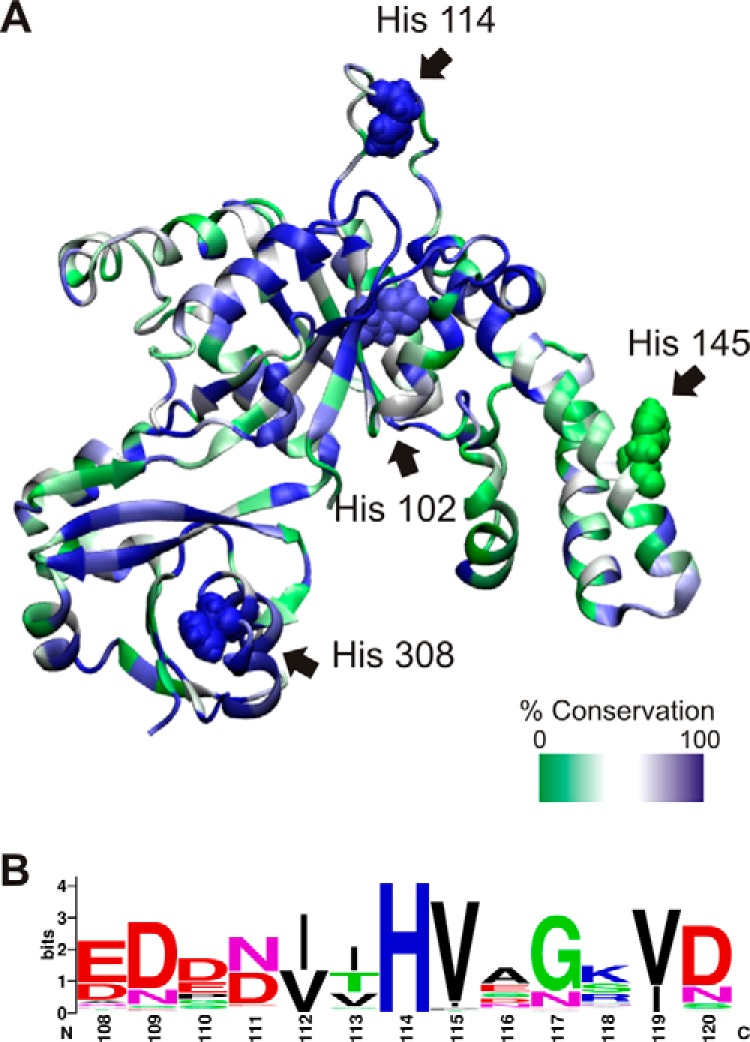
E. coli YchF contains four histidines. A, homology model of E. coli YchF generated with SWISS-MODEL using H. influenzae YchF (PDB ID 1JAL) as template and the amino acid sequence of E. coli YchF (UniProt accession code P0ABU2), represented as a ribbon diagram. The level of conservation of each residue is depicted using a green/white/blue color scale based on the alignment of YchF from 116 bacterial species. The four histidines present in E. coli YchF are indicated and shown as a space-filling model: His-102 (76% conserved), His-114 (98%), His-145 (17%), and His-308 (100%). B, sequence logo of the His-114-containing loop (numbering according to E. coli YchF) based on the multiple-sequence alignment in A.
MD Simulations of YchF
Structural information obtained by x-ray crystallography provides only limited insight into the dynamic features of a molecular system. We therefore used MD simulations to assess the potential of His-102 and His-114 to participate in the catalysis of ATP hydrolysis by YchF. To this end, we constructed three homology models of E. coli YchF, in complex with ATP or ADP or in the apo state, and performed 50-ns MD simulations (see “Experimental Procedures”). The stability of these models during simulations was scored by measuring the r.m.s.d. of the backbone atoms with respect to the initial structure throughout the whole simulation (Fig. 4A). After an initial increase in r.m.s.d. of ∼2–3 Å, depending on the particular simulation, the models appeared to stabilize after 5 ns. The flexibility of each amino acid in the different YchF complexes was determined by calculating the r.m.s.f. of each α-carbon over the final 40 ns of each simulation. The r.m.s.f. profiles vary only marginally among complexes, indicating that, on the whole, the dynamics of each structure is similar (Fig. 4B). Interestingly, in all three simulations, His-102 showed very small r.m.s.f. values. In fact, the side chain of His-102 is pointing away from the nucleotide-binding pocket due to a stable hydrogen bond between Nδ1 of His-102 and Hϵ1/Hϵ2 of Tyr-204 (the hydrogen bond flips between Hϵ1 and Hϵ2 around 22 ns during the YchF·ATP·Mg2+ simulation) (Fig. 5). This interaction is also maintained when YchF is bound to ADP and in its apo state (data not shown).
FIGURE 4.
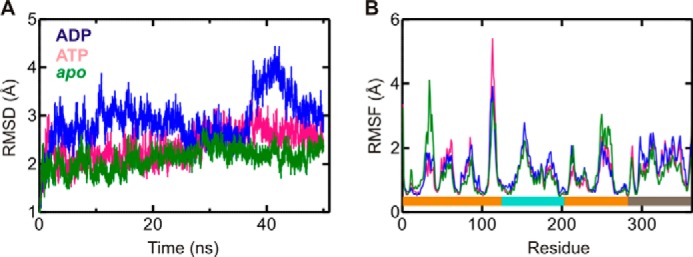
Structural dynamics of YchF during MD simulations. A, r.m.s.d. of E. coli YchF in complex with ATP (pink) or ADP (blue) or in its apo state (green) with respect to its initial conformation. r.m.s.d. values were calculated using all backbone atoms. B, Cα r.m.s.f. for E. coli YchF in complex with ATP (pink) or ADP (blue) or in its apo state (green). Colored bars at the bottom indicate the different domains of the protein: G-domain (orange), A-domain (teal), and TGS domain (gray).
FIGURE 5.
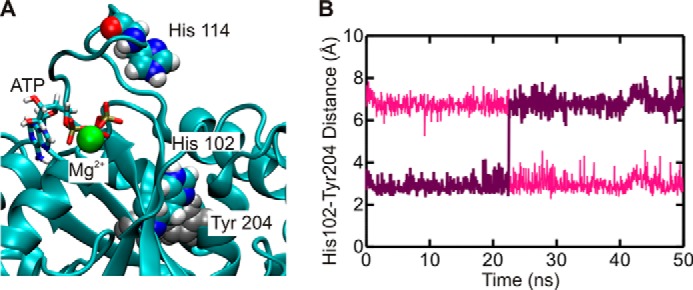
His-102 forms a stable interaction with Tyr-204. A, YchF is shown in complex with ATP after 50 ns of MD simulations. The locations of His-102 and Tyr-204, involved in a stable interaction, are indicated. B, hydrogen bond distance between Nδ1 of His-102 and Hϵ2 (pink) or Hϵ1 (purple) of Tyr-204.
A closer look at His-114 reveals that it is located in an extended loop structure (residues 105–121) within the G-domain. This loop displays greater r.m.s.f. values during simulations in the order ATP > ADP > apo (Fig. 4B). The observed high level of flexibility is consistent with the fact that electron density for this loop is observed in only one of the five experimentally determined structures of YchF/human OLA1 available in the PDB (IDs 2DBY, 1JAL, 1NI3, 2DWQ, and 2OHF) (9, 10). This prompted us to reference this loop as the “flexible” loop. Interestingly, a visual comparison of loop conformations before and after 50 ns of simulation revealed that the flexible loop is found in a more open conformation in the YchF·ADP·Mg2+ complex and in a more closed conformation (closer to the nucleotide-binding pocket) in the YchF·ATP·Mg2+ complex or apo state (Fig. 6A). To quantify the movement of the flexible loop during simulation, the distances between the α-carbons of His-114 and Ser-16 were measured and plotted over the simulation time (0–50 ns) (Fig. 6B). Ser-16 is a conserved amino acid in the P-loop responsible for coordinating the magnesium ion and showed essentially no fluctuations in its position during the 50-ns simulations. A similar plot was constructed for each of the remaining histidines (His-102, His-145, and His-308). None of these measurements revealed any nucleotide-dependent changes in dynamics over the course of the simulations (Fig. 7).
FIGURE 6.
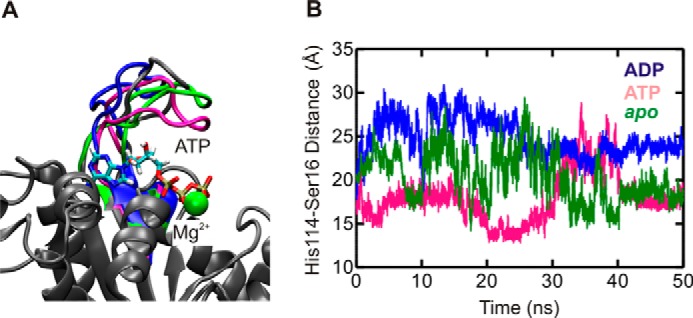
MD simulations reveal different conformations of the flexible loop. A, final structures of YchF·ATP·Mg2+ (pink), YchF·ADP·Mg2+ (blue), and apo-YchF (green) after 50 ns of simulation aligned with pre-simulation structures (gray). B, distance between the α-carbons of His-114 and Ser-16 over a 50-ns simulation of YchF·ATP·Mg2+ (pink), YchF·ADP·Mg2+ (blue), and apo-YchF (green).
FIGURE 7.
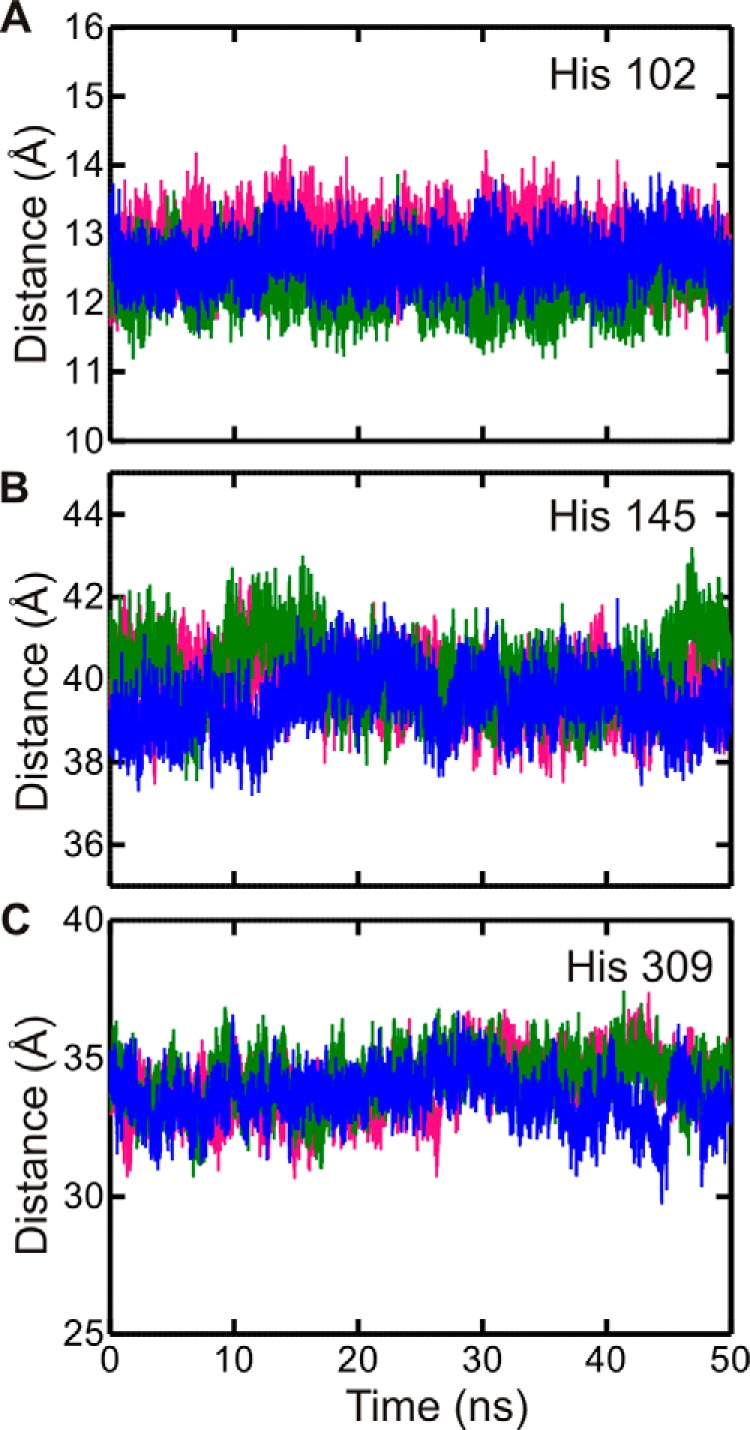
His-102, His-145, and His-308 do not show nucleotide-dependent conformations. Shown is the distance between the α-carbons of His-102 (A), His-145 (B), or His-308 (C) and Ser-16 of the P-loop during a 50-ns MD simulation. YchF·ATP·Mg2+ is shown in pink, YchF·ADP·Mg2+ in blue, and apo-YchF in green.
To evaluate the distribution of different conformations explored by the flexible loop, the distances plotted in Fig. 6B were binned and plotted as a histogram, counting the number of times the Cα–Cα distance fell into a particular distance bin (Fig. 8). Analysis of the three 50-ns MD trajectories (YchF·ATP·Mg2+, YchF·ADP·Mg2+, and apo-YchF) revealed distinct distribution profiles, indicating that the presence and identity of the bound nucleotide influence the preference for different conformations of the flexible loop. In the YchF·ATP·Mg2+ MD simulation, the flexible loop favors at least three different Cα16–Cα114 distances centered near 14, 17, or 23 Å. In comparison, the YchF·ADP·Mg2+ trajectory favors distances of ∼24 and 27 Å, which appear to be sampled by the YchF·ATP·Mg2+ simulation as well. Notably, the Cα16–Cα114 distance rarely drops below 20 Å in the YchF·ADP·Mg2+ simulation, whereas YchF·ATP·Mg2+ mainly samples distances <20 Å during the 50-ns simulation. In contrast, the apo state MD simulation of YchF exhibits a broad distribution with a maximum around 17 Å and a long tail extending toward 30 Å. The broad distribution of Cα16–Cα114 distances observed for the apo state simulation is consistent with the rapid fluctuations observed in the time-dependent analysis (Fig. 6B).
FIGURE 8.
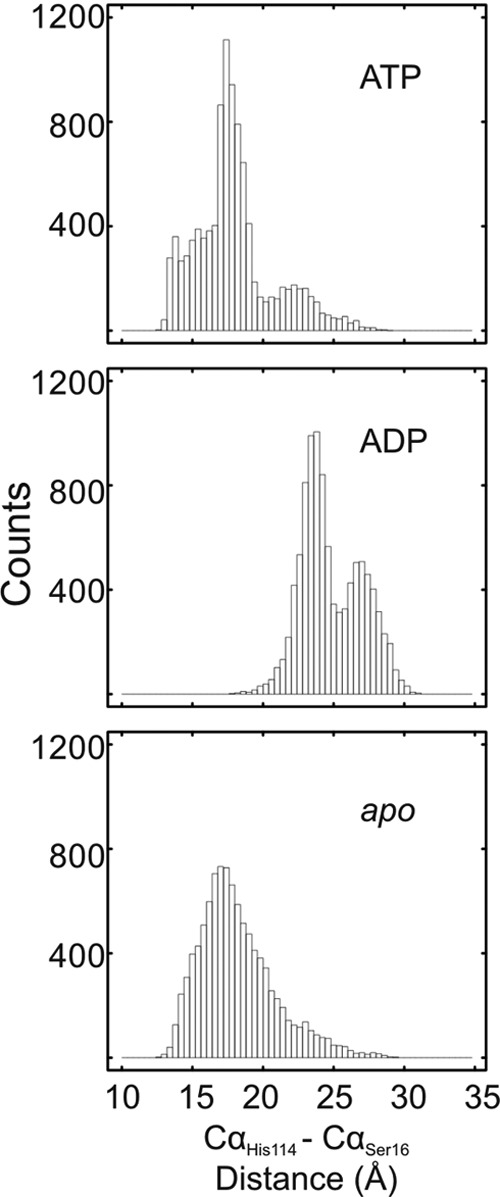
Nucleotide-dependent conformations of the flexible loop in YchF. Shown are histograms of the Cα–Cα (His-114 to Ser-16) distances (bin size of 0.4 Å) measured during 50-ns MD simulations of the YchF·ATP·Mg2+, YchF·ADP·Mg2+, and apo-YchF models.
These observations clearly support a nucleotide-sensitive behavior of the flexible loop, with primarily open conformations of the loop in the ADP-bound form of YchF, which convert into primarily closed conformations in the ATP-bound and apo states of YchF. Although the flexible loop exists primarily in closed conformations in the ATP-bound and apo states, it is still capable of sampling long Cα16–Cα114 distances characteristic of the open conformation sampled by the YchF·ADP·Mg2+ simulation (Fig. 8). This suggests a potential for interconversion between the conformations, enabling nucleotide-dependent molecular switching in YchF.
Nucleotide-binding Properties of YchF Variants
Our MD simulations in conjunction with the reported biochemical data suggest that His-114 is most likely involved in the catalysis of ATP hydrolysis by YchF and that it is located in a highly mobile, nucleotide-sensing structural element. To confirm a role for His-114 in catalysis, we constructed four single-amino acid substitution variants of YchF containing Ala, Phe, Arg, or Lys (YchF(H114A), YchF(H114F), YchF(H114R), and YchF(H114K)) in place of His-114. First, we confirmed that introducing these substitutions did not alter the overall structure of the protein using CD spectroscopy (Fig. 9). We then assessed adenine nucleotide binding in each variant using equilibrium fluorescence titrations to exclude any effect of these substitutions on the nucleotide-binding properties. The TGS domain of YchF contains a single Trp residue, which was previously used to determine the equilibrium binding constants (KD) for the interaction of YchF and ADP/ATP (11). Upon monitoring the Trp fluorescence between 295 and 400 nm, a decrease in fluorescence could be observed following the addition of ADP or ATP (Fig. 10, A and C). The fluorescence decrease at 337 nm was plotted as a function of nucleotide concentration and fitted with a hyperbolic equation to obtain a KD (Fig. 10, B and D). All YchF variants tested were capable of binding adenine nucleotides with an affinity comparable with WT YchF (Table 2). Interestingly, although WT YchF and YchF(H114K) did not show a signal change upon the addition of ATP (11), all other variants did. The KD values for all tested mutants are summarized in Table 2 and are in the low micromolar range, similar to values obtained for ADP.
FIGURE 9.
CD spectroscopy of WT YchF and variants. The CD spectra of WT YchF (blue), YchF(H114A) (orange), YchF(H114R) (purple), YchF(H102Q) (green), and YchF(L76Q) (black) were recorded in a 1-mm cell. deg, degrees.
FIGURE 10.

YchF(H114A) binds adenine nucleotide di- and triphosphates. Shown are equilibrium fluorescence titrations of 1 μm YchF(H114A) with increasing concentrations of nucleotides. Tyr and Trp in YchF were excited at 280 nm, and fluorescence emission spectra were detected from 295 to 400 nm in the presence of increasing concentrations of ADP (A) or ATP (C). Fluorescence intensities measured at 337 nm are plotted against the concentrations of ADP (B) or ATP (D). KD values were obtained by fitting the data in B and D with a hyperbolic function.
TABLE 2.
Equilibrium dissociation constants for YchF variants and adenine nucleotides
| YchF |
KD |
||
|---|---|---|---|
| ADP | ATP | AMPPNPa | |
| μm | μm | μm | |
| WT YchF | 14 ± 5 | No signal change | 9 ± 8 |
| YchF(H114A) | 9 ± 4 | 2.5 ± 2.0 | ND |
| YchF(H114F) | 8 ± 5 | 5 ± 2 | ND |
| YchF(H114R) | 8 ± 7 | 3 ± 2 | ND |
| YchF(H114K) | 10 ± 5 | No signal change | ND |
a AMPPNP, adenosine 5′-(β,γ-imido)triphosphate; ND, not determined.
Kinetics of Nucleotide Binding
On the basis of our observation in the MD simulations that nucleotide binding triggered different conformations within YchF and in particular the His-114-containing flexible loop, we performed fluorescence-based rapid kinetics experiments using fluorescent analogs (mant) of the respective nucleotides. Dissociation of the mant-nucleotide resulted in a decrease in mant fluorescence, whereas the respective association experiments yielded fluorescence time courses with increasing fluorescence (Fig. 11).
FIGURE 11.

Pre-steady-state kinetics of adenine nucleotide binding and dissociation for WT YchF and YchF(H114A). Representative time courses of the dissociation of a YchF·mant-nucleotide complex (1 μm) in the presence of excess unlabeled nucleotide (100 μm) are shown in the left panels. Representative time courses of mant-nucleotide (5 μm) association with YchF (1 μm) are shown in the right panels, with the concentration dependence of kapp on mant-nucleotide association with YchF shown in the insets. The kapp values were calculated by one-, two-, or three-exponential fitting of the time courses. A, WT YchF; B, YchF(H114A). A.U., arbitrary units.
The dissociation of mant-ADP from YchF was fit with a one-exponential function, yielding a koff of 30 ± 2 s−1. Association time courses were also best fit with a one-exponential function, and the value for the association rate constant (kon) was 3.3 ± 0.5 μm−1 s−1 (Fig. 11A). The two determined rate constants can be used to calculate the value of the respective equilibrium binding constant (KD = 9 ± 1 μm), which is comparable with values (4 ± 2 μm) reported previously by Becker et al. (11) and is consistent with the values obtained from the amplitude changes of nucleotide association (Fig. 12). Compared with the catalytically inactive variant YchF(H114A), both the rate constants for mant-ADP association and dissociation were slightly faster (2-fold), yielding an equilibrium binding constant for the nucleotide that is similar to the WT enzyme (Table 3).
FIGURE 12.
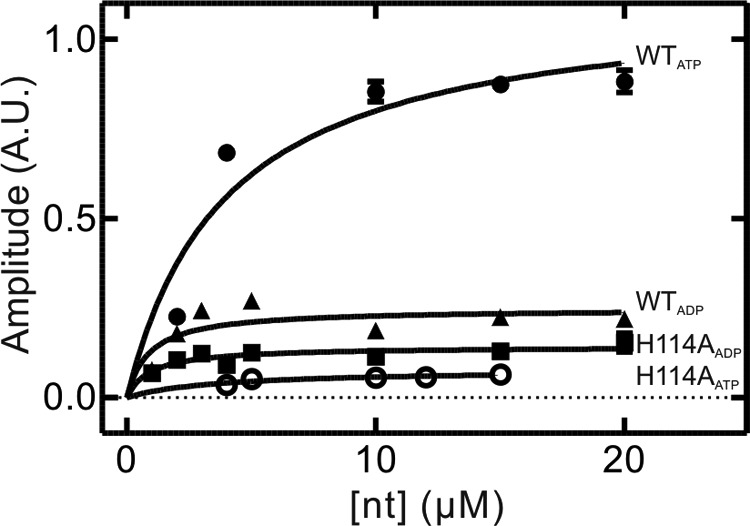
KD determination using amplitude plots. Shown are amplitudes of the overall signal change observed in stopped-flow time courses plotted as a function of nucleotide concentration. Lines represent hyperbolic fits to obtain equilibrium binding constants (KD) for mant-ATP binding to WT YchF (●) and YchF(H114A) (○) and for mant-ADP binding to WT YchF (▴) and YchF(H114A) (■). A.U., arbitrary units.
TABLE 3.
Summary of experimentally determined rate constants of WT YchF and variants for binding to adenine nucleotides
| Constant | WT YchF | YchF(H114A) |
|---|---|---|
| mant-ADP | ||
| kon (μm−1 s−1) | 3.3 ± 0.5 | 8.0 ± 0.4 |
| koff (s−1) | 30 ± 2 | 58 ± 9 |
| koff (kon plot)(s−1) | 26 ± 2 | 45 ± 4 |
| KD (μm) | 9 ± 1 | 7 ± 1 |
| KD (konplot) (μm) | 8 ± 1 | 6 ± 1 |
| KD (Amp plot) (μm) | 3 ± 1 | 1.0 ± 0.4 |
| mant-ATP | ||
| kon1 (μm−1 s−1) | 0.59 ± 0.04 | 0.9 ± 0.2 |
| kon2 (s−1) | 0.15 ± 0.01 | 0.05 ± 0.02 |
| kon3 (s−1) | 1.0 ± 0.2 | |
| koff1 (s−1) | 5 ± 2 | 14 ± 3 |
| koff1 (kon plot) (s−1) | 3.4 ± 0.5 | 9 ± 2 |
| koff2 (s−1) | 0.33 ± 0.05 | 0.02a |
| koff3 (s−1) | 0.24a | |
| KD (Amp plot) (μm) | 4 ± 1 | 4 ± 2 |
a Calculated from KD.
In contrast, the dissociation of mant-ATP from YchF was best fit with a two-exponential function, yielding two rate constants (koff1 = 5 ± 2 s−1 and koff2 = 0.33 ± 0.05 s−1) (Fig. 11A), whereas the association of mant-ATP and YchF was best fit with a three-exponential function. In the latter case, only one of the apparent rate constants exhibited the linear concentration dependence expected for a bimolecular reaction (Fig. 11A). The two remaining rates observed were not concentration-dependent, suggesting two conformational changes as a result of nucleotide binding. These data are consistent with a three-step linear binding mechanism in which binding is followed by two conformational changes. The determined association and dissociation rate constants are summarized in Table 3. To determine which observed rate constant (koff1 or koff2) is associated with mant-ATP dissociation as opposed to a conformational change, we compared the determined values with those obtained from the kon plot (y axis intercept). The koff value determined from the kon plot (3.4 ± 0.5 s−1) suggests that the faster step (5 ± 2 s−1) represents dissociation of the nucleotide and that the 10-fold slower step (0.33 ± 0.05 s−1) is associated with a conformational change. Although the pre-steady-state analysis revealed a three-step binding process, the corresponding nucleotide dissociation experiments showed two-phase kinetics. This suggests that one of the dissociation steps is either extremely fast and cannot be observed using the current system or that the rate constants for two of the steps are of the same magnitude and cannot be discriminated. Therefore, we calculated the missing rate constant (koff3) based on the KD value obtained from the amplitude plot (Fig. 12) using the equation KD = (koff1 × koff2 × koff3)/(kon1 × kon2 × kon3). The resulting value (koff3 = 0.24 s−1) is comparable with the value for koff2 and therefore supports the interpretation that dissociation precedes two conformational changes that cannot be kinetically resolved.
Interestingly, the observed dissociation kinetics of mant-ATP from YchF(H114A) varied from that of WT YchF (see above) in that it was best fit with a one-exponential equation, yielding a single rate constant for nucleotide dissociation that was ∼3-fold faster than the fastest step observed for WT YchF dissociation (Table 3). A similar reduction in complexity of the binding mechanism is also reflected in the observed association kinetics. Time courses obtained from mant-ATP binding to YchF(H114A) were best fit with a two-exponential function instead of a three-exponential function as in the case of WT YchF. Consistent with a two-step binding mechanism, only one of the observed apparent rate constants was concentration-dependent and yielded a 2-fold higher kon than compared with WT YchF (Fig. 11B). Similar to WT YchF, the observed association kinetics for mant-ATP suggest a two-step binding mechanism, whereas the dissociation seems to follow a one-step mechanism for YchF(H114A). Using the equation KD = (koff1 × koff2)/(kon1 × kon2) and the KD value obtained from the amplitude plot, the missing rate constant (koff2) can be calculated as 0.02 s−1. Given the very slow conversion to the second state and the rapid dissociation, the majority of the nucleotide-bound complex will exist in the initial binding state. Given the small overall fluorescence change associated with the conformational change, this might prevent detection due to a too small signal change.
Intrinsic Nucleotide Hydrolysis Activity
To confirm biochemically that His-114 is required for ATP hydrolysis by YchF, we determined the ability of variants YchF(H114A), YchF(H114F), YchF(H114R), and YchF(H114K) to hydrolyze ATP. Similar to the experiments shown in Fig. 2A, the rate of ATP hydrolysis was determined by thin-layer chromatography (Table 4). Although WT YchF and all variants were able to bind adenine nucleotides with similar affinities, the ATP hydrolysis rates were strongly affected. No YchF-dependent ATP hydrolysis could be detected for YchF(H114A), YchF(H114F), YchF(H114R), and YchF(H114K). This strongly suggests that His-114 is indeed important for efficient catalysis of ATP hydrolysis by YchF.
TABLE 4.
ATPase activities of WT YchF and variants
| YchF | Intrinsic kATPase | +70S (6 μm) kATPase 70S |
|---|---|---|
| min−1 | min−1 | |
| WT YchF | 0.20 ± 0.02a | 1.32 ± 0.13a |
| WT YchF | 0.10 ± 0.01 | 1.24 ± 0.04 |
| YchF(H114A) | <0.001 | 0.18 ± 0.03 |
| YchF(H114R) | <0.001 | 0.16 ± 0.02 |
| YchF(H114F) | <0.001 | |
| YchF(H114K) | <0.001 | |
| YchF(H102Q) | 0.06 ± 0.01 | 1.24 ± 0.03 |
| YchF(L76Q) | 0.05 ± 0.01 |
a At 37 °C
To investigate a putative role of the other conserved His located in the G-domain, we also constructed a variant (YchF(H102Q)) in which it is replaced with a side chain that is able to maintain the hydrogen bond observed in the MD simulations (see above). The observed intrinsic rate of ATP hydrolysis was unaffected by this substitution compared with WT YchF (Table 4), supporting our assumption that His-102 does not participate in catalysis.
Based on the fact that YchF is a member of the class of HAS-NTPases (Table 1), we wanted to determine whether reverting the Leu directly following the G3 motif, a hallmark for this class of hydrolytic enzymes, to the canonical Gln found in Ras would improve catalysis of ATP hydrolysis. We therefore constructed a variant of YchF with the native Leu (Fig. 1) replaced with Gln at position 76 (YchF(L76Q)) and determined the intrinsic ATPase activity, which was slightly lower than for WT YchF (Table 4).
Ribosome-stimulated ATPase Activity
To score the role of His-114 against a biologically significant function of YchF, we determined the rate of ATP hydrolysis for two of the His-114 variants (YchF(H114A) and YchF(H114R)) and YchF(H102Q) in the presence of 6 μm purified 70S ribosomes, which have been shown previously to stimulate the ATPase activity of WT YchF by 10-fold (11). Under these conditions, the rate of ATP hydrolysis by the YchF(H102Q) variant was identical to that observed for WT YchF (kATPase 70S = 1.24 ± 0.04 and 1.24 ± 0.03 min−1, respectively), reflecting a rate enhancement of 10-fold compared with the intrinsic activity (Table 4). Interestingly and consistent with a role for His-114 in catalysis, the rate of ATP hydrolysis for the two tested variants was 7–8-fold slower (YchF(H114A), kATP 70S = 0.18 ± 0.03 min−1; and YchF(H114R), 0.16 ± 0.02 min−1). To further investigate if this reduction is due to an effect on the catalytic step or, for example, a change in affinity of these variants for the 70S ribosome, we determined the Michaelis-Menten parameters (kcat and Km) (Table 5) for this reaction as described (11). These parameters strongly support a role of His-114 during catalysis of ATP hydrolysis by YchF, as the substitution of His-114 with Ala and Arg reduced kcat by 5- and 10-fold, respectively.
TABLE 5.
Michaelis-Menten kinetic parameters of E. coli YchF(H114) variants
| YchF | Km | kcat |
|---|---|---|
| μm | min−1 | |
| WT YchFa | 7.7 ± 1.1 | 3.1 ± 0.2 |
| YchF(H114A) | 9 ± 7 | 0.7 ± 0.2 |
| YchF(H114R) | 13 ± 12 | 0.3 ± 0.2 |
a From Ref. 11.
Discussion
Although YchF hydrolyzes ATP more efficiently than GTP, it was originally classified as a GTPase and might have retained certain functional aspects associated with GTPases. Furthermore, it belongs to the enzymatically poorly understood class of HAS-ATPases. Understanding their enzymatic properties and molecular mechanism is of fundamental importance. YchF is of particular interest because this highly conserved ATPase is found in all domains of life and shares >45% sequence identity with its human homolog, which is implicated in breast cancer (28).
His-114 Is Important for ATP Hydrolysis
To address the question regarding the molecular mechanism of nucleotide hydrolysis in the highly conserved HAS-ATPase YchF, we conducted a combined biochemical and in silico study. The pH dependence of the ATPase activity of YchF revealed that catalysis depends on a single ionizable group, with a pKa consistent with a catalytic His residue. Although YchF from E. coli contains four histidines, only two of them are located in the same domain as the nucleotide-binding pocket. Interestingly, both residues are highly conserved. MD simulations of an E. coli YchF homology model revealed that one of these histidines (His-114) is located in a flexible loop that visits different conformations depending on whether ATP, ADP, or no nucleotide is bound. In contrast, His-102 is engaged in a highly stable hydrogen bond with Tyr-204, locking it in an orientation that renders it inaccessible for catalysis. Furthermore, its movement is restricted due to a vast number of surrounding hydrophobic residues, excluding His-102 as a candidate for participation in catalysis of ATP hydrolysis. To biochemically test the hypothesis that His-114 has a role in catalysis, we determined the ATPase activity of several YchF variants bearing substitutions at position 114. Replacing His-114 with Ala, Phe, Arg, and Lys completely abolished the intrinsic ATPase activity, whereas replacing His-102 with Gln had no effect. Interestingly, in the 116 bacterial species examined, two species showed a substitution of His-114 with either Lys or Arg (Wigglesworthia glossinidia and S. pneumoniae, respectively). However, the flexible loop is poorly conserved in W. glossinidia YchF and contains a four-amino acid insert in S. pneumoniae YchF, suggesting that this loop may have a different conformation or function than that in E. coli YchF. Together with the fact that substitution of His-114 does not affect nucleotide binding, our findings strongly support a catalytic role of His-114 during ATP hydrolysis.
YchF Undergoes Nucleotide-dependent Conformational Changes
GTPases typically function as molecular switches, existing in at least two nucleotide-dependent conformations. YchF has evolved as a functional ATPase, and it is unclear if it has retained the ability to switch between two states, as no such behavior has been reported previously. Our pre-steady-state measurements suggest that YchF indeed behaves as a molecular switch. Differential analysis of the kinetic mechanism of mant-ADP and mant-ATP binding revealed that ADP binds to YchF in a single-step binding process, but the binding of ATP involves two observed conformational changes. This difference in binding mechanisms likely reflects different conformations of ATP- and ADP-bound YchF.
Different conformations of YchF·ADP and YchF·ATP are also supported by the differential behavior of the flexible loop in our MD simulations of YchF·ATP·Mg2+ and YchF·ADP·Mg2+. In the YchF·ATP·Mg2+ complex, the flexible loop is in an overall closed conformation wrapped over the bound nucleotide, with an average Cα16–Cα114 distance of ∼15 Å and the ability to sample at least two sub-states (Fig. 13). On the other hand, the conformations closest to the nucleotide-binding pocket are essentially not accessed by the loop in the YchF·ADP·Mg2+ simulation, resulting in an open conformation (average Cα16–Cα114 distance of 25 Å) (Fig. 13). In the apo state, the flexible loop is able to sample all states (Figs. 6B and 13), consistent with a model that would limit the conformational space of the flexible loop in response to the binding of a particular nucleotide.
FIGURE 13.

Nucleotide binding induces conformational changes in WT YchF. Binding of ADP results in the open conformation of the flexible loop containing the conserved His-114 essential for catalysis, whereas interaction with ATP causes a conformational change in the flexible loop leading to a closed conformation and ultimately to the catalytically competent state (ATPase activation).
Although the mechanism of ADP binding was essentially unaffected by substitution of His-114 with Ala, a marked difference was observed when mant-ATP interacted with WT YchF or YchF(H114A). In the YchF(H114A) variant, only a single conformational change was observed, as opposed to two for the WT enzyme. These results indicate that upon binding ATP, WT YchF undergoes at least two conformational changes, one of which relies on His-114. Because His-114 is located in the flexible loop and is essential for catalyzing ATP hydrolysis, one conformational change observed during ATP binding is likely a movement of the flexible loop that causes ATPase activation (Fig. 13) by bringing the catalytic His into the proximity of the bound ATP. Using steered MD simulations, it is possible to place His-114 within 7 Å of the γ-phosphate into a position similar to the catalytic Glu-282 in MnmE (data not shown). This conformation of the loop does not require unfavorable Φ and Ψ angles of the peptide backbone, further supporting the ability of the flexible loop to reach a catalytically active conformation.
Catalytic Mechanism of ATP Hydrolysis
The catalytic mechanisms of only a few HAS-GTPases have been proposed to date, including those for MnmE, YqeH, RbgA, and FeoB (13, 15, 39, 40). Of these, FeoB does not appear to use a catalytic side chain to align an attacking water molecule; MnmE and YqeH provide a catalytic residue from helix α2; and RbgA provides a catalytic residue from a flexible linker that is located in a position analogous to helix α2. Compared with most HAS-GTPases, helix α2 in YchF is shifted away from the nucleotide-binding pocket, and helix α4 is located adjacent to the nucleotide-binding pocket instead. This is similar to RbgA, in which helix α2 is also displaced away from the nucleotide-binding pocket. YchF and RbgA may therefore share similar solutions with respect to catalyzing ATP hydrolysis. In fact, RbgA has also been shown to possess a catalytic His residue in a flexible loop region (40). We suspect that upon ATPase activation, the flexible loop of YchF undergoes a conformational change to place the catalytic His residue in the active site at a position analogous to that of either the catalytic Gln in Ras-like GTPases or the catalytic Glu in MnmE. This raises the question of the role that the conserved His-114 plays during catalysis in YchF. The fact that the ATPase activity drops at pH <6.5, where His will likely be protonated, suggests that His-114 is not involved in the stabilization of the evolving negative charge on the γ-phosphate during hydrolysis. It rather supports a role of His-114 either in positioning and/or activating the hydrolytic water during the nucleophilic attack. This would be consistent with the role and the pH dependence for GTPase activation observed in EF-Tu, where experimental conditions prevented analysis below a pH of 6.5 (41).
Furthermore, a role in positioning the catalytic water is consistent with our observation that the ribosome stimulates the ATPase activity of YchF(H114R) to a 10-fold lower rate (kcat) than WT YchF. It suggests that besides the catalytic His-114, additional catalytic residues are provided either in trans directly by the ribosome or in cis by YchF upon interaction with the 70S ribosome, which, in addition to the contribution of His-114, could stabilize the charge developing on the γ-phosphate during hydrolysis. We therefore suspect that the interaction with the ribosome will stabilize the ATPase-activated conformation of the flexible loop in YchF, locking His-114 in a catalytically active position and in turn providing the observed additional 10-fold rate enhancement of ATP hydrolysis in the presence of ribosomes.
In summary, we have for the first time identified a single amino acid (His-114) that is critical for ATP hydrolysis by E. coli YchF, located in a highly flexible loop able to adopt nucleotide-dependent conformations. The highly conserved His-114 is important for sensing the presence of the γ-phosphate in the bound nucleotide and contributes critically to catalysis of ATP hydrolysis by YchF.
Acknowledgments
We thank Ute Kothe for critical reading of the manuscript and Fan Mo and Harland Brandon for technical assistance.
This work was supported by Canadian Institutes of Health Research (CIHR) Operating Grants MOP 114938, 103033, 89353, and 84917 and Canada Foundation for Innovation (CFI) Grant 202588 (to H.-J. W.), Alberta Innovates Technology Futures New Investigator Award, iCORE Strategic Chair Program (to H.-J. W.) and postgraduate scholarships (to E. M. and K. S. R.), and resource allocations from Compute Canada and WestGrid. The authors declare that they have no conflicts of interest with the contents of this article.
- EF
- elongation factor
- HAS
- hydrophobic amino acid-substituted
- AMPPCP
- adenosine 5′-(β,γ-methylene)triphosphate
- mant
- N-methylanthraniloyl
- MD
- molecular dynamics
- PDB
- Protein Data Bank
- GMPPNP
- guanosine 5′-(β,γ-imido)triphosphate
- r.m.s.d.
- root mean square deviation
- r.m.s.f.
- root mean square fluctuation.
References
- 1. Bourne H. R., Sanders D. A., McCormick F. (1990) The GTPase superfamily: a conserved switch for diverse cell functions. Nature 348, 125–132 [DOI] [PubMed] [Google Scholar]
- 2. Verstraeten N., Fauvart M., Versées W., Michiels J. (2011) The universally conserved prokaryotic GTPases. Microbiol. Mol. Biol. Rev. 75, 507–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caldon C. E., March P. E. (2003) Function of the universally conserved bacterial GTPases. Curr. Opin. Microbiol 6, 135–139 [DOI] [PubMed] [Google Scholar]
- 4. Bourne H. R., Sanders D. A., McCormick F. (1991) The GTPase superfamily: conserved structure and molecular mechanism. Nature 349, 117–127 [DOI] [PubMed] [Google Scholar]
- 5. Sprang S. R. (1997) G protein mechanisms: insights from structural analysis. Annu. Rev. Biochem. 66, 639–678 [DOI] [PubMed] [Google Scholar]
- 6. Scheffzek K., Ahmadian M. R., Kabsch W., Wiesmüller L., Lautwein A., Schmitz F., Wittinghofer A. (1997) The Ras-RasGAP complex: structural basis for GTPase activation and its loss in oncogenic Ras mutants. Science 277, 333–338 [DOI] [PubMed] [Google Scholar]
- 7. Sprang S. R. (1997) G proteins, effectors and GAPs: structure and mechanism. Curr. Opin. Struct. Biol. 7, 849–856 [DOI] [PubMed] [Google Scholar]
- 8. Mishra R., Gara S. K., Mishra S., Prakash B. (2005) Analysis of GTPases carrying hydrophobic amino acid substitutions in lieu of the catalytic glutamine: implications for GTP hydrolysis. Proteins 59, 332–338 [DOI] [PubMed] [Google Scholar]
- 9. Teplyakov A., Obmolova G., Chu S. Y., Toedt J., Eisenstein E., Howard A. J., Gilliland G. L. (2003) Crystal structure of the YchF protein reveals binding sites for GTP and nucleic acid. J. Bacteriol. 185, 4031–4037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Koller-Eichhorn R., Marquardt T., Gail R., Wittinghofer A., Kostrewa D., Kutay U., Kambach C. (2007) Human OLA1 defines an ATPase subfamily in the Obg family of GTP-binding proteins. J. Biol. Chem. 282, 19928–19937 [DOI] [PubMed] [Google Scholar]
- 11. Becker M., Gzyl K. E., Altamirano A. M., Vuong A., Urban K., Wieden H. J. (2012) The 70S ribosome modulates the ATPase activity of Escherichia coli YchF. RNA Biol. 9, 1288–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tomar S. K., Kumar P., Prakash B. (2011) Deciphering the catalytic machinery in a universally conserved ribosome binding ATPase YchF. Biochem. Biophys. Res. Commun. 408, 459–464 [DOI] [PubMed] [Google Scholar]
- 13. Scrima A., Wittinghofer A. (2006) Dimerisation-dependent GTPase reaction of MnmE: how potassium acts as GTPase-activating element. EMBO J. 25, 2940–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anand B., Surana P., Bhogaraju S., Pahari S., Prakash B. (2009) Circularly permuted GTPase YqeH binds 30S ribosomal subunit: implications for its role in ribosome assembly. Biochem. Biophys. Res. Commun. 386, 602–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ash M. R., Maher M. J., Guss J. M., Jormakka M. (2011) The initiation of GTP hydrolysis by the G-domain of FeoB: insights from a transition-state complex structure. PLoS ONE 6, e23355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marlovits T. C., Haase W., Herrmann C., Aller S. G., Unger V. M. (2002) The membrane protein FeoB contains an intramolecular G protein essential for Fe(II) uptake in bacteria. Proc. Natl. Acad. Sci. U.S.A. 99, 16243–16248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Foucher A. E., Reiser J. B., Ebel C., Housset D., Jault J. M. (2012) Potassium acts as a GTPase-activating element on each nucleotide-binding domain of the essential Bacillus subtilis EngA. PLoS ONE 7, e46795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Achila D., Gulati M., Jain N., Britton R. A. (2012) Biochemical characterization of ribosome assembly GTPase RbgA in Bacillus subtilis. J. Biol. Chem. 287, 8417–8423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Danese I., Haine V., Delrue R. M., Tibor A., Lestrate P., Stevaux O., Mertens P., Paquet J. Y., Godfroid J., De Bolle X., Letesson J. J. (2004) The Ton system, an ABC transporter, and a universally conserved GTPase are involved in iron utilization by Brucella melitensis 16M. Infect. Immun. 72, 5783–5790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guerrero C., Tagwerker C., Kaiser P., Huang L. (2006) An integrated mass spectrometry-based proteomic approach: quantitative analysis of tandem affinity-purified in vivo cross-linked protein complexes (QTAX) to decipher the 26 S proteasome-interacting network. Mol. Cell. Proteomics 5, 366–378 [DOI] [PubMed] [Google Scholar]
- 21. Fernebro J., Blomberg C., Morfeldt E., Wolf-Watz H., Normark S., Normark B. H. (2008) The influence of in vitro fitness defects on pneumococcal ability to colonize and to cause invasive disease. BMC Microbiol. 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gradia D. F., Rau K., Umaki A. C., de Souza F. S., Probst C. M., Correa A., Holetz F. B., Avila A. R., Krieger M. A., Goldenberg S., Fragoso S. P. (2009) Characterization of a novel Obg-like ATPase in the protozoan Trypanosoma cruzi. Int. J. Parasitol. 39, 49–58 [DOI] [PubMed] [Google Scholar]
- 23. Zhang J., Rubio V., Lieberman M. W., Shi Z. Z. (2009) OLA1, an Obg-like ATPase, suppresses antioxidant response via nontranscriptional mechanisms. Proc. Natl. Acad. Sci. U.S.A. 106, 15356–15361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang J. W., Rubio V., Zheng S., Shi Z. Z. (2009) Knockdown of OLA1, a regulator of oxidative stress response, inhibits motility and invasion of breast cancer cells. J. Zhejiang Univ. Sci. B 10, 796–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wenk M., Ba Q., Erichsen V., MacInnes K., Wiese H., Warscheid B., Koch H. G. (2012) A universally conserved ATPase regulates the oxidative stress response in Escherichia coli. J. Biol. Chem. 287, 43585–43598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheung M. Y., Xue Y., Zhou L., Li M. W., Sun S. S., Lam H. M. (2010) An ancient P-loop GTPase in rice is regulated by a higher plant-specific regulatory protein. J. Biol. Chem. 285, 37359–37369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen Y. C., Chung Y. T. (2011) A conserved GTPase YchF of Vibrio vulnificus is involved in macrophage cytotoxicity, iron acquisition, and mouse virulence. Int. J. Med. Microbiol. 301, 469–474 [DOI] [PubMed] [Google Scholar]
- 28. Matsuzawa A., Kanno S., Nakayama M., Mochiduki H., Wei L., Shimaoka T., Furukawa Y., Kato K., Shibata S., Yasui A., Ishioka C., Chiba N. (2014) The BRCA1/BARD1-interacting protein OLA1 functions in centrosome regulation. Mol. Cell 53, 101–114 [DOI] [PubMed] [Google Scholar]
- 29. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 31. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Humphrey W., Dalke A., Schulten K. (1996) VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38 [DOI] [PubMed] [Google Scholar]
- 34. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom emperical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 35. Glykos N. M. (2006) Software news and updates. Carma: a molecular dynamics analysis program. J. Comput. Chem. 27, 1765–1768 [DOI] [PubMed] [Google Scholar]
- 36. Voet D. V., Voet J. G. (eds) (2004) Biochemistry, 3rd Ed., John Wiley & Sons, New York [Google Scholar]
- 37. Noel J. P., Hamm H. E., Sigler P. B. (1993) The 2.2 Å crystal structure of transducin-α complexed with GTPγS. Nature 366, 654–663 [DOI] [PubMed] [Google Scholar]
- 38. Coleman D. E., Berghuis A. M., Lee E., Linder M. E., Gilman A. G., Sprang S. R. (1994) Structures of active conformations of Giα1 and the mechanism of GTP hydrolysis. Science 265, 1405–1412 [DOI] [PubMed] [Google Scholar]
- 39. Anand B., Surana P., Prakash B. (2010) Deciphering the catalytic machinery in 30S ribosome assembly GTPase YqeH. PLoS ONE 5, e9944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gulati M., Jain N., Anand B., Prakash B., Britton R. A. (2013) Mutational analysis of the ribosome assembly GTPase RbgA provides insight into ribosome interaction and ribosome-stimulated GTPase activation. Nucleic Acids Res. 41, 3217–3227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Daviter T., Wieden H. J., Rodnina M. V. (2003) Essential role of histidine 84 in elongation factor Tu for the chemical step of GTP hydrolysis on the ribosome. J. Mol. Biol. 332, 689–699 [DOI] [PubMed] [Google Scholar]