

Background: Haloacid dehalogenase (HAD)-like hydrolases represent the largest superfamily of phosphatases.
Results: Biochemical, structural, and evolutionary studies of the 10 uncharacterized soluble HADs from Saccharomyces cerevisiae provided insight into their substrates, active sites, and evolution.
Conclusion: Evolution of novel substrate specificities of HAD phosphatases shows no strict correlation with sequence divergence.
Significance: Our work contributes to a better understanding of an important model organism.
Keywords: crystal structure, enzyme catalysis, phosphatase, Saccharomyces cerevisiae, substrate specificity, haloacid dehalogenase
Abstract
The haloacid dehalogenase (HAD)-like enzymes comprise a large superfamily of phosphohydrolases present in all organisms. The Saccharomyces cerevisiae genome encodes at least 19 soluble HADs, including 10 uncharacterized proteins. Here, we biochemically characterized 13 yeast phosphatases from the HAD superfamily, which includes both specific and promiscuous enzymes active against various phosphorylated metabolites and peptides with several HADs implicated in detoxification of phosphorylated compounds and pseudouridine. The crystal structures of four yeast HADs provided insight into their active sites, whereas the structure of the YKR070W dimer in complex with substrate revealed a composite substrate-binding site. Although the S. cerevisiae and Escherichia coli HADs share low sequence similarities, the comparison of their substrate profiles revealed seven phosphatases with common preferred substrates. The cluster of secondary substrates supporting significant activity of both S. cerevisiae and E. coli HADs includes 28 common metabolites that appear to represent the pool of potential activities for the evolution of novel HAD phosphatases. Evolution of novel substrate specificities of HAD phosphatases shows no strict correlation with sequence divergence. Thus, evolution of the HAD superfamily combines the conservation of the overall substrate pool and the substrate profiles of some enzymes with remarkable biochemical and structural flexibility of other superfamily members.
Introduction
Characterization of proteins with unknown functions is one of the major challenges to modern biology (1). Global genome and metagenome sequencing efforts have already produced millions of new sequences, from which 30–40% of the genes have no known function (2–4). Even for the two best characterized model organisms, Escherichia coli and Saccharomyces cerevisiae, ∼20% of their genes have no function assigned or have only a general function predicted (e.g. putative hydrolase), and experimental data are available for only 54% of E. coli proteins (5, 6). In addition, a substantial and growing number of genes have inaccurate functional annotations. Focused analyses of new sequences for members of 37 protein families deposited in 2005 have shown that about 40% of these proteins remain misannotated (7, 8). Our knowledge gap also includes over 1,000 of the known enzyme activities in the Enzyme Classification (of the 4,997 EC numbers) that have no associated gene sequence (orphan enzymes) (9, 10).
To infer gene function, complementary computational and experimental approaches and their combinations have been used, including sequence analysis, comparative genomics, gene expression and disruption, protein interaction, and protein structure, but ultimately all depend on experimental testing (3, 11–21). Recently, the Computational Bridge to Experiments (COMBREX) consortium coordinated a community of computational and experimental scientists to generate functional predictions for the most interesting families of unknown proteins and to carry out experimental characterization (22). Another large scale project, the Enzyme Function Initiative (EFI) merges experimental approaches with computationally based function predictions (23). The EFI combines bioinformatics, protein structure analysis, and enzymology to assign reliable functions to unknown enzymes from microbial genomes. Both projects have considerable potential to accelerate functional characterization of unknown proteins.
Most enzymes are organized into families of sequence-related proteins whose members catalyze the same or similar reactions but have evolved different substrate preferences and specific biological functions. These families represent a challenge for functional annotation because their catalytic activities and substrates are likely to be very similar. One such family are haloacid dehalogenase (HAD)2-like hydrolases, which represent one of the largest enzyme superfamilies found in all organisms, with 479,051 sequences in databases (InterPro IPR023214) and 33 major families (24, 25). Most genomes are predicted to contain multiple HAD-like proteins, including 28 genes in E. coli, 45 genes in S. cerevisiae, and 183 genes in humans (26). This superfamily was originally named after the haloacid dehalogenases, but it is dominated by putative phosphatases (∼79%) and ATPases (∼20%) and also includes phosphonatases and phosphomutases (25–27). Specifically, the HAD-like phosphatases are a family of diverse enzymes that are responsible for the majority of metabolic phosphomonoester hydrolysis reactions in all kingdoms of life (26, 28). Extensive sequence comparisons show that the HAD-like hydrolases are defined by the presence of four conserved sequence motifs with the characteristic N-terminal motif I containing two Asp residues (DXD), whereas motifs II and III contain a highly conserved Thr (or Ser) and Lys, respectively (24, 25). Most HAD-like hydrolases contain a highly conserved α/β core domain with a mobile cap domain, which can be inserted between motifs I and II (type I or C1) or between motifs II and III (type II or C2) of the core domain (25, 29). Type III HAD-like hydrolases have no cap domain (C0).
HAD-like phosphatases can perform different cellular roles, including primary and secondary metabolism, regulation of enzyme activity or protein assembly, cell housekeeping, and nutrient uptake (26). To date, many HAD-like phosphatases from different organisms have been characterized both biochemically and structurally, including the phosphoserine phosphatase SerB from Methanococcus jannaschii (30), phosphoglycolate phosphatase TA0175 from Thermoplasma acidophilum (31), UMP nucleotidase NagD from E. coli (32), and inorganic pyrophosphatase BT2127 from Bacteroides thetaiotaomicron (33). In particular, phosphoprotein phosphatase activity has been demonstrated for several eukaryotic HAD-like hydrolases: human CTDP1 (or FCP1; Ser(P) phosphatase) (34), Drosophila and mammalian Eyes absent (Tyr(P) phosphatase) (35–37), and mammalian chronophin (Ser(P) phosphatase) (38, 39). In a previous study, we have experimentally characterized the substrate specificities of the 19 soluble HAD-like phosphatases from E. coli and demonstrated that most of the E. coli HADs show remarkably broad and overlapping substrate profiles, being active against phosphorylated carbohydrates, nucleotides, organic acids, and coenzymes (40). A phylogenetic analysis of the E. coli HADs suggested that their secondary activities might have no direct physiological function, but they could comprise a reservoir for the evolution of phosphatases with novel specificities.
The genome of the yeast S. cerevisiae contains at least 45 genes encoding the predicted HAD-like hydrolases (IPR023214), which include 19 membrane proteins (mostly ATPases), seven HAD-like domains in large proteins, and 19 soluble, stand-alone HAD-like proteins (supplemental Table 1). From the last group, the enzymatic activity and specific substrates have previously been experimentally demonstrated for two deoxyglucose-6-phosphate phosphatases (DOG1 and DOG2), two glycerol-3-phosphate phosphatases (GPP1 and GPP2), DNA 3′-phosphatase TPP1, phosphomannomutase SEC53, phosphatidylglycerophosphatase GEP4, pyrimidine 5′-nucleotidase SDT1, and protein fructosamine 6-phosphatase MDP-1 (41–47).
Here, we present the results of biochemical and structural studies of soluble HAD-like hydrolases from S. cerevisiae with a focus on the 10 uncharacterized HADs. Eight previously uncharacterized HADs had phosphatase activity. Additional substrates were found for the five known S. cerevisiae HAD phosphatases. Collectively, these were active against 2-phosphoglycolate, thiamine monophosphate, pyridoxal phosphate, phosphoserine, glycerol 1-phosphate (Gly-1-P), nucleotides, and phosphorylated peptides. The crystal structures of four S. cerevisiae HADs provided insight into the molecular basis of their substrate specificity. Although the S. cerevisiae and E. coli HADs share low overall sequence similarities, these enzymes show remarkable conservation of their biochemical activities.
Experimental Procedures
Gene Cloning, Protein Purification, and Mutagenesis
The genes encoding 15 selected yeast HADs (Table 1) were amplified by PCR from S. cerevisiae genomic DNA and cloned into a modified pET15b vector (Novagen) as described previously (40). Purification of proteins for screening and biochemical characterization was performed as described previously (48). The oligomeric state of purified proteins was determined using gel filtration analysis on a Superdex 200 10/300 column (GE Healthcare). Site-directed mutagenesis of YKR070W was performed using a protocol based on the QuikChange site-directed mutagenesis kit (Stratagene) as described previously (48). The presence of mutations was verified by DNA sequencing, and the mutant proteins were overexpressed and purified in the same manner as the wild-type YKR070W.
TABLE 1.
15 purified soluble HAD-like hydrolases from S. cerevisiae and their phosphatase activity against pNPP
| Gene name | ORF name | Database annotation | Reported or predicted substrate | pNPP hydrolysisa |
|---|---|---|---|---|
| units/mg | ||||
| Previously characterized HADs | ||||
| DOG1 | Yhr044c | 2-Deoxyglucose 6-phosphate phosphatase 1 | 2-Deoxyglucose 6-phosphateb | 0.9 |
| DOG2 | Yhr043c | 2-Deoxyglucose 6-phosphate phosphatase 2 | 2-Deoxyglucose 6-phosphateb | 0.6 |
| HOR2 | Yer062c | Glycerol-1-phosphate phosphohydrolase 2 | Glycerol 1-phosphatec | 1.1 |
| RHR2 | Yil053w | Glycerol-1-phosphate phosphohydrolase 1 | Glycerol 1-phosphatec | 1.3 |
| SDT1 | Ygl224c | Pyrimidine 5′-nucleotidase | 5′-UMP, NMNd | 9.2 |
| Uncharacterized HADs | ||||
| PHM8 | Yer037w | Phosphate metabolism protein 8 | Lysophosphatidic acide | 17.4 |
| PHO13 | Ydl236w | 4-Nitrophenyl phosphatase | p-Nitrophenyl phosphate | 203.8 |
| SER2 | Ygr208w | Phosphoserine phosphatase | Phosphoserine (predicted) | 1.3 |
| UTR4 | Yel038w | Enolase-phosphatase E1 | 5-(Methylthio)-2,3-dioxopentyl-P (predicted) | 0.7 |
| YCR015C | Ycr015c | UPF0655 protein YCR015C | No information | 0.3 |
| YKR070W | Ykr070w | Uncharacterized protein | No information | 17.1 |
| YKL033W | Ykl033w | Uncharacterized hydrolase | No information | NDf |
| YMR130W | Ymr130w | Uncharacterized protein | No information | 0.02 |
| YNL010W | Ynl010w | Uncharacterized phosphatase | No information | 0.8 |
| YOR131C | Yor131c | Uncharacterized hydrolase | No information | 9.4 |
Enzymatic Screens and Assays
Purified yeast HADs were initially screened for the presence of phosphatase activity against the general phosphatase substrates pNPP and acetyl phosphate as described previously (40). Secondary phosphatase screens with 93 phosphorylated metabolites (supplemental Table 2) were then performed to identify the preferred in vitro substrates for these proteins (40). Phosphatase activity against phosphorylated metabolites and phosphopeptides was measured spectrophotometrically using the malachite green reagent or a mild phosphate detection method (48). The dependence of phosphatase activity on divalent metal cations was determined using saturating concentrations of the indicated substrates and cations (5 mm Mg2+ or 0.5 mm for other ions), whereas the pH dependence was determined using a mixed buffer system (MEGA buffer) (49). For the determination of kinetic parameters (Km and kcat), enzymatic assays were performed using a range of substrate concentrations (0.05–10 mm) or a range of metal ion concentrations (0.005–2.5 mm) in the presence of saturating substrate concentrations (0.5–0.8 mm). Kinetic parameters were calculated by non-linear regression analysis of raw data to fit to the Michaelis-Menten function using GraphPad Prism Software (version 4.00 for Windows, GraphPad Software, San Diego, CA). A heat map of HAD activity was performed using the gplots heatmap.2 function, whereas hierarchical clustering of HADs (based on substrate profiles) was calculated using Euclidian distance, and groups were clustered using the complete linkage method (R Foundation for Statistical Computing).
HPLC Methods
An HPLC-based protocol was used for the analysis of phosphohydrolase activity against pseudouridine monophosphate (Ψ-UMP), which was generated using commercially available pseudouridine triphosphate (TriLink Biotechnologies) and purified Maf nucleotide pyrophosphatases from yeast (YOR111W) or Bacillus subtilis (BSU28050) (50). The reactions were performed in a reaction mixture (20 μl final volume) containing 50 mm HEPES-K (pH 7.5), 10 mm CoCl2, 1 mm pseudouridine triphosphate (Ψ-UTP), and 2.5 μg of YOR111W or BSU28050 for Ψ-UTPase reactions (to generate Ψ-UMP) followed by the addition of purified yeast HADs (PHM8, SDT1, or YKL033W-A; 5 μg) for Ψ-UMPase assays. The reactions were carried out at 30 °C for 2 h and analyzed using reversed phase chromatography on a Varian ProStar HPLC system equipped with a Varian Pursuit C18 column as described previously for Maf proteins (50). Standard solutions of Ψ-UTP and Ψ were used to confirm the identity of the observed peaks and products.
The S. cerevisiae haploid mutant ycr015cΔ (genotype BY4741; Mat a; his3D1; leu2D0; met15D0; ura3D0; YCR015c::kanMX4) was obtained from the Euroscarf collection, and the YCR015c deletion was verified by PCR. The ycr015cΔ and the corresponding wild type strain BY4741 were grown at 30 °C in a synthetic defined medium without thiamine (SD −thiamine) that consisted of yeast nitrogen base without amino acids, without ammonium sulfate, and without thiamine (1.9 g/liter; ForMedium), −Ura DO supplement (0.77 g/liter; Clontech), and uracil (180 mg/liter; Sigma) with glucose at 2% (w/v) final concentration. Cells were harvested when A600 reached 0.6, frozen in liquid nitrogen, and stored at −80 °C. Cell pellets were resuspended in 0.5 ml of 7.2% (v/v) perchloric acid and sonicated. The sonicate was held on ice for 15 min with periodic vortex mixing and then cleared by centrifugation at 4 °C (2,000 × g, 15 min). Thiamine and its phosphates were analyzed by oxidation to thiochrome derivatives followed by HPLC with fluorometric detection (51). The oxidation reagent was a freshly prepared solution of 12.14 mm potassium ferricyanide in 3.35 m NaOH. Samples or standards (160 μl) were mixed with 15 μl of methanol; 100 μl of oxidation agent was added and mixed for 60 s, and 100 μl of 1.43 m phosphoric acid was then added. The standards (thiamine, TMP, and thiamine pyrophosphate (TPP) dissolved in 0.1 m HCl) were made up in 7.2% (v/v) perchloric acid, 0.25 m NaOH (1:1, v/v). Samples (25 μl) were separated on an Alltima HP C18 amide column (150 × 4.6 mm, 5 μm, 190 Å, Alltech). The mobile phase (1 ml/min) consisted of a gradient of potassium phosphate (140 mm, pH 7.00), 12% methanol (v/v) (buffer A) to 70% (v/v) methanol (buffer B). Runs began with 100% buffer A; within 10 min, the ratio A/B reached 50:50, becoming 0:100 in the following 5 min.
Analysis of Protein-Protein Interactions
The physical interactions of the S. cerevisiae YKR070W were analyzed using an affinity tagging and purification mass spectrometry approach essentially as described previously (52). The C-terminally GFP-tagged YKR070W was purified using anti-GFP MicroBeads, and the associated proteins were analyzed using a high performance linear trap quadrupole Orbitrap Velos Pro mass spectrometer (Thermo Scientific, Waltham, MA). After filtering the data set with a confidence score of 95% probability and 2 or more peptides, we were able to retain 119 protein-protein interactions. This data set was merged with previously known interactions extracted from the literature and public databases (BioGRID, MINT, IntAct, and DIP), resulting in 127 unique protein-protein interactions. These were then subjected to the GO Slim Mapper deployed in SGD (53) to group the interacting proteins into specific processes.
Protein Crystallization
The S. cerevisiae HAD-like proteins were crystallized at room temperature using the sitting or hanging drop vapor diffusion protocols. The crystals of RHR2 were grown in a crystallization solution containing 100 mm Bistris propane buffer (pH 7.0) and 2.5 m ammonium sulfate. Mercury-labeled crystals were obtained by overnight soaking of crystals in 10 mm HgCl2 followed by cryoprotection in 3.6 m ammonium sulfate and flash freezing in liquid nitrogen. The selenomethionine-labeled YKR070W (54) was crystallized in a solution containing 100 mm potassium/sodium phosphate (pH 6.2), and 26% (w/v) polyethylene glycol (PEG) 3350. The crystals were treated with paratone oil as the cryoprotectant and flash-frozen in liquid nitrogen. The crystals of the YKR070W complex with glycerol 3-phosphate (Gly-3-P) were obtained by co-crystallization in a solution containing 100 mm HEPES-K (pH 7.5), 1.2 m sodium citrate, 0.2 mm MgCl2, and 4 mm Gly-3-P (added to the crystallization sitting drop prior to set-up). The SDT1 crystals were grown using native protein in a solution containing 100 mm BisTris (pH 6.5) and 25% (w/v) PEG 2000 MME (monomethylether). Prior to freezing, 25% (v/v) ethylene glycol was added for cryoprotection. Mercury labeling was performed by adding 6 mm HgCl2 to the crystallization drop followed by a 4-h incubation.
Data Collection, Structure Determination, and Refinement
Single-wavelength anomalous diffraction (SAD) data sets were collected for the RHR2 (Hg-SAD) and YKR070W (Se-SAD) crystals at 12.66 keV at the Structural Biology Center at the Advanced Photon Source (19-ID) (55). The low resolution data sets were collected on mercury-labeled SDT1 crystals (data not shown) using a Rigaku Micromax-007 HF generator equipped with a CR Raxis-4++ detector and producing chromium Kα radiation, whereas the refinement of the SDT1 structure was performed with native crystal data sets collected using copper Kα radiation. Data collection, integration, and scaling on all data were performed with the HKL3000 suite of programs (56). The initial phases of RHR2, SDT1, and YKR070W were determined by SAD phasing, and the initial protein models were built using the HKL3000 software package (57–62). A summary of the crystallographic data can be found in Table 2. The structure of the YKR070W·Gly-3-P complex was solved by molecular replacement using the program MOLREP from the CCP4 program suite and the YKR070W structure as a search model (60, 63). All protein models required substantial rebuilding and refining using the program COOT in order to obtain the final models (57). The models were refined against all reflections in the resolution range except for a randomly selected 5% of reflections, which were used for monitoring Rfree. The quality of the structures was checked using the validation tools included in the programs COOT and Molprobity (64). The final refinement statistics are shown in Table 2.
TABLE 2.
Kinetic parameters of purified HAD phosphatases from S. cerevisiae
Assays were performed in the presence of 5 mm Mg2+.
| Protein | Variable substrate | Km | kcat | kcat/Km |
|---|---|---|---|---|
| mm | s−1 | s−1 m−1 | ||
| DOG1 | 2-Deoxyglucose-6-P | 1.39 ± 0.07 | 99.6 ± 2.1 | 7.1 ± 104 |
| Mannose-6-P | 5.72 ± 0.14 | 43.4 ± 0.6 | 8.1 ± 103 | |
| DOG2 | 2-Deoxyglucose-6-P | 0.77 ± 0.04 | 17.0 ± 0.5 | 2.2 ± 104 |
| Mannose-6-P | 1.1 ± 0.1 | 13.8 ± 0.5 | 1.3 ± 104 | |
| Fructose-1-P | 1.8 ± 0.1 | 29.0 ± 0.5 | 1.6 ± 104 | |
| HOR2 | Gly-3-P | 3.02 ± 0.12 | 122 ± 2 | 4.0 ± 104 |
| Gly-1-P | 4.72 ± 0.15 | 114 ± 1 | 2.4 ± 104 | |
| PHM8 | AMP | 2.1 ± 0.1 | 31.5 ± 0.3 | 1.5 ± 104 |
| CMP | 0.063 ± 0.002 | 19.8 ± 0.1 | 3.2 ± 105 | |
| GMP | 0.21 ± 0.01 | 57.6 ± 0.7 | 2.7 ± 105 | |
| IMP | 0.99 ± 0.1 | 32.8 ± 0.8 | 3.3 ± 104 | |
| UMP | 0.083 ± 0.002 | 15.1 ± 0.1 | 1.8 ± 105 | |
| XMP | 0.52 ± 0.02 | 28.6 ± 0.6 | 5.5 ± 104 | |
| PHO13 | P-glycolate | 0.061 ± 0.003 | 67.4 ± 0.8 | 1.1 ± 106 |
| RHR2 | Gly-3-P | 2.81 ± 0.28 | 79.0 ± 2.7 | 2.8 ± 104 |
| Gly-1-P | 2.95 ± 0.46 | 77.7 ± 3.4 | 2.6 ± 104 | |
| SDT1 | UMP | 0.028 ± 0.004 | 0.64 ± 0.02 | 2.3 ± 104 |
| CMP | 0.073 ± 0.003 | 0.78 ± 0.01 | 1.1 ± 104 | |
| XMP | 0.24 ± 0.03 | 0.42 ± 0.01 | 1.7 ± 103 | |
| Tyr(P) | 1.19 ± 0.15 | 0.79 ± 0.03 | 6.6 ± 102 | |
| NMN | 1.56 ± 0.14 | 0.66 ± 0.03 | 4.2 ± 102 | |
| SER2 | Ser(P) | 0.08 ± 0.01 | 117 ± 4 | 1.4 ± 106 |
| YCR015C | Thiamine-P | 0.019 ± 0.002 | 20.4 ± 0.4 | 1.2 ± 106 |
| YKL033W | XMP | 1.26 ± 0.06 | 1.9 ± 0.1 | 1.5 ± 103 |
| FMN | 0.19 ± 0.01 | 0.20 ± 0.01 | 0.8 ± 103 | |
| YKR070W | Glu-6-P | 0.22 ± 0.02 | 35.1 ± 0.8 | 1.6 ± 105 |
| Glu-6-P (Mn2+) | 0.06 ± 0.01 | 7.9 ± 0.1 | 1.4 ± 105 | |
| 2-Deoxyglucose-6-P | 0.59 ± 0.03 | 46.2 ± 0.6 | 7.8 ± 104 | |
| 2-Deoxyribose-5-P | 0.31 ± 0.01 | 27.3 ± 0.4 | 8.8 ± 104 | |
| Fructose-1,6-bisP | 0.65 ± 0.02 | 40.1 ± 0.4 | 6.2 ± 104 | |
| Fructose-6-P | 1.04 ± 0.05 | 67.4 ± 0.6 | 6.5 ± 104 | |
| Mannose-6-P | 0.67 ± 0.03 | 39.6 ± 0.6 | 5.9 ± 104 | |
| Ribose-5-P | 0.75 ± 0.03 | 49.0 ± 0.4 | 6.5 ± 104 | |
| Erythrose-4-P | 0.33 ± 0.07 | 29.5 ± 1.6 | 9.0 ± 104 | |
| Gly-3-P | 0.28 ± 0.02 | 44.1 ± 0.8 | 1.6 ± 105 | |
| Gly-1-P | 0.39 ± 0.05 | 35.6 ± 1.3 | 9.2 ± 104 | |
| Gly-2-P | 1.22 ± 0.04 | 12.2 ± 0.2 | 1.0 ± 104 | |
| YNL010W | Gly-1-P | 1.12 ± 0.06 | 35.8 ± 0.5 | 3.2 ± 104 |
| Erythrose-4-P | 2.84 ± 0.11 | 26.3 ± 0.4 | 0.9 ± 104 | |
| Sorbitol-6-P | 0.26 ± 0.02 | 0.70 ± 0.03 | 2.7 ± 103 | |
| YOR131C | PLP | 0.07 ± 0.01 | 3.0 ± 0.1 | 4.2 ± 104 |
| Imido-diP | 0.69 ± 0.05 | 3.0 ± 0.1 | 4.4 ± 103 |
Accession Numbers
The atomic coordinates and structure factors have been deposited in the Protein Data Bank with accession codes 2QLT (RHR2), 3NUQ (SDT1), 3KC2 (the wild type YKR070W), and 3RF6 (the YKR070W D19A in complex with Gly-3-P).
Results
The Complement of Soluble HADs in S. cerevisiae
The S. cerevisiae genome encodes at least 45 HAD-like hydrolases, of which 19 proteins have been predicted to be membrane-bound ATPases (PEDANT database) (supplemental Table 1). Previously characterized yeast HADs include the RNA polymerase II subunit A C-terminal domain phosphatase FCP1 (YMR277W); the phosphatidate phosphatase PAH1 (YMR165C); the three trehalose-phosphate synthase-phosphatases TPS2 (YDR074W), TSL1 (YML100W), and TPS3 (YMR261C); the PAH1 protein phosphatase NEM1 (YHR004C); the DNA 3′-phosphatase TPP1 (YMR156C); the mitochondrial phosphatidylglycerophosphatase GEP4 (YHR100C); the phosphomannomutase SEC53 (YFL045C); the pyrimidine- and NMN-specific 5′-nucleotidases ISN1 (YOR155C) and SDT1 (YGL224C); and the protein-fructosamine-6-phosphatase MDP-1 (YER134C) (43–47, 65–70). In addition, specific substrates have been identified for two pairs of paralogous HADs, namely the 2-deoxyglucose-6-phosphate phosphatases DOG1 and DOG2 (92% sequence identity) and the glycerol-1-phosphate phosphohydrolases RHR2 (GPP1) and HOR2 (GPP2) (95% sequence identity) (41, 42). Although purified PHM8 protein has been shown to specifically hydrolyze lysophosphatidic acid, the reported specific activities were at a low nanomolar level (up to 3 nmol min−1 mg−1 protein) (71). Sequence analysis of the 15 selected HAD-like hydrolases confirmed the presence of conserved HAD motifs in all of these proteins (supplemental Fig. 1). Based on the cap domain architecture, they mostly belong to the HAD C1 group (the cap domain is inserted between the first and second HAD motifs) with only two C2 proteins (the cap domain is located between the second and third HAD motifs) (Fig. 1). According to a previous sequence analysis (25), the yeast HADs of the C1 group can be further divided into five families, including PSP, Epo, BGPM, Dehr, and Eno. Members of each family that are likely to be (co)orthologs of the respective yeast HADs were also detected in E. coli and, for three of the families, in humans as well (Fig. 1).
FIGURE 1.
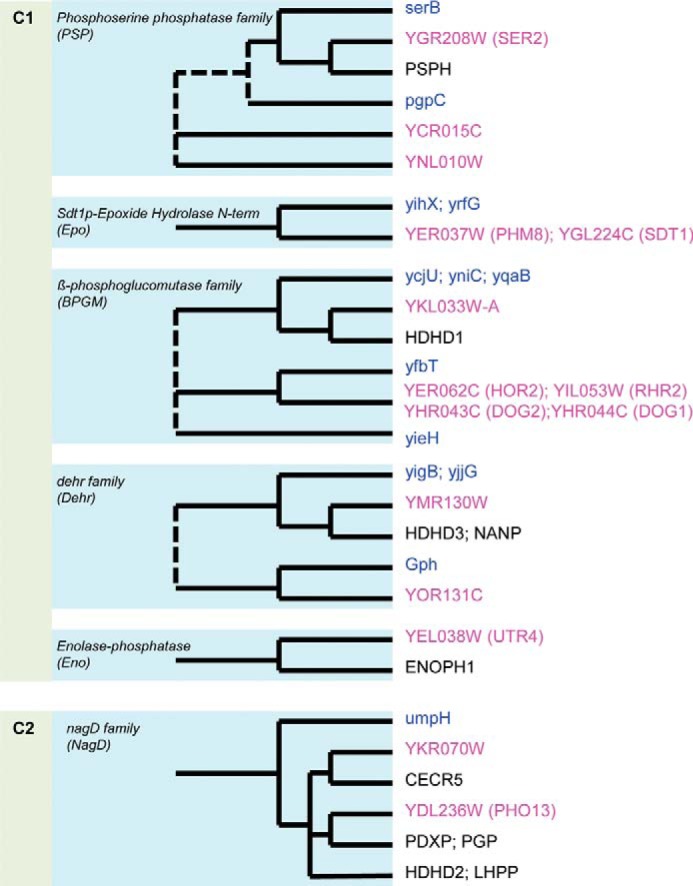
Sequence-based clustering of the 15 soluble S. cerevisiae HADs and their homologs. Schematic trees, based on the COG, KOG, and OrthoMCL data (25, 132, 133), show the relationships between HADs from S. cerevisiae (pink labels), humans (black labels), and E. coli (blue labels). Dashed lines, uncertain relationships.
Screening of Purified Yeast HADs for Phosphatase Activity
We expressed in E. coli and purified 10 uncharacterized S. cerevisiae HAD-like proteins as well as five HAD enzymes with identified substrates (DOG1, DOG2, HOR2, RHR2, and SDT1) to confirm these activities and to explore the possibility of their having additional activities (Table 1). The proteins were affinity-purified to over 95% homogeneity and first analyzed for Mg2+-dependent phosphatase activity using the generic phosphatase substrate p-nitrophenyl phosphate (pNPP) (21). Except for YKL033W, most yeast HADs showed readily detectable Mg2+-dependent hydrolysis of pNPP (Table 1), with the highest activity observed for PHO13 (∼200 μmol min−1 mg−1 protein), which has been annotated as a pNPP phosphatase (72). Like the E. coli HADs (40), the majority of S. cerevisiae HADs also hydrolyzed the small phosphodonor substrate acetyl phosphate (data not shown).
Several HAD domain-containing proteins have protein phosphatase activity (37–39, 73). Therefore, we screened the purified yeast HADs against a library of 65 synthetic oligopeptides containing Ser(P), Thr(P), or Tyr(P) (supplemental Table 3). The sequences of the 46 phosphopeptides include the most common protein phosphorylation sites found in the S. cerevisiae phosphoproteome, whereas the remaining 19 phosphopeptides represent common protein phosphatase substrates related to various protein kinases (74, 75). Four S. cerevisiae HADs showed Mg2+-dependent protein phosphatase activity, including SDT1 (Tyr(P)- and Ser(P)-containing peptides), YKR070W (Tyr(P)), YNL010W (Tyr(P) and Ser(P)), and YOR131C (Tyr(P) and Ser(P)) (supplemental Table 4). The targeted peptide sequences are present in the S. cerevisiae MAPKs HOG1, FUS3, and KSS1; in the regulatory proteins REG1 and MMF1; and in subunits of several complexes (Pat1p, RIF1, and NMD2). Although the presence of protein phosphatase activity has not yet been reported for E. coli HADs (25, 40), this activity has been proposed for several microbial effector proteins with HAD domains from the pathogenic bacteria Porphyromonas gingivalis and Coxiella burnetii (76, 77).
Substrate Profiles of Purified Yeast HADs
To identify potential natural substrates of the S. cerevisiae HADs, purified proteins were screened for the presence of Mg2+-dependent phosphatase activity against a set of 93 phosphorylated compounds (supplemental Table 2) representing all main divisions of natural phosphometabolomes (nucleotides and phosphorylated carbohydrates, organic acids, and amino acids) (78). These assays confirmed the previously reported substrates for DOG1 and DOG2 (2-deoxyglucose-6-phosphate), RHR2 (GPP1), and HOR2 (GPP2) (Gly-3-P) as well as for SDT1 (UMP and CMP) (Fig. 2). Both DOG1 and DOG2 exhibited significant but lesser activity toward mannose 6-phosphate, glucose 6-phosphate, fructose 1-phosphate, and ribose 5-phosphate, whereas RHR2 and HOR2 were also active against Gly-1-P (Fig. 2). The enzymes possessed higher catalytic activity and lower Km toward the major substrates (2-deoxyglucose-6-phosphate and Gly-3-P) than reported previously (41, 42, 45) (Table 3 and supplemental Table 5). Recently, nicotinamide mononucleotide (NMN) has been reported to be another physiological substrate for SDT1 (70). Indeed, our in vitro assays with purified SDT1 revealed high activity of this protein toward this substrate, which was comparable with that using CMP and XMP (Table 3). However, SDT1 showed lower Km for nucleoside monophosphates compared with NMN.
FIGURE 2.

Phosphatase activity of yeast HAD-like hydrolases. A, substrate profiles. Reaction mixtures contained 1 mm phosphorylated substrate and 1 μg of purified HAD. 2dGlu6P, 2-deoxyglucose-6-phosphate; 2dRib5P, 2-deoxyribose-5-phosphate; 2-PGA, 2-phosphoglycerate; 3P-GlyAld, 3-phosphoglyceraldehyde; 3-PGA, 3-phosphoglycerate; 6P-Glu, 6-phosphogluconate; Ery4P, erythrose-4P; Fru1P, fructose 1-phosphate; Fru1,6bisP, fructose-1,6-bisP; Fru6P, fructose 6-phosphate; Glu6P, glucose 6-phosphate; GluA6P, glucosamine 6-phosphate; bGlu1P, β-glucose-1-phosphate; Glu1,6bisP, glucose-1,6-bisphosphate; Gly-1-P, glycerol 1-phosphate; Gly-2P, glycerol 2-phosphate; Man1P, mannose 1-phosphate; Man6P, mannose 6-phosphate; NMN, nicotinamide mononucleotide; PEP, phosphoenolpyruvate; P-Arg, phosphoarginine; P-glycolate, 2-phosphoglycolate; PLP, pyridoxal 5-phosphate; Rib5P, ribose 5-phosphate; Ribu5P, ribulose 5-phosphate; Suc6P, sucrose 6-phosphate. Error bars, S.D. B, hierarchical clustering of 13 yeast HADs using Euclidian distance measurements. The horizontal x axis clusters HADs into groups based on the similarity of their substrate profiles (phosphatase activities from A). Similarities between objects were calculated using Euclidian distance. Activity levels (μmol/min/mg) are color-coded as shown (gray indicates zero activity).
TABLE 3.
Crystallographic data collection and model refinement statistics for the structures of S. cerevisiae HADs: RHR2, UTR4, and YKR070W
| RHR2 | SDT1 | YKR070W | YKR070W·Gly-3-P | |
|---|---|---|---|---|
| Data collection | ||||
| PDB code | 2QLT | 3NUQ | 3KC2 | 3RF6 |
| Space group | P212121 | P21212 | C2 | P212121 |
| Unit cell (Å) | a = 40, b = 56, c = 98 | a = 58, b = 65, c = 68 | a = 144, b = 67, c = 77, β = 110º | a = 60, b = 72, c = 195 |
| Wavelength (Å) | 0.9794 | 1.54 | 0.9794 | 0.9794 |
| Resolution (Å) | 22.9–1.60 | 23.4–1.60 | 40–1.55 | 35.2–1.70 |
| No. of unique reflections | 29,314 | 32,965 | 99,266 | 93,836 |
| Average redundancy | 14.4 | 5.7 | 3.55 | 6.0 |
| Rmergea (%) | 0.094 (0.51) | 0.077 (0.35) | 0.063 (0.74) | 0.066 (0.93) |
| Completeness (%) | 99.9 (100) | 95.5 (65.3) | 99.6 (98.9) | 99.8 (100) |
| I/C | 47.7 (4.43) | 14.9 (2.40) | 24.3 (1.90) | 30 (1.80) |
| Refinement statistics | ||||
| Rcryst (%) | 16.8 | 19.4 | 14.5 | 16.4 |
| Rfree (%) | 19.5 | 22.6 | 16.9 | 19.4 |
| Protein residues/solvent | 250/248 | 280/136 | 679/880 | 691/545 |
| Ligands | 1Ca2+/1Cl − /4Hg2+/4SO42 − /2EDO | 1Na+/2 Cl−/1PEG/ 2EDO | 3 Mg2+/4PO42 − | 3Mg2+/2G3P/ 2FLC |
| r.m.s. deviation from target values | ||||
| Bond lengths (Å) | 0.014 | 0.012 | 0.016 | 0.021 |
| Bond angles (degrees) | 1.55 | 1.40 | 1.51 | 1.53 |
| Average B factors (Å2) | ||||
| Protein whole chains | 19.7 | 17.23 | 10.81 | 33.16 |
| Solvent | 29.0 | 34.30 | 22.40 | 36.13 |
| Ligands | 15.4/23.5/23.5/29.0/ 30.1/35.3 | 13.6/58.3/58.9/ 19.5 | 13.8/23.5 | 27.7/34.1/50.5 |
| Ramachandran plotb (%) | 91.7/8.3/—c | 93.4/6.6/— | 92.7/7.3/— | 92.3/7.7/— |
a Numbers in parentheses are values for the highest resolution bin.
b Ramachandran plot statistics; favored/allowed/outlier.
c —, no outlier.
SER2 is annotated as a putative phosphoserine phosphatase based on sequence similarity to known Ser(P) phosphatases from the HAD superfamily. The S. cerevisiae SER2 shows 31.9% sequence identity to the E. coli SerB, but phosphoserine phosphatase activity of SER2 has not yet been verified experimentally. Our screens confirmed the Ser(P) phosphatase activity in purified SER2, which exhibited high catalytic activity against Ser(P) (Fig. 2, Table 3, and supplemental Table 5). SER2 and SerB had similar Km values for Ser(P), but SER2 was at least 2 times more active than SerB (Table 3) (40). The high catalytic efficiency of SER2 is consistent with its cellular function, because this enzyme catalyzes the last step in the biosynthesis of serine from carbohydrates (79).
We screened the remaining HADs and revealed phosphatase activity in seven proteins. Two enzymes showed nucleotidase activity (PHM8 and YKL033W), two had glycerol phosphatase activity (YNL010W and YKR070W), and enzymes were found with phosphohydrolase activity against 2-phosphoglycolate (P-glycolate; PHO13), thiamine monophosphate (thiamine-P; YCR015C), or pyridoxal 5-phosphate (PLP; YOR131C) (Fig. 2). In addition to glycerol phosphates, YKR070W was also active against phosphorylated metabolites with 4–6 carbon atoms as well as toward fructose 1,6-bisphosphate (Fig. 2). We could not detect phosphatase activity for UTR4 and YMR130W, although both proteins appear to be properly folded (based on their circular dichroism spectra; data not shown). This suggests that these (predicted) enzymes are specific for substrates that are missing in the substrate library used in this work. UTR4 is predicted to be involved in l-methionine biosynthesis, and its phosphatase domain is predicted to catalyze dephosphorylation of 5-(methylthio)-2,3-dioxopentyl phosphate (UniProt P32626). The natural substrate for YMR130W is not known. Other HAD members have β-phosphoglucomutase or dehalogenase activity, but none was detected for YMR130W (data not shown).
Similar to the E. coli HADs (40), phosphatase activities of yeast HADs against natural substrates had slightly acidic or neutral pH optima (pH 6.5–7.5) and were strictly dependent on the addition of a divalent metal cation, with Mg2+, Mn2+, and Co2+ being the preferred metal ions for most enzymes (supplemental Table 6). In contrast to E. coli HADs, Ni2+ also supported phosphatase activity of PHO13, PHM8, SDT1, and SER2, whereas Zn2+ was found to be inefficient for all tested yeast HADs. The yeast HADs required higher concentrations of Mg2+ for maximal activity (Kd = 0.03–1.4 mm, depending on substrate used, mostly 0.2–0.5 mm), whereas Mn2+, Co2+, and Ni2+ were saturating at lower concentrations (Kd = 0.1–80 μm) (supplemental Table 6). Mg2+ was the best metal cofactor for most yeast HADs except for YKR070W, which showed a preference for Co2+. Like E. coli HADs, the yeast HADs usually showed higher Km in the presence of Mg2+ and lower Km with other metal ions (supplemental Table 5). In addition, several yeast HADs (DOG1, DOG2, PHM8, SER2, YKL033W, YKR070W, and YOR131C) exhibited sigmoidal substrate saturation curves, suggesting positive cooperativity in substrate binding with Hill coefficients nH = 1.4–1.8 (supplemental Table 5). This is in line with the presence of dimers in these protein preparations (supplemental Table 5). Thus, both E. coli and yeast HADs exhibit notable promiscuity toward divalent metal cations and positive cooperativity in substrate binding.
YKR070W, a Broad Substrate Range Mitochondrial Phosphatase
YKR070W is annotated as an unknown protein localized in mitochondria and has no homologs in E. coli. YKR070W shares 29% sequence identity with the uncharacterized human HAD-like protein CECR5, which is also localized in mitochondria and is associated with cat eye syndrome, a developmental disorder (UniProt Q9BXW7). Mitochondria are ubiquitous organelles that carry out many crucial processes in eukaryotic cells, including ATP production, gluconeogenesis, pentose phosphate pathway, and NAD metabolism (80). As shown in Fig. 2 and Table 3, purified YKR070W is a broad substrate range phosphatase with high activity against several phosphorylated carbohydrates and glycerol phosphates, which represent various intermediates of the respiratory, gluconeogenetic, and pentose phosphate pathways as well as NAD metabolism. Analysis of the YKR070W kinetic parameters revealed low Km values mostly below 1 mm, suggesting that this enzyme might contribute to the homeostasis of phosphorylated metabolites (Table 3 and supplemental Table 5). The intracellular levels of phosphorylated metabolites have to be tightly regulated because high levels are toxic and cause DNA damage and growth inhibition (81–84). In our previous work on E. coli HAD phosphatases, we proposed that several enzymes (YniC, YfbT, YbiV, YidA, YjjG, YihX, and YigL) might have a detoxification function in E. coli and experimentally demonstrated this role for one protein (YniC) (40). Recently, the phosphosugar detoxification role has been demonstrated for E. coli YigL (85). In S. cerevisiae, the phosphatase activity of YKR070W against of a broad range of phosphorylated metabolites might represent a similar molecular mechanism for fast attenuation of phosphosugar stress by reducing their levels and promoting the efflux of dephosphorylated products.
Another potential in vivo role of YKR070W might be associated with its ability to dephosphorylate Tyr(P)-containing phosphopeptides demonstrated in this work (supplemental Table 4). Using affinity tagging and purification mass spectrometry, we identified a high number of protein-protein interactions of YKR070W with yeast proteins involved in metabolism, protein folding, translation, ribosomal RNA processing, transport, and phosphorylation, including the HAD-like glycerol-3-phosphatase HOR2 (Fig. 3). Presently, 36 protein phosphatase genes are known in S. cerevisiae, including 21 Ser(P)/Thr(P)-specific phosphatases and 14 Tyr(P)-specific phosphatases, none of which is related to the HAD superfamily (86, 87). Mitochondrial localization has been demonstrated for the three yeast PP2C-like (Ser(P)/Thr(P)-specific) protein phosphatases PTC5 (YOR090C), PTC6 (AUP1, YCR079W), and PTC7 (YHR076W) (88) (Saccharomyces Genome Database). Human mitochondria have been shown to contain several non-HAD-related protein Ser(P)/Tyr(P)-specific protein phosphatases, including PP2Cm (PPM1K, PP2C family), PGAM5, and the Tyr(P)-specific protein phosphatase PTPMT1, which play important roles in ATP production, insulin secretion, and cell death regulation (89–91). Thus, YKR070W might also function as a protein phosphatase in S. cerevisiae.
FIGURE 3.

The protein-protein interaction map of YKR070W. The map was determined using an affinity tagging and purification mass spectrometry. Edges represent protein interactions, whereas the color-coded nodes indicate interacting proteins (or preys).
PHM8 and YKL033W-A, Novel Yeast Nucleotidases
Previously, PHM8 (phosphate metabolism protein 8) has been reported to exhibit low nanomolar phosphatase activity against lysophosphatidic acid (71). However, this protein shares significant sequence similarity with the S. cerevisiae nucleotidase SDT1 (41.5% identity), and our screens with purified PHM8 revealed a much higher phosphatase activity against nucleotides, in the order CMP > XMP > GMP = UMP (Fig. 2 and Table 3) (92). With CMP as substrate, the enzyme showed hyperbolic saturation, whereas with XMP, sigmoidal saturation was observed with the Hill coefficient nH = 1.4–1.8, indicating positive cooperativity between the PHM8 subunits in XMP binding. This finding is consistent with the oligomeric state of PHM8 in solution, which is dimeric, as indicated by size exclusion chromatography (observed Mr of 64,000 predicted monomer Mr of 39,000).
Phosphatase screens with purified YKL033W-A identified XMP as the best in vitro substrate for this protein (Fig. 2). YKL033W-A is annotated as a protein of unknown function (Saccharomyces Genome Database) or uncharacterized hydrolase (UniProt Q86ZR7) with low sequence similarity to both SDT1 and PHM8 (20 and 19% sequence identity, respectively). Comparison of the catalytic parameters shows that YKL033W-A is a less efficient XMP phosphatase, due to a low kcat value and high apparent Km (Table 3). Lower catalytic efficiencies of YKL033W-A observed with XMP and other substrates suggest that these could be secondary activities for this protein, and its primary substrate was not included in our screen. The other new yeast nucleotidase PHM8 (YER037W) exhibited a higher level of nucleotidase activity with a substrate profile similar to that of SDT1 (Fig. 2). These two phosphatases have different roles in yeast cells, with SDT1 shown to be responsible for the production of nicotinamide riboside and nicotinic acid riboside as well as for removal of toxic 6- or 5-modified pyrimidines, whereas PHM8 is a nucleotidase involved in autophagy and ribose salvage (45, 92).
Presently, over 100 types of RNA modifications have been characterized, with pseudouridine (Ψ) being the most abundant modified base found both in non-coding RNAs and mRNAs (93, 94). Pseudouridine stabilizes the structure of transfer RNA and ribosomal RNA, enhancing their function (95). In S. cerevisiae and humans, the formation of pseudouridine in mRNA is regulated by environmental signals, suggesting a mechanism for the rapid regulation of protein synthesis and regulated rewiring of the genetic code (93). Because of the abundance of pseudouridine, many organisms have evolved various pseudouridine-metabolizing enzymes (96). Pseudouridine monophosphate (Ψ-UMP or pseudouridine 5′-phosphate) produced in the course of RNA degradation can be phosphorylated by promiscuous kinases to Ψ-UTP, which needs to be removed from the cellular nucleotide pool to prevent its uncontrolled incorporation into new RNAs (97, 98). Recently, we have identified the presence of Ψ-UTP pyrophosphatase activity in several Maf proteins, including yeast YOR111W and human ASMTL-Maf, which produced Ψ-UMP and pyrophosphate as reaction products (50). In the present work, we found that the addition of purified YKL033W-A, PHM8, or SDT1 to a reaction mixture with a Maf protein (yeast YOR111W or BSU28050 from B. subtilis) and Ψ-UTP resulted in dephosphorylation of the produced Ψ-UMP and formation of pseudouridine (Ψ) (Fig. 4). These results suggest that the yeast nucleotidases YKL033W-A, PHM8, and SDT1 possess pseudouridine 5′-phosphatase activity and together with YOR111W might constitute a pathway for the detoxification of Ψ-UTP and Ψ-UMP in S. cerevisiae. A similar pathway comprising the human proteins ASMTL-Maf and pseudouridine 5′-phosphatase HDHD1 can also be proposed for humans (50, 99). HDHD1 also belongs to the HAD superfamily and shows low sequence similarity to YKL033W-A (35% sequence identity), PHM8 (19% sequence identity), and SDT1 (17% sequence identity).
FIGURE 4.
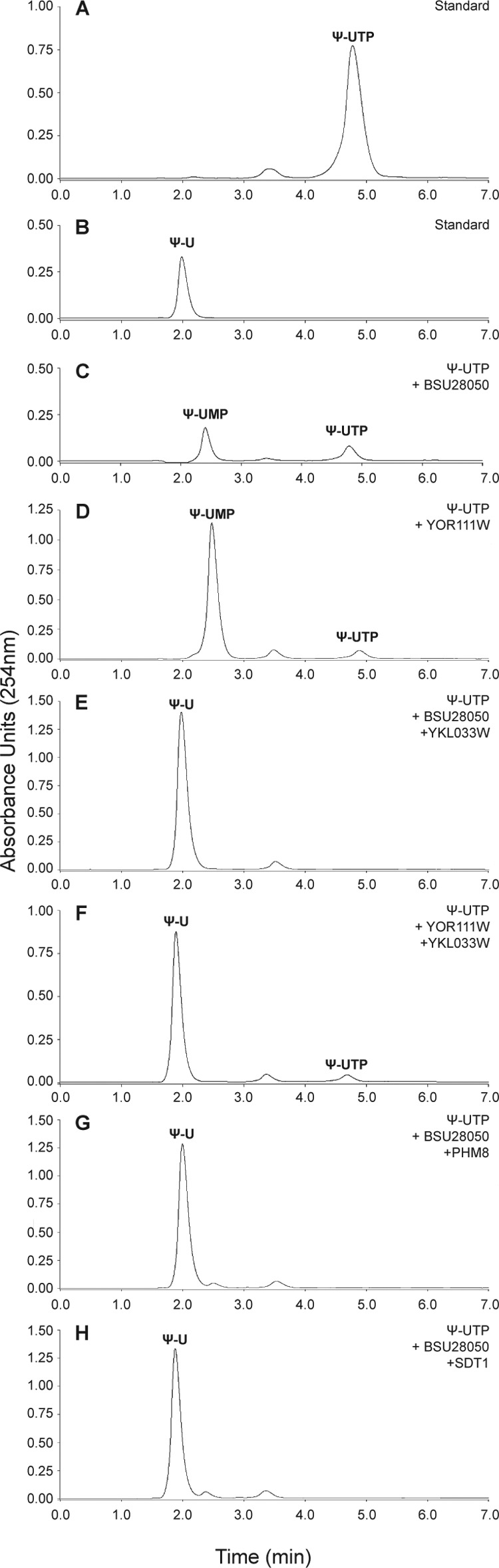
Phosphohydrolase activity of purified yeast HADs against pseudouridine monophosphate. HPLC profiles show the dephosphorylation of Ψ-UMP, a reaction product of hydrolysis of Ψ-UTP by the Maf proteins BSU28050 (C) and YOR111W (D), by purified yeast HADs: YKL033W (E and F), PHM8 (G), and SDT1 (H). A and B are nucleotide standards.
PHO13, a Phosphoglycolate Phosphatase
PHO13 is annotated as 4-nitrophenyl phosphatase (UniProt P19881) or as an alkaline phosphatase active against pNPP and phosphorylated histone II-a and casein (YDL236W, Saccharomyces Genome Database), with the latter annotation based on experiments with a partially purified protein (100). However, our results demonstrated that the recombinantly expressed and purified PHO13 has a neutral pH optimum for pNPP hydrolysis and exhibits no dephosphorylation activity against phosphopeptides. Furthermore, our screens revealed that purified PHO13 is highly active against P-glycolate, with micromolar activity toward several secondary substrates, including Gly-2-P, imidodiphosphate, Gly-1-P, Gly-3-P, 3-phosphoglycerate, and phosphoenolpyruvate (PEP) (Fig. 2). In all organisms, 2-phosphoglycolate is produced during the repair of DNA apurinic/apyrimidinic (AP) sites as well as during photorespiration in plants. The AP sites are the most frequent DNA lesions that can be formed spontaneously or as a consequence of the removal of damaged bases during DNA repair (101). The AP sites are primarily repaired via the base excision repair and nucleotide excision repair pathways (102). These pathways produce various intermediates with blocked 3′-ends, including those with 3′-phosphoglycolate termini, which are removed through a 3′-phosphodiesterase activity of AP endonucleases, producing 2-phosphoglycolate as one of the products (101, 102). In E. coli, there are two major AP endonucleases (exonuclease III and endonuclease IV), whereas the HAD-like P-glycolate phosphatase Gph has been identified as a housekeeping enzyme responsible for the hydrolysis of produced 2-phosphoglycolate (103). In S. cerevisiae, the major endonuclease responsible for the repair of AP sites is APN1 (YKL114C), which has several catalytic activities, including 3′-phosphodiesterase activity, which excise the 3′-phosphoglycolate group (101). Our results show that PHO13 exhibits high catalytic efficiency toward 2-phosphoglycolate in vitro, suggesting that in S. cerevisiae, this enzyme might be involved in the hydrolysis of 2-phosphoglycolate produced by the major AP endonuclease APN1 in the base excision repair pathway.
It has been reported that deletion or inactivation of PHO13 improved growth and production of ethanol by S. cerevisiae from d-xylose (104–106). Based on a higher phosphatase activity of crude extracts from S. cerevisiae cells overexpressing PHO13 against xylulose 5-phosphate, it has been proposed that PHO13 dephosphorylates xylulose 5-phosphate, creating a futile cycle with xylulokinase (106). We could not test this hypothesis because xylulose 5-phosphate is not commercially available. Another possible explanation for the increased ethanol production by the PHO13 deletion strains is based on the fact that 2-phosphoglycolate is a competitive inhibitor of Gly-3-P dehydrogenase, an enzyme involved in the production of glycerol in S. cerevisiae (GPD1 and GPD2) and other organisms (107). Deletion of PHO13 might increase the intracellular level of 2-phosphoglycolate in S. cerevisiae cells, thereby inhibiting the activity of GPD1 and GPD2 and increasing carbon flow in the ethanol production branch.
YCR015C, a Thiamine Monophosphate Phosphatase
YCR015C is annotated as a protein of unknown function (UPF0655; UniProt: P25616), and our screens with purified YCR015C revealed the presence of high phosphatase activity against thiamine monophosphate (TP) (Fig. 2 and Table 3). Like most prokaryotes and plants, S. cerevisiae can synthesize thiamine (vitamin B1) and its active form TPP de novo (108, 109). The two separate branches of the TPP biosynthetic pathway generate the thiazole and pyrimidine moieties, which are then joined to produce TP. In S. cerevisiae and plants, TP is dephosphorylated by an unknown phosphatase producing thiamine, which is then pyrophosphorylated by the thiamine pyrophosphokinase THI80 (YOR143C) to form TPP (108). The dephosphorylation of TP has been proposed to be catalyzed by nonspecific phosphatase(s) located in the cytosol (108). Given that purified YCR015C showed low Km for TP in vitro (19 μm) (Table 3), it might represent the missing phosphatase involved in the final stage of TPP synthesis. However, our HPLC analyses revealed no significant increase in the TP level in extracts of the YCR015C deletion strain (supplemental Fig. 2), suggesting that there might be another phosphatase(s) in S. cerevisiae that dephosphorylates TP. Alternatively, YCR015C might dephosphorylate “damaged” forms of TP, as has been recently demonstrated for the S. cerevisiae Nudix hydrolase YJR142W, which is up to 60-fold more active against the toxic TPP degradation products oxy- and oxo-TPP compared with TPP (51). Further studies using “damaged” TP forms are required to test this hypothesis.
YOR131C, a Pyridoxal Phosphate Phosphatase
YOR131C is annotated as a putative uncharacterized hydrolase (UniProt Q12486). Our enzymatic screens with purified YOR131C revealed the presence of phosphatase activity against pyridoxal 5-phosphate (PLP), which is the active form of vitamin B6, and Tyr(P)-containing peptides (Fig. 2). The biochemically characterized human PLP phosphatase PDXP (also known as chronophin, CIN) shows less than 20% sequence identity with YOR131C, in contrast to 30% identity with the yeast phosphoglycolate phosphatase PHO13. This discrepancy once again demonstrates that at low sequence similarity (roughly less than 30% identity with HADs), homology-based prediction of enzyme substrate specificity (as opposed to the general type of the catalyzed reaction) often results in erroneous functional annotations. PLP is the active form of vitamin B6, which is an essential cofactor in all organisms involved in a broad variety of enzymatic reactions (110). The intracellular level of PLP is mainly controlled by its synthesis, binding to enzymes, and degradation by phosphatases (111, 112). Given that purified YOR131C showed high catalytic activity and low Km to PLP (Table 3), we propose that this protein is the missing PLP phosphatase that is involved in PLP catabolism in S. cerevisiae (EC 3.1.3.74).
The human PLP phosphatase PDXP (CIN) has been shown to also be active as a Ser(P)-specific protein phosphatase that directly dephosphorylates cofilin, a key regulator of actin polymerization that is present in all eukaryotes (38, 113, 114). The activity of human cofilin is regulated by phosphorylation (inactivation) at the conserved Ser-3 by specific kinases and dephosphorylation (activation) by the unrelated phosphatases CIN and SSH (38, 115, 116). In our assays, the S. cerevisiae PLP phosphatase YOR131C was active against phosphorylated peptides containing Tyr(P) or Ser(P), suggesting that it might also function as a protein phosphatase (supplemental Table 4). However, systematic mutational analysis of the S. cerevisiae cofilin indicated that in contrast to vertebrates, yeast cofilin appears not to be phosphorylated at the conserved N-terminal Ser residue (Ser-4), whereas the YOR131C deletion strain was found to be viable (117, 118). Thus, if YOR131C also functions as a protein phosphatase in S. cerevisiae, it is likely that this enzyme targets different (not cofilin) proteins.
YNL010W, a Novel Yeast Glycerol Phosphate Phosphatase
Several yeast HAD-like hydrolases were found to be capable of dephosphorylating glycerol phosphates, including both the known (RHR2/GPP1 and HOR2/GPP2) and newly characterized (YNL010W, PHO13, and YKR070W) phosphatases (Fig. 2). The two known yeast glycerol phosphate phosphatases (RHR2 and HOR2) dephosphorylated Gly-3-P as the preferred substrate but were also able to hydrolyze Gly-1-P, albeit with lower activities. Gly-1-P and Gly-3-P are enantiomeric isomers, of which Gly-1-P is typically found in archaea, whereas Gly-3-P is present in bacteria and eukaryotes (119). However, a recent study has demonstrated the presence of Gly-1-P dehydrogenase in B. subtilis (120), indicating that Gly-1-P is not exclusive to archaea. Our screens have identified a novel glycerol phosphate phosphatase in S. cerevisiae, YNL010W, which showed a preference for Gly-1-P in vitro but was also active against Gly-3-P (Fig. 2). Glycerol is the main compatible solute in S. cerevisiae, which is important for osmoregulation, stress response, carbon metabolism, redox balance, and lipid synthesis (121). HOR2 and RHR2 are required for glycerol synthesis and are involved in responses to osmotic, anaerobic, and oxidative stress (122). Our results suggest that YNL010W might contribute to these processes and may also have additional functions in S. cerevisiae. It has been shown that deletion of both gene copies of YNL010W leads to an increase in glycogen accumulation (123). This effect might be due to the ability of YNL010W to dephosphorylate erythrose-4-P (Fig. 2) or Ser(P)/Tyr(P)-containing peptides (supplemental Table 4).
Structural Analysis of Yeast HAD Phosphatases
The high resolution crystal structures of the three yeast HAD phosphatases RHR2 (GPP1, 1.60 Å resolution), SDT1 (1.60 Å), and YKR070W (1.55 Å) were determined by the SAD method (Table 2). In addition, the crystal structure of the yeast UTR4 has been solved by the Joint Center for Structural Genomics (PDB code 2G80, resolution 2.28 Å). The crystal structures of four yeast HADs revealed the general topology of the HAD hydrolase fold, which forms a three-layer αβα sandwich with the α,β core Rossmannoid domain containing a six-stranded parallel β-sheet flanked by five or more α-helices on both sides (Fig. 5). The crystal structures also revealed the presence of a cap domain, which can be classified as type C1 in RHR2, UTR4, and SDT1 (inserted between HAD motifs 1 and 2) or type C2 in YKR070W (inserted between HAD motifs 2 and 3) (Fig. 5). The cap domains of the first three proteins represent a five- or six-helix bundle, whereas YKR070W has an α,β three-layer sandwich cap domain of similar size to that of the core domain (Fig. 5). Previous analysis of the available structures of HAD-like hydrolases indicated that ∼60% of them are likely to form dimers or higher oligomers, whereas the other proteins appeared to be monomeric (124). In the human PLP/protein phosphatase chronophin, homodimerization is essential for the proper positioning of a conserved His residue in the substrate specificity loop (124). The structures of four yeast HADs suggested a monomeric state for RHR2, SDT1, and UTR4 and a dimeric state for YKR070W with the cap domain of one protomer extending/reaching close to the core domain of the second protomer (Fig. 5).
FIGURE 5.
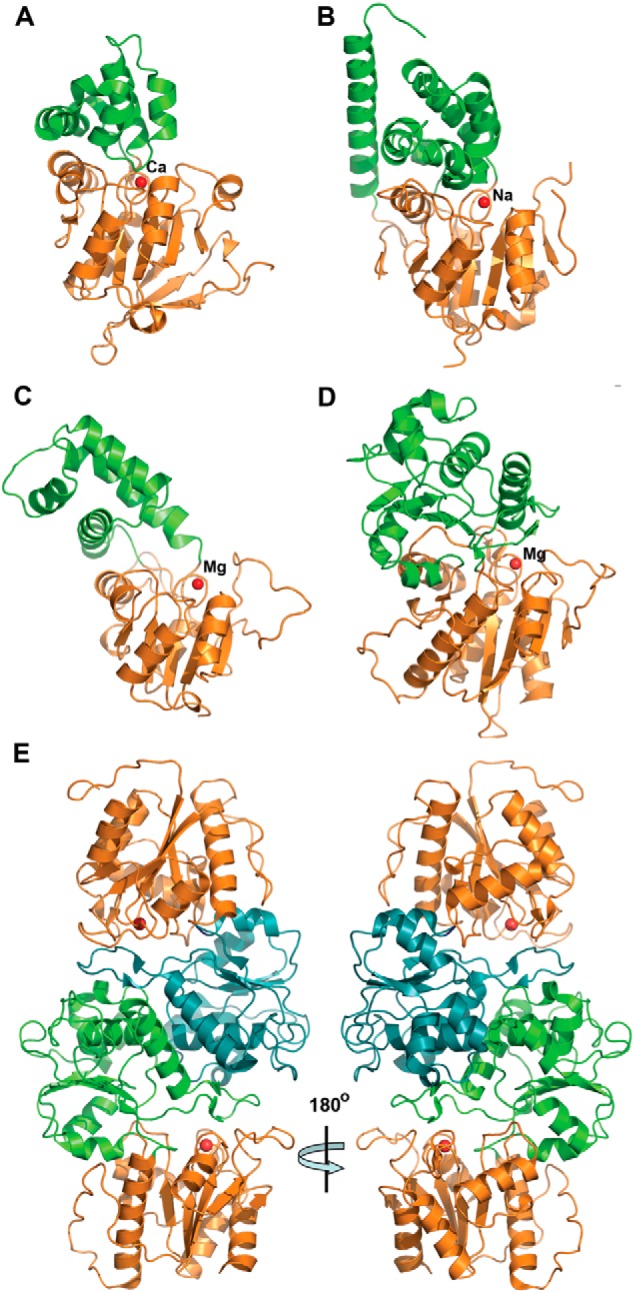
Crystal structures of four S. cerevisiae HADs. Overall structures of the protomers: RHR2 (A), SDT1 (B), UTR4 (C), and YKR070W (D). All enzymes show a two-domain organization with the Rossmann-like protein core (orange ribbons) and mostly α-helical cap domain (green ribbons). In all structures, the position of potential active site is indicated by the bound metal ion (shown as a red sphere and labeled). E, two protomers of YKR070W form a tight dimer through the interaction of their cap domains (colored green and cyan, two views related by a 180° rotation).
Despite low sequence similarity between the four yeast HADs (up to 21% sequence identity), the superposition of their core Rossmann-like domains revealed high conservation of the overall fold (average r.m.s. deviation of ∼2.4 Å). The conservation of the structural elements that form the active site loops, the central β-sheet region, and the “squiggle” (formed by a 6-residue helical turn, which is located after the first strand of the central β-sheet) was even higher, with an average r.m.s. deviation of 0.9 Å signifying the importance of these elements for catalytic activity of HAD phosphatases (Fig. 6A). In contrast, the cap domains of the four yeast HADs showed high structural diversity, which is consistent with different substrate specificities of these enzymes (Fig. 2).
FIGURE 6.
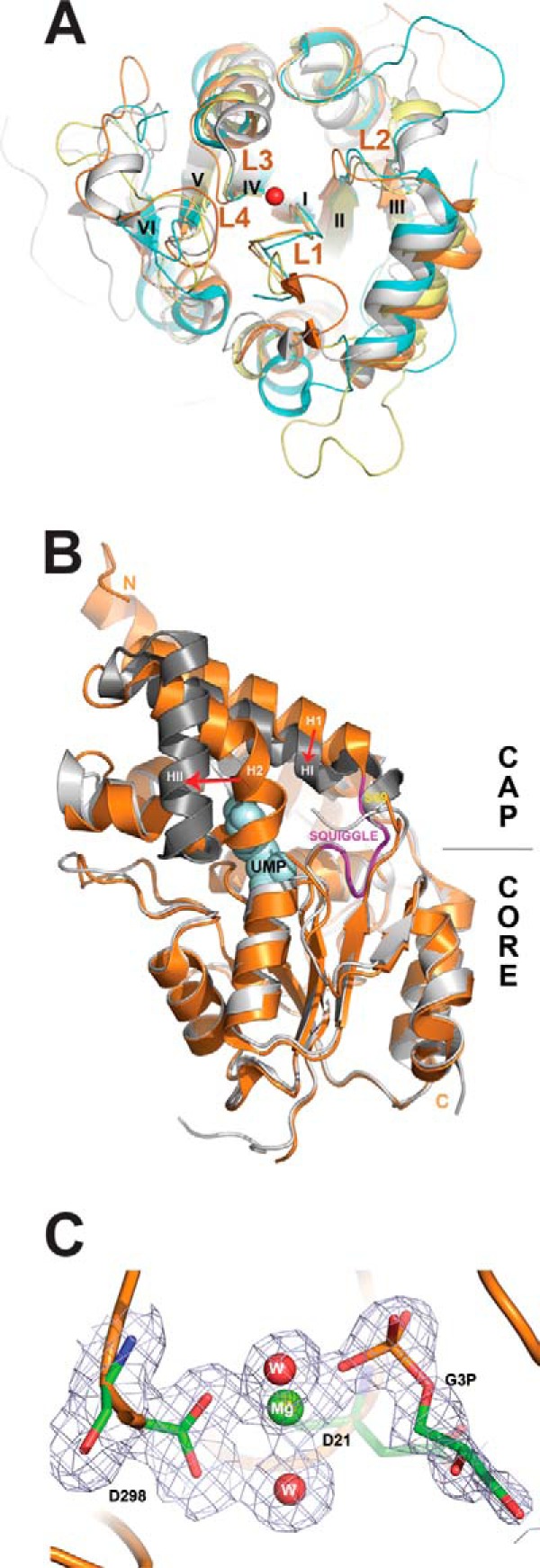
Structural analysis of S. cerevisiae HADs. A, structural superposition of core domains of the four yeast HADs: RHR2 (gray), SDT1 (cyan), UTR4 (magenta), and YKR070W (orange). The overlay shows close alignment of the structural elements involved in catalysis, including the active site loops (L1–L4) and bound metal ions (the Mg2+ ion from the YKR070W structure is shown as a red sphere). The conserved structural elements squiggle and flap are positioned close to loop L1 (labeled). For clarity, the cap domains of these HADs are not shown, whereas the β-strands are numbered with Roman numerals. B, structural superposition of the apo-form and UMP binary complex of SDT1. We have determined the 1.70 Å structure of the apo-form of SDT1 (PDB 3NUQ; orange ribbon), whereas the structure of the SDT1-substrate complex with UMP is available from PDB (3OPX; shown as a gray ribbon with UMP shown as a space-filled model). Superimposition of these two structures revealed a similar overall structural fold (r.m.s. deviation 1.43 Å) with significant structural changes in the cap domain (indicated by red arrows). C, close-up view of the active site of YKR070W with bound Gly-3-P with 2Fo − Fc electron density map (gray) contoured at 1.0 σ. The bound Gly-3-P and residues are shown as sticks, the Mg2+ ion as a green sphere, and two water molecules as red spheres. The molecule of Gly-3-P was omitted during the map calculation.
In HAD C1 proteins, the cap domains are inserted in the “flap,” allowing large conformational changes, including opening and closing of the active site (25). The closed active site is represented by the RHR2 structure showing the cap domain positioned close to the core domain, whereas the UTR4 structure revealed the active site in an open conformation (Fig. 5). In addition, the comparison of the SDT1 structure with the recently determined structure of this protein in complex with substrate (UMP) (125) displayed significant structural changes in its cap domain upon substrate binding (Fig. 6B). The major change is associated with the cap domain helices H1 and H2 with the initiation point located near Ser-69 (labeled) at the border of the squiggle region. Ser-69 is the last residue of the 6-residue squiggle structure, which undergoes conformational change of 3.5 Å during the open-closed state transitions. This triggers a ∼3-Å movement of helix H1 (orange) to the new position HI (dark gray) and a 30° swing of the helix H2, moving its end 13 Å away (to the HII position). In contrast, the cap domain of the HAD C2 proteins, including YKR070W, is inserted into a rigid part of the core domain (between HAD motifs II and III). Accordingly, the structures of YKR070W, in complex with Gly-3-P (a substrate) or phosphate (a product), revealed limited conformational changes (Fig. 5).
A Dali search for four yeast HADs identified many similar structures of characterized and uncharacterized HAD-like hydrolases from different organisms that show low sequence similarity to yeast HADs (13–25% sequence identity). For RHR2, the top two characterized structural neighbors include the inorganic pyrophosphatase BT2127 from B. thetaiotaomicron (Z-score 20.7–21.3, r.m.s. deviation 3.0 Å, PDB codes 3QUB and 3QU2) and the 2-deoxyglucose 6-phosphatase YniC from E. coli (Z-score 20.9, r.m.s. deviation 3.0 Å, 18% sequence identity, PDB code 1TE2); for UTR4, the haloacid dehalogenase L-DEX from Pseudomonas sp. YL (Z-score 17.2, r.m.s. deviation 3.7, 17% sequence identity, PDB code 1JUD) and the inorganic pyrophosphatase BT2127 from B. thetaiotaomicron (Z-score 14.8, r.m.s. deviation 3.7 Å, 13% sequence identity, PDB code 3QU2); for SDT1, the haloacid dehalogenase L-DEX from Pseudomonas sp. YL (Z-score 17.2, r.m.s. deviation 3.4 Å, 15% sequence identity, PDB code 1JUD) and the human N-acylneuraminate-9-phosphatase NANP (Z-score 16.4, r.m.s. deviation 3.1 Å, 20% sequence identity, PDB code 2W4M); for YKR070W, human and murine PLP/protein phosphatase chronophin (Z-score 25.6, r.m.s. deviation 2.5 Å, 19% sequence identity, PDB code 2P69; Z-score 25.4, r.m.s. deviation 2.3 Å, 18% identity, PDB code 4BX2). Thus, the results of the Dali search indicate that the four yeast HADs exhibit overall structural similarity to other HAD-like phosphatases, which is not related to their substrate specificity.
The location of the HAD active sites is indicated by the position of a metal ion (Mg2+, Ca2+, or Na+) which is bound at the bottom of the active site close to the side chains of conserved catalytic residues of the signature HAD motif (Fig. 7). The active site volumes calculated using CASTp (126) were found to be 460 Å3 for RHR2, 966–1003 Å3 for YKR070W, 1737 Å3 for UTR4, and 1698–2096 Å3 for SDT1. Thus, it appears that the HAD active site volumes show no correlation with their substrate promiscuity, which is the highest in YKR070W. The active sites of the four yeast HADs include four loops that support the catalytic residues. Loop 1 accommodates the Asp nucleophile, followed 2 residues downstream by the second Asp, which functions as a general acid-base. Loops 2 and 3 support the phosphoryl group binding residues Thr (or Ser) and Lys, whereas the two Mg2+-binding carboxylates are located on loop 4 (Fig. 7). The RHR2 active site also revealed the presence of a bound sulfate ion, whereas a phosphate was found in the YKR070W structure, suggesting that these groups mimic the position of the phosphate product (Fig. 7). In YKR070W, the Mg2+ ion adopts an octahedral coordination geometry, being coordinated by the backbone carbonyl oxygen of Asp-21, the side chain carboxylates of Asp-19 and Asp-298, the phosphate group oxygen atom, and two water molecules (all bond lengths are less than 2.12 Å). The phosphate ion is coordinated by direct hydrogen bond interactions with the side chain atoms of Asp-19 (OD1-PO3, 2.6 Å), Asp-21 (OD2-PO4, 2.6 Å), Thr-52 (O-PO3, 2.7 Å), Lys246 (NZ-PO1, 2.9 Å), and the main chain amide groups of Asn-53 (N-PO4, 2.7 Å), and Asp-21 (N-PO3, 2.8 Å) (Fig. 7).
FIGURE 7.

Close-up view of the active sites of four yeast HADs. A, RHR2; B, UTR4; C, SDT1; D, YKR070W. The active site residues are shown as sticks and labeled, whereas the metal ions are shown as magenta spheres. The active sites of RHR2 (A) and YKR070W (D) show the presence of bound sulfate or phosphate ions, respectively (shown as sticks).
The role of the HAD motifs and other conserved residues of the core and cap domains of YKR070W were analyzed using site-directed mutagenesis (Fig. 8 and supplemental Table 7). Alanine replacement mutagenesis of the HAD motif residues in YKR070W produced inactive or almost inactive proteins (D19A, D21A, T52A, K246A, D298A, and D303A). In addition, a greatly reduced phosphatase activity was found in the YKR070W mutant proteins with mutations in Asn-53 and Asp-206, which are located in the core and cap domains, respectively, indicating that residues from both domains are important for the YKR070W activity (Fig. 8 and supplemental Table 7). Mutations in the other residues of the YKR070W core and cap domains generated proteins with reduced or wild type activity (Lys-28, Arg-62, Phe-132, Asp-163, Asn-204, and Trp-209). Conservative amino acid replacements (e.g. F132Y, W209Y, or W209F) typically had less significant effects on the YKR070W activity and kinetic parameters compared with non-conservative mutations (e.g. K28E, R62E, or W209A) (Fig. 8 and supplemental Table 7).
FIGURE 8.
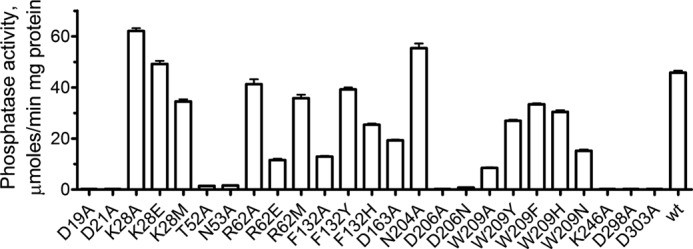
Site-directed mutagenesis of YKR070W; phosphatase activity of the purified YKR070W mutant proteins. The assays contained 1 mm Gly-3-P as substrate and 5 mm Mg2+. Results are means ± S.D. (error bars) from at least two independent determinations.
The Structure of the YKR070W Dimer in Complex with Gly-3-P Reveals a Composite Substrate Binding Site
To provide insight into the molecular mechanisms of substrate selectivity of yeast HADs, we crystallized the inactive YKR070W D19A protein in the presence of Gly-3-P. This crystal structure revealed the presence of two molecules of Gly-3-P and two Mg2+ ions per dimer bound to the active site, representing a structure of an enzyme-substrate (Michaelis) complex (Fig. 9). The positions of the Gly-3-P phosphate group and Mg2+ are very similar to those in the YKR070W structure in complex with phosphate (r.m.s. deviation 0.6 and 0.3 Å for the phosphate and Mg2+ ions, respectively) (Fig. 7). The phosphate moiety of Gly-3-P is positioned near the Mg2+ ion (2.1 Å) at the bottom of the active site pocket and is also coordinated by the side chains of Thr-52 (2.3 Å) and Lys-246 (2.7 Å) as well as by the main chain amide group of Asn-53 (2.5 Å) (Figs. 6C and 9). The glycerol moiety of Gly-3-P is bound mostly within the cap domain in a large cavity formed by the side chains of Thr-101, Asn-162, Asp-163, His-165, Asn-204, and Trp-209 (Fig. 9). The C2-OH group of Gly-3-P forms hydrogen bonds with the side chain ND2 nitrogen of Asn-204 (3.2 Å) and OD2 oxygen of Asp-206 (2.8 Å), whereas the C1-OH is 4 Å away from the side chains of Asp-163 and Trp-209 and interacts with a water molecule coordinated by Asp-163 (OD2), Asn-204 (ND2), and the main chain amide of Gln-220. Therefore, the substrate-binding site of the YKR070W pocket can potentially accommodate longer substrates, in line with the high activity of this phosphatase against phosphorylated C4, C5, and C6 carbohydrates (Fig. 2).
FIGURE 9.

Close-up view of the active site of the YKR070W dimer with the bound Gly-3-P (A and B, two views related by a 180° rotation). The core and cap domains of one protomer are colored in orange and light green, respectively, with amino acid side chains shown as sticks along a protein ribbon, whereas the ribbon and Phe-132 of the second protomer are colored in gray. The side chain of Phe-132 of one protomer contributes to the substrate binding site of another protomer.
The structure of the YKR070W dimer revealed that the side chain of Phe-132 from one protomer contributes to the substrate binding site of another protomer and is positioned near the side chains of Asp-163, His-165, and Trp-209 (3.3–3.7 Å) and just 5.2 Å away from the C1 hydroxyl group of bound Gly-3-P (Fig. 9). This suggests that binding of the substrate to one active site of the YKR070W dimer can be transmitted to the second active site through the Phe-132 side chain, creating an opportunity for allosteric interaction of the two active sites. Therefore, the structure of YKR070W proposes a potential molecular mechanism for positive substrate cooperativity in dimeric and tetrameric HAD-like hydrolases based on the conformational change induced by substrate binding to one subunit and transmitted (through Phe-132 in YKR070W) to another subunit. This is consistent with the observed positive cooperativity of substrate binding by purified YKR070W with 2-deoxyribose-5-phosphate as substrate (kH = 1.5). Substrate saturation experiments with 2-deoxyribose-5-phosphate and purified YKR070W mutant proteins revealed a significant reduction both in cooperativity (kH = 1.1–1.2) and substrate binding (Km = 0.4–0.5 mm) in the F132A, F132Y, and F132H proteins (kH = 1.1–1.2) compared with the wild type YKR070W (kH = 1.4 and Km = 0.3 mm). Although a recent analysis of the available structures of HAD dimers has led to the suggestion that the adjacent HAD protomers do not share active site residues (124), all of these structures show the presence of a conserved aromatic residue homologous to the YKR070W Phe-132 positioned close to the substrate binding site of another protomer. These HAD proteins include the mouse chronophin (Phe-152, PDB code 4BX3) and HDHD2 (Tyr-124, PDB code 2HO4), human LHPP (Tyr-132, PDB code 2X4D), PH1952 from Pyrococcus horikoshii (Tyr-132, PDB code 1ZJJ), and AF0374 from Archaeoglobus fulgidus (Phe-128, PDB code 3QGM). Thus, in dimeric HADs, each of the protomers can contribute an aromatic residue to the substrate binding site of the other protomer, providing a potential molecular mechanism of cooperativity in dimeric HAD-like hydrolases.
Discussion
Biochemical characterization of previously unexplored HAD superfamily hydrolases from S. cerevisiae revealed the presence of phosphatase activity against the general phosphatase substrate pNPP in nine proteins and activity against specific phosphorylated substrates for eight proteins (Table 1 and Fig. 2). Thus, the in vitro or in vivo substrates have been experimentally identified or can be reliably predicted for 25 of the 26 soluble S. cerevisiae HADs, with the exception of YMR130W, for which we demonstrated only low phosphatase activity against pNPP. Most of the soluble S. cerevisiae HADs are phosphomonoesterases active against a broad range of C2–C12 metabolites, including phosphorylated carbohydrates, nucleotides, organic acids, glycerols, and cofactors (Fig. 10) and supplemental Table 8). The identified in vitro substrates of previously uncharacterized S. cerevisiae HAD phosphatases suggest that these enzymes have a role in cell metabolism (SER2, YCR015C, and YNL010W), regulation (PHM8, SDT1, and YKL033W), or housekeeping (PHO13 and YOR131C).
FIGURE 10.

Substrate diversity of S. cerevisiae and E. coli HADs; the molecular structures of positive HAD substrates. Light yellow background, positive substrates common for both organisms; light blue background, substrates shown to be positive only for S. cerevisiae HADs; light pink background, substrates shown to be positive only for E. coli HADs.
The S. cerevisiae HAD family includes both highly specific (e.g. SER2 and YCR015C) and promiscuous phosphatases (e.g. YKR070W and DOG2), which show high activity against multiple substrates (Fig. 2). Previously, pronounced substrate promiscuity has been demonstrated for HAD phosphatases from E. coli and other organisms (28, 32, 40). This broad substrate specificity apparently contributes to the key role of these enzymes in the hydrolysis of a broad range of phosphomonoesters as well as the “house-cleaning” functions. Enzyme substrate promiscuity (also known as substrate ambiguity) is likely to be the starting point for the evolution of new enzymes through gene duplication followed by subfunctionalization (substrate specialization) of the diverging enzymes (28, 127). Beneficial promiscuous activities in the promiscuous repertoire can be selected and improved through mutations, initially without losing the primary enzyme activity (127). In the evolutionary biological context, this mode of evolution represents a special case of subfunctionalization (128, 129), whereby the diverging paralogs not only retain but enhance some of the multiple activities of the common ancestor. The S. cerevisiae complement of HAD phosphatases provides at least three examples of gene duplication that are compatible with this general model of enzyme evolution but reflect different stages of subfunctionalization. The glycerol phosphatases HOR2 and RHR2 share 95% sequence identity and have almost identical substrate profiles, suggesting a recent gene duplication without much sequence or functional divergence (Fig. 2). In contrast, despite nearly as high sequence conservation, the 2-deoxyglucose-6-phosphatases DOG1 and DOG2 (92% sequence identity) have already diverged, with DOG2 having evolved a preference toward fructose 1-phosphate but still retaining high activities toward 2-deoxyglucose-6-phosphate and mannose 6-phosphate (Fig. 2 and Table 3). Finally, the nucleotidases PHM8 and SDT1 appear to represent older duplication and greater divergence because they show lower sequence similarity to each other (41.5% sequence identity) and prefer different (although similar) substrates, CMP and UMP, respectively (Fig. 2). The third nucleotidase, YKL033W, shares only 20% sequence identity with PHM8/SDT1 and has a different substrate profile, suggesting that it evolved from an even older gene duplication event or developed a preference for nucleotide substrates independently (through convergent evolution).
The only other organism with thoroughly characterized HADs is E. coli, which encodes a comparable number of soluble HADs (23 proteins) but only five membrane-bound HADs (compared with 19 in S. cerevisiae) (40). Although the orthologous HADs from yeast and E. coli share low overall sequence similarities (30% sequence identity at most), a comparison of the top preferred substrates for these enzymes identified seven common metabolites (Ser(P), P-glycolate, PLP, 2-deoxyglucose-6-phosphatase, fructose 1-phosphate, UMP, and trehalose-6-P). Thus, in the evolution of the HAD superfamily phosphatases, conservation of substrate specificity of orthologous enzymes does not require a particularly high level of sequence conservation. Furthermore, analysis of substrates that support at least moderate activity of HADs from both organisms (>0.2 μmol/min/mg of protein) revealed comparable numbers of substrates (40 in yeast and 44 in E. coli) with 28 common metabolites (70%), which include phosphorylated carbohydrates, nucleotides, organic acids, FMN, and PLP (Fig. 10 and supplemental Table 8). We propose that these metabolites represent the primary pool of potential substrates of the HAD superfamily that are likely to be conserved in most free living organisms with comparable numbers of HADs. This pool of secondary activities is likely to represent the reservoir for evolution of novel phosphatases.
In this work, we also identified several substrates that have not been reported for E. coli HADs and might represent recent additions to the yeast HAD substrate pool, including 3-phosphoglycerate, thiamine-P, and phosphopeptides (Fig. 10 and supplemental Table 8). Four yeast HADs (YOR131C, YNL010W, YKR070W, and SDT1) were found to be active against phosphorylated (Tyr(P) and Ser(P)) peptides, adding novel potential protein phosphatases to the previously identified Ser(P)/Thr(P) phosphatases NEM1 and FCP1. HAD-like hydrolases with protein phosphatase activity have previously been identified in several eukaryotes (37–39, 73) but not in E. coli, suggesting that this activity might be a recent addition to the substrate repertoire in the evolution of the HAD superfamily. In contrast, E. coli encodes two HADs that are involved in a bacteria-specific pathway of cell wall lipopolysaccharide biosynthesis (YrbI and GmhB) (130, 131), which are absent in S. cerevisiae. Overall, E. coli has more HAD superfamily enzymes that are active against phosphorylated carbohydrates (seven compared with three in yeast), whereas S. cerevisiae has more HADs with a preference toward P-glycerols (three compared with none in E. coli).
Using the information from the COG and OrthoMCL databases combined with the results of the previous sequence analysis of the HAD superfamily (25, 132, 133), we established several co-orthologous groups for yeast, E. coli, and human HADs and compared their substrate specificity (Fig. 1 and Table 4). As shown in Fig. 1, yeast and E. coli HADs belong to six co-orthologous groups. The two pairs of recently evolved paralogous yeast HADs, DOG1/DOG2 and HOR2/RHR2 (92 and 95% sequence identity, respectively), which belong to the same co-orthologous group (Fig. 1), share similar substrate profiles, although, as pointed out above, the preferred substrates differ for DOG1 and DOG2 (Fig. 2). However, the E. coli HAD phosphatase YfbT, to which all of these yeast enzymes appear to be co-orthologous, has a different substrate profile with a preference for fructose 1-phosphate (40). The BPGM group of phosphatases from both organisms are active against 2-deoxyglucose-6-phosphatase, fructose 1-phosphate, and Gly-3-P, whereas the Epo group enzymes are mostly nucleotidases (Table 4). Thus, substrate preferences of these HAD-like phosphatases correlate with their sequence similarity at the family level. Similarly, the PSP group HADs from S. cerevisiae (SER2), human (PSPH), and E. coli (SerB) were found to be Ser(P) phosphatases, suggesting that this activity is highly conserved in many organisms. In contrast, the S. cerevisiae P-glycolate phosphatase PHO13 (YDL236W) from the NagD family has several human orthologs active against P-glycolate (PGP) and PLP (PDXP), whereas the E. coli ortholog NagD is a UMP phosphatase, and the more diverged human paralog LHPP is a P-lysine phosphatase (Table 4). This distribution of activities implies that the P-glycolate phosphatase activity probably emerged early in the evolution of eukaryotes, but its paralogs can quickly change their substrate specificity. Moreover, the three E. coli PLP phosphatases YbhA, YigL, and Cof belong to the Cof family, which also includes the yeast phosphomannomutase SEC53 (YFL045C) (not shown).
TABLE 4.
Co-orthologous groups and substrates of HADs from S. cerevisiae, E. coli, and humans
The preferred HAD substrates are indicated in parentheses. The 15 characterized yeast HADs and their COG groups are shown together with the orthologous HADs from E. coli and humans. 2d-glucose-6P, 2-deoxyglucose 6-phosphate phosphatase 1.
| Family | S. cerevisiae | E. coli | Human |
|---|---|---|---|
| PSP | SER2 (Ser(P)) | SerB (Ser(P)) | PSPH (Ser(P)) |
| YNL010W (Gly-1-P) | No | No | |
| YCR015C (thiamine-P) | No | No | |
| Epo | PHM8 (CMP), SDT1 (UMP) | YihX (Glu-1-P), YrfG (GMP) | No |
| BPGM | DOG1 (2d-glucose-6P), DOG2 (2d-glucose-6P), HOR2 (Gly-3-P), RHR2 (Gly-3-P) | YfbT (fructose-1-P) | No |
| YKL033W (XMP) | YniC (2d-glucose-6P), YqaB (fructose-1-P), YcjU (P-glucomutase) | HDHD1 (pseudouridine 5′-phosphate) | |
| Dehr | YOR131C (PLP) | Gph (P-glycolate) | No |
| YMR130W | YjjG (UMP) YigB (FMN) | HDHD3, NANP (N-acylneuraminate 9-phosphatase) | |
| Eno | YEL038W/UTR4 (5-(methylthio)-2,3-dioxopentyl-P) | No | ENOPH1 (5-(methylthio)-2,3-dioxopentyl-P) |
| NagD | PHO13 (P-glycolate), YKR070W (Glu-6-P) | NagD (UMP)a | LHPP (Lys(P)), PGP (P-glycolate), PDXP (PLP), CERC5, HDHD2 |
a Data from Ref. 32.
The variation of enzyme substrate preferences within several families of HADs from yeast, humans, and E. coli indicates that the evolution of substrate specificity does not necessarily follow sequence divergence, although, as pointed out above, in some cases, even highly diverged orthologs retain the ancestral specificity. In addition, our analysis indicates that the HAD phosphatases from different families can convergently evolve to catalyze the dephosphorylation of the same substrate (e.g. P-glycolate). Thus, in general, sequence similarity-based classification of HAD-like phosphatases cannot accurately predict their preferred substrates, emphasizing the importance of biochemical experiments for functional annotation of these enzymes. The presence of large numbers of paralogous HADs with (often) low sequence similarity in many organisms suggests a high rate of evolution of these genes. HAD phosphatases can quickly evolve new biochemical functions and acquire novel biological roles using a broad pool of secondary substrates. The substrate pools of the HAD phosphatases are closely similar in E. coli and yeast even if, in many cases, the specificities of enzymes within the same co-orthologous family are not. The biochemical promiscuity of the HAD phosphatases together with their structural flexibility and catalytic efficiency underlies their dominant role in metabolic dephosphorylation in all kingdoms of life.
Author Contributions
E. K., G. B., R. F., and A. K. designed, performed, and analyzed the experiments shown in Tables 1 and 2; Figs. 2, 4, and 8; and supplemental Tables 5–8. B. N., E. E., K. T., A. S., A. J., and A. M. E. designed, performed, and analyzed experiments shown in Figs. 5, 6, 7, and 9, and supplemental Table 3. K. S. M., Y. I. W., and E. V. K. designed, performed, and analyzed the experiments shown in Table 3, Fig. 1, and supplemental Fig. 1. A. D. H., G. H., R. Z., and V. C. L. designed, performed, and analyzed the experiments shown in supplemental Fig. 2. K. J. and M. B. designed, performed, and analyzed the experiments shown in Fig. 3. A. E. M., E. V. K., and A. F. Y. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank all members of the Centre for Structural Proteomics in Toronto for help in conducting these experiments. Dr. Zhaolei Zhang (Donnelly Centre for Cellular and Biomolecular Research, University of Toronto) is thanked for help with the design of the S. cerevisiae phosphopeptide library.
This work was supported, in whole or in part, by National Institutes of Health Grant GM094585 (to A. J.). This work was also supported by the Government of Canada through Genome Canada, the Ontario Genomics Institute, and the Ontario Research Fund (Grants 2009-OGI-ABC-1405 and ORF-GL2-01-004 to A. F. Y.), by the United States Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02-06CH11357 (to A. J.), and by U. S. National Science Foundation Grant IOS-1025398 (to A. D. H.). The authors declare that they have no conflict of interest with the content of this article.

This article contains supplemental Tables 1–8 and Figs. 1 and 2.
The atomic coordinates and structure factors (codes 2QLT, 3NUQ, 3KC2, and 3RF6) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- HAD
- haloacid dehalogenase
- Gly-1-P
- glycerol 1-phosphate
- Gly-2-P
- glycerol 2-phosphate
- Gly-3-P
- glycerol 3-phosphate
- pNPP
- p-nitrophenyl phosphate
- P-glycolate
- 2-phosphoglycolate
- PLP
- pyridoxal 5-phosphate
- Ψ-UMP
- pseudouridine monophosphate
- Ψ-UTP
- pseudouridine triphosphate
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- Bistris propane
- 1,3-bis[tris(hydroxymethyl)methylamino]propane
- SAD
- single-wavelength anomalous diffraction
- AP
- apurinic/apyrimidinic
- TP
- thiamine monophosphate
- r.m.s.
- root mean square
- PDB
- Protein Data Bank.
References
- 1. Galperin M. Y., Koonin E. V. (2010) From complete genome sequence to “complete” understanding? Trends Biotechnol. 28, 398–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Galperin M. Y., Koonin E. V. (2004) “Conserved hypothetical” proteins: prioritization of targets for experimental study. Nucleic Acids Res. 32, 5452–5463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Osterman A., Overbeek R. (2003) Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 7, 238–251 [DOI] [PubMed] [Google Scholar]
- 4. Hanson A. D., Pribat A., Waller J. C., de Crécy-Lagard V. (2010) “Unknown” proteins and “orphan” enzymes: the missing half of the engineering parts list and how to find it. Biochem. J. 425, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Frishman D. (2007) Protein annotation at genomic scale: the current status. Chem. Rev. 107, 3448–3466 [DOI] [PubMed] [Google Scholar]
- 6. Blaby-Haas C. E., de Crécy-Lagard V. (2011) Mining high-throughput experimental data to link gene and function. Trends Biotechnol. 29, 174–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schnoes A. M., Brown S. D., Dodevski I., Babbitt P. C. (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol. 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brenner S. E. (1999) Errors in genome annotation. Trends Genet. 15, 132–133 [DOI] [PubMed] [Google Scholar]
- 9. Karp P. D. (2004) Call for an enzyme genomics initiative. Genome Biol. 5, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sorokina M., Stam M., Médigue C., Lespinet O., Vallenet D. (2014) Profiling the orphan enzymes. Biol. Direct 9, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galperin M. Y., Koonin E. V. (2000) Who's your neighbor? New computational approaches for functional genomics. Nat. Biotechnol. 18, 609–613 [DOI] [PubMed] [Google Scholar]
- 12. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Green M. L., Karp P. D. (2004) A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito T., Tashiro K., Muta S., Ozawa R., Chiba T., Nishizawa M., Yamamoto K., Kuhara S., Sakaki Y. (2000) Toward a protein-protein interaction map of the budding yeast: a comprehensive system to examine two-hybrid interactions in all possible combinations between the yeast proteins. Proc. Natl. Acad. Sci. U.S.A. 97, 1143–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Uetz P., Giot L., Cagney G., Mansfield T. A., Judson R. S., Knight J. R., Lockshon D., Narayan V., Srinivasan M., Pochart P., Qureshi-Emili A., Li Y., Godwin B., Conover D., Kalbfleisch T., Vijayadamodar G., Yang M., Johnston M., Fields S., Rothberg J. M. (2000) A comprehensive analysis of protein-protein interactions in Saccharomyces cerevisiae. Nature 403, 623–627 [DOI] [PubMed] [Google Scholar]
- 16. Brown P. O., Botstein D. (1999) Exploring the new world of the genome with DNA microarrays. Nat. Genet. 21, 33–37 [DOI] [PubMed] [Google Scholar]
- 17. DeRisi J. L., Iyer V. R., Brown P. O. (1997) Exploring the metabolic and genetic control of gene expression on a genomic scale. Science 278, 680–686 [DOI] [PubMed] [Google Scholar]
- 18. Winzeler E. A., Shoemaker D. D., Astromoff A., Liang H., Anderson K., Andre B., Bangham R., Benito R., Boeke J. D., Bussey H., Chu A. M., Connelly C., Davis K., Dietrich F., Dow S. W., El Bakkoury M., Foury F., Friend S. H., Gentalen E., Giaever G., Hegemann J. H., Jones T., Laub M., Liao H., Liebundguth N., Lockhart D. J., Lucau-Danila A., Lussier M., M'Rabet N., Menard P., Mittmann M., Pai C., Rebischung C., Revuelta J. L., Riles L., Roberts C. J., Ross-MacDonald P., Scherens B., Snyder M., Sookhai-Mahadeo S., Storms R. K., Véronneau S., Voet M., Volckaert G., Ward T. R., Wysocki R., Yen G. S., Yu K., Zimmermann K., Philippsen P., Johnston M., Davis R. W. (1999) Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906 [DOI] [PubMed] [Google Scholar]
- 19. Christendat D., Yee A., Dharamsi A., Kluger Y., Savchenko A., Cort J. R., Booth V., Mackereth C. D., Saridakis V., Ekiel I., Kozlov G., Maxwell K. L., Wu N., McIntosh L. P., Gehring K., Kennedy M. A., Davidson A. R., Pai E. F., Gerstein M., Edwards A. M., Arrowsmith C. H. (2000) Structural proteomics of an archaeon. Nat. Struct. Biol. 7, 903–909 [DOI] [PubMed] [Google Scholar]
- 20. Norin M., Sundström M. (2002) Structural proteomics: developments in structure-to-function predictions. Trends Biotechnol. 20, 79–84 [DOI] [PubMed] [Google Scholar]
- 21. Kuznetsova E., Proudfoot M., Sanders S. A., Reinking J., Savchenko A., Arrowsmith C. H., Edwards A. M., Yakunin A. F. (2005) Enzyme genomics: application of general enzymatic screens to discover new enzymes. FEMS Microbiol. Rev. 29, 263–279 [DOI] [PubMed] [Google Scholar]
- 22. Anton B. P., Chang Y. C., Brown P., Choi H. P., Faller L. L., Guleria J., Hu Z., Klitgord N., Levy-Moonshine A., Maksad A., Mazumdar V., McGettrick M., Osmani L., Pokrzywa R., Rachlin J., Swaminathan R., Allen B., Housman G., Monahan C., Rochussen K., Tao K., Bhagwat A. S., Brenner S. E., Columbus L., de Crécy-Lagard V., Ferguson D., Fomenkov A., Gadda G., Morgan R. D., Osterman A. L., Rodionov D. A., Rodionova I. A., Rudd K. E., Söll D., Spain J., Xu S. Y., Bateman A., Blumenthal R. M., Bollinger J. M., Chang W. S., Ferrer M., Friedberg I., Galperin M. Y., Gobeill J., Haft D., Hunt J., Karp P., Klimke W., Krebs C., Macelis D., Madupu R., Martin M. J., Miller J. H., O'Donovan C., Palsson B., Ruch P., Setterdahl A., Sutton G., Tate J., Yakunin A., Tchigvintsev D., Plata G., Hu J., Greiner R., Horn D., Sjolander K., Salzberg S. L., Vitkup D., Letovsky S., Segre D., DeLisi C., Roberts R. J., Steffen M., Kasif S. (2013) The COMBREX project: design, methodology, and initial results. PLoS Biol. 11, e1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gerlt J. A., Allen K. N., Almo S. C., Armstrong R. N., Babbitt P. C., Cronan J. E., Dunaway-Mariano D., Imker H. J., Jacobson M. P., Minor W., Poulter C. D., Raushel F. M., Sali A., Shoichet B. K., Sweedler J. V. (2011) The Enzyme Function Initiative. Biochemistry 50, 9950–9962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koonin E. V., Tatusov R. L. (1994) Computer analysis of bacterial haloacid dehalogenases defines a large superfamily of hydrolases with diverse specificity: application of an iterative approach to database search. J. Mol. Biol. 244, 125–132 [DOI] [PubMed] [Google Scholar]
- 25. Burroughs A. M., Allen K. N., Dunaway-Mariano D., Aravind L. (2006) Evolutionary genomics of the HAD superfamily: understanding the structural adaptations and catalytic diversity in a superfamily of phosphoesterases and allied enzymes. J. Mol. Biol. 361, 1003–1034 [DOI] [PubMed] [Google Scholar]
- 26. Allen K. N., Dunaway-Mariano D. (2009) Markers of fitness in a successful enzyme superfamily. Curr. Opin. Struct. Biol. 19, 658–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aravind L., Galperin M. Y., Koonin E. V. (1998) The catalytic domain of the P-type ATPase has the haloacid dehalogenase fold. Trends Biochem. Sci. 23, 127–129 [DOI] [PubMed] [Google Scholar]
- 28. Pandya C., Farelli J. D., Dunaway-Mariano D., Allen K. N. (2014) Enzyme promiscuity: engine of evolutionary innovation. J. Biol. Chem. 289, 30229–30236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Selengut J. D. (2001) MDP-1 is a new and distinct member of the haloacid dehalogenase family of aspartate-dependent phosphohydrolases. Biochemistry 40, 12704–12711 [DOI] [PubMed] [Google Scholar]
- 30. Wang W., Kim R., Jancarik J., Yokota H., Kim S. H. (2001) Crystal structure of phosphoserine phosphatase from Methanococcus jannaschii, a hyperthermophile, at 1.8 Å resolution. Structure 9, 65–71 [DOI] [PubMed] [Google Scholar]
- 31. Kim Y., Yakunin A. F., Kuznetsova E., Xu X., Pennycooke M., Gu J., Cheung F., Proudfoot M., Arrowsmith C. H., Joachimiak A., Edwards A. M., Christendat D. (2004) Structure- and function-based characterization of a new phosphoglycolate phosphatase from Thermoplasma acidophilum. J. Biol. Chem. 279, 517–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tremblay L. W., Dunaway-Mariano D., Allen K. N. (2006) Structure and activity analyses of Escherichia coli K-12 NagD provide insight into the evolution of biochemical function in the haloalkanoic acid dehalogenase superfamily. Biochemistry 45, 1183–1193 [DOI] [PubMed] [Google Scholar]
- 33. Huang H., Patskovsky Y., Toro R., Farelli J. D., Pandya C., Almo S. C., Allen K. N., Dunaway-Mariano D. (2011) Divergence of structure and function in the haloacid dehalogenase enzyme superfamily: Bacteroides thetaiotaomicron BT2127 is an inorganic pyrophosphatase. Biochemistry 50, 8937–8949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Archambault J., Pan G., Dahmus G. K., Cartier M., Marshall N., Zhang S., Dahmus M. E., Greenblatt J. (1998) FCP1, the RAP74-interacting subunit of a human protein phosphatase that dephosphorylates the carboxyl-terminal domain of RNA polymerase IIO. J. Biol. Chem. 273, 27593–27601 [DOI] [PubMed] [Google Scholar]
- 35. Tootle T. L., Silver S. J., Davies E. L., Newman V., Latek R. R., Mills I. A., Selengut J. D., Parlikar B. E., Rebay I. (2003) The transcription factor Eyes absent is a protein tyrosine phosphatase. Nature 426, 299–302 [DOI] [PubMed] [Google Scholar]
- 36. Li X., Oghi K. A., Zhang J., Krones A., Bush K. T., Glass C. K., Nigam S. K., Aggarwal A. K., Maas R., Rose D. W., Rosenfeld M. G. (2003) Eya protein phosphatase activity regulates Six1-Dach-Eya transcriptional effects in mammalian organogenesis. Nature 426, 247–254 [DOI] [PubMed] [Google Scholar]
- 37. Rayapureddi J. P., Kattamuri C., Steinmetz B. D., Frankfort B. J., Ostrin E. J., Mardon G., Hegde R. S. (2003) Eyes absent represents a class of protein tyrosine phosphatases. Nature 426, 295–298 [DOI] [PubMed] [Google Scholar]
- 38. Gohla A., Birkenfeld J., Bokoch G. M. (2005) Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat. Cell Biol. 7, 21–29 [DOI] [PubMed] [Google Scholar]
- 39. Seifried A., Knobloch G., Duraphe P. S., Segerer G., Manhard J., Schindelin H., Schultz J., Gohla A. (2014) Evolutionary and structural analyses of mammalian haloacid dehalogenase-type phosphatases AUM and chronophin provide insight into the basis of their different substrate specificities. J. Biol. Chem. 289, 3416–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuznetsova E., Proudfoot M., Gonzalez C. F., Brown G., Omelchenko M. V., Borozan I., Carmel L., Wolf Y. I., Mori H., Savchenko A. V., Arrowsmith C. H., Koonin E. V., Edwards A. M., Yakunin A. F. (2006) Genome-wide analysis of substrate specificities of the Escherichia coli haloacid dehalogenase-like phosphatase family. J. Biol. Chem. 281, 36149–36161 [DOI] [PubMed] [Google Scholar]
- 41. Randez-Gil F., Blasco A., Prieto J. A., Sanz P. (1995) DOGR1 and DOGR2: two genes from Saccharomyces cerevisiae that confer 2-deoxyglucose resistance when overexpressed. Yeast 11, 1233–1240 [DOI] [PubMed] [Google Scholar]
- 42. Norbeck J., Pâhlman A. K., Akhtar N., Blomberg A., Adler L. (1996) Purification and characterization of two isoenzymes of dl-glycerol-3-phosphatase from Saccharomyces cerevisiae: identification of the corresponding GPP1 and GPP2 genes and evidence for osmotic regulation of Gpp2p expression by the osmosensing mitogen-activated protein kinase signal transduction pathway. J. Biol. Chem. 271, 13875–13881 [DOI] [PubMed] [Google Scholar]
- 43. Osman C., Haag M., Wieland F. T., Brügger B., Langer T. (2010) A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 29, 1976–1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kepes F., Schekman R. (1988) The yeast SEC53 gene encodes phosphomannomutase. J. Biol. Chem. 263, 9155–9161 [PubMed] [Google Scholar]
- 45. Nakanishi T., Sekimizu K. (2002) SDT1/SSM1, a multicopy suppressor of S-II null mutant, encodes a novel pyrimidine 5′-nucleotidase. J. Biol. Chem. 277, 22103–22106 [DOI] [PubMed] [Google Scholar]
- 46. Vance J. R., Wilson T. E. (2001) Uncoupling of 3′-phosphatase and 5′-kinase functions in budding yeast. Characterization of Saccharomyces cerevisiae DNA 3′-phosphatase (TPP1). J. Biol. Chem. 276, 15073–15081 [DOI] [PubMed] [Google Scholar]
- 47. Fortpied J., Maliekal P., Vertommen D., Van Schaftingen E. (2006) Magnesium-dependent phosphatase-1 is a protein-fructosamine-6-phosphatase potentially involved in glycation repair. J. Biol. Chem. 281, 18378–18385 [DOI] [PubMed] [Google Scholar]
- 48. Kuznetsova E., Xu L., Singer A., Brown G., Dong A., Flick R., Cui H., Cuff M., Joachimiak A., Savchenko A., Yakunin A. F. (2010) Structure and activity of the metal-independent fructose-1,6-bisphosphatase YK23 from Saccharomyces cerevisiae. J. Biol. Chem. 285, 21049–21059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Heering H. A., Weiner J.H., Armstrong F. A. (1997) Direct detection and measurement of electron relays in a multicentered enzyme: voltammetry of electrode-surface films of E. coli fumarate reductase, an iron-sulfur flavoprotein. J. Am. Chem. Soc. 119, 11628–11638 [Google Scholar]
- 50. Tchigvintsev A., Tchigvintsev D., Flick R., Popovic A., Dong A., Xu X., Brown G., Lu W., Wu H., Cui H., Dombrowski L., Joo J. C., Beloglazova N., Min J., Savchenko A., Caudy A. A., Rabinowitz J. D., Murzin A. G., Yakunin A. F. (2013) Biochemical and structural studies of conserved Maf proteins revealed nucleotide pyrophosphatases with a preference for modified nucleotides. Chem. Biol. 20, 1386–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Goyer A., Hasnain G., Frelin O., Ralat M. A., Gregory J. F., 3rd, Hanson A. D. (2013) A cross-kingdom Nudix enzyme that pre-empts damage in thiamin metabolism. Biochem. J. 454, 533–542 [DOI] [PubMed] [Google Scholar]
- 52. Graef M., Friedman J. R., Graham C., Babu M., Nunnari J. (2013) ER exit sites are physical and functional core autophagosome biogenesis components. Mol. Biol. Cell 24, 2918–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Costanzo M. C., Engel S. R., Wong E. D., Lloyd P., Karra K., Chan E. T., Weng S., Paskov K. M., Roe G. R., Binkley G., Hitz B. C., Cherry J. M. (2014) Saccharomyces genome database provides new regulation data. Nucleic Acids Res. 42, D717–D725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang R. G., Skarina T., Katz J. E., Beasley S., Khachatryan A., Vyas S., Arrowsmith C. H., Clarke S., Edwards A., Joachimiak A., Savchenko A. (2001) Structure of Thermotoga maritima stationary phase survival protein SurE: a novel acid phosphatase. Structure 9, 1095–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rosenbaum G., Alkire R. W., Evans G., Rotella F. J., Lazarski K., Zhang R. G., Ginell S. L., Duke N., Naday I., Lazarz J., Molitsky M. J., Keefe L., Gonczy J., Rock L., Sanishvili R., Walsh M. A., Westbrook E., Joachimiak A. (2006) The Structural Biology Center 19ID undulator beamline: facility specifications and protein crystallographic results. J. Synchrotron Radiat. 13, 30–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 57. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 58. Schneider T. R., Sheldrick G. M. (2002) Substructure solution with SHELXD. Acta Crystallogr. D Biol. Crystallogr. 58, 1772–1779 [DOI] [PubMed] [Google Scholar]
- 59. Cowtan K. D., Main P. (1993) Improvement of macromolecular electron-density maps by the simultaneous application of real and reciprocal space constraints. Acta Crystallogr. D Biol. Crystallogr. 49, 148–157 [DOI] [PubMed] [Google Scholar]
- 60. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 61. Terwilliger T. C. (2003) SOLVE and RESOLVE: automated structure solution and density modification. Methods Enzymol. 374, 22–37 [DOI] [PubMed] [Google Scholar]
- 62. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 63. Lebedev A. A., Vagin A. A., Murshudov G. N. (2008) Model preparation in MOLREP and examples of model improvement using x-ray data. Acta Crystallogr. D Biol. Crystallogr. 64, 33–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Davis I. W., Murray L. W., Richardson J. S., Richardson D. C. (2004) MOLPROBITY: structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 32, W615–W619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Kobor M. S., Archambault J., Lester W., Holstege F. C., Gileadi O., Jansma D. B., Jennings E. G., Kouyoumdjian F., Davidson A. R., Young R. A., Greenblatt J. (1999) An unusual eukaryotic protein phosphatase required for transcription by RNA polymerase II and CTD dephosphorylation in S. cerevisiae. Mol. Cell 4, 55–62 [DOI] [PubMed] [Google Scholar]
- 66. Han G. S., Wu W. I., Carman G. M. (2006) The Saccharomyces cerevisiae Lipin homolog is a Mg2+-dependent phosphatidate phosphatase enzyme. J. Biol. Chem. 281, 9210–9218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Vandercammen A., François J., Hers H. G. (1989) Characterization of trehalose-6-phosphate synthase and trehalose-6-phosphate phosphatase of Saccharomyces cerevisiae. Eur. J. Biochem. 182, 613–620 [DOI] [PubMed] [Google Scholar]
- 68. Su W. M., Han G. S., Carman G. M. (2014) Yeast Nem1-Spo7 protein phosphatase activity on Pah1 phosphatidate phosphatase is specific for the Pho85-Pho80 protein kinase phosphorylation sites. J. Biol. Chem. 289, 34699–34708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Itoh R., Saint-Marc C., Chaignepain S., Katahira R., Schmitter J. M., Daignan-Fornier B. (2003) The yeast ISN1 (YOR155c) gene encodes a new type of IMP-specific 5′-nucleotidase. BMC Biochem. 4, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bogan K. L., Evans C., Belenky P., Song P., Burant C. F., Kennedy R., Brenner C. (2009) Identification of Isn1 and Sdt1 as glucose- and vitamin-regulated nicotinamide mononucleotide and nicotinic acid mononucleotide [corrected] 5′-nucleotidases responsible for production of nicotinamide riboside and nicotinic acid riboside. J. Biol. Chem. 284, 34861–34869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reddy V. S., Singh A. K., Rajasekharan R. (2008) The Saccharomyces cerevisiae PHM8 gene encodes a soluble magnesium-dependent lysophosphatidic acid phosphatase. J. Biol. Chem. 283, 8846–8854 [DOI] [PubMed] [Google Scholar]
- 72. Kaneko Y., Toh-e A., Banno I., Oshima Y. (1989) Molecular characterization of a specific p-nitrophenylphosphatase gene, PHO13, and its mapping by chromosome fragmentation in Saccharomyces cerevisiae. Mol. Gen. Genet. 220, 133–139 [DOI] [PubMed] [Google Scholar]
- 73. Kim Y., Gentry M. S., Harris T. E., Wiley S. E., Lawrence J. C., Jr., Dixon J. E. (2007) A conserved phosphatase cascade that regulates nuclear membrane biogenesis. Proc. Natl. Acad. Sci. U.S.A. 104, 6596–6601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ficarro S. B., McCleland M. L., Stukenberg P. T., Burke D. J., Ross M. M., Shabanowitz J., Hunt D. F., White F. M. (2002) Phosphoproteome analysis by mass spectrometry and its application to Saccharomyces cerevisiae. Nat. Biotechnol. 20, 301–305 [DOI] [PubMed] [Google Scholar]
- 75. Chi A., Huttenhower C., Geer L. Y., Coon J. J., Syka J. E., Bai D. L., Shabanowitz J., Burke D. J., Troyanskaya O. G., Hunt D. F. (2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2193–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moffatt C. E., Inaba H., Hirano T., Lamont R. J. (2012) Porphyromonas gingivalis SerB-mediated dephosphorylation of host cell cofilin modulates invasion efficiency. Cell Microbiol. 14, 577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lifshitz Z., Burstein D., Schwartz K., Shuman H. A., Pupko T., Segal G. (2014) Identification of novel Coxiella burnetii Icm/Dot effectors and genetic analysis of their involvement in modulating a mitogen-activated protein kinase pathway. Infect. Immun. 82, 3740–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Nobeli I., Ponstingl H., Krissinel E. B., Thornton J. M. (2003) A structure-based anatomy of the E. coli metabolome. J. Mol. Biol. 334, 697–719 [DOI] [PubMed] [Google Scholar]
- 79. Melcher K., Entian K. D. (1992) Genetic analysis of serine biosynthesis and glucose repression in yeast. Curr. Genet. 21, 295–300 [DOI] [PubMed] [Google Scholar]
- 80. Newmeyer D. D., Ferguson-Miller S. (2003) Mitochondria: releasing power for life and unleashing the machineries of death. Cell 112, 481–490 [DOI] [PubMed] [Google Scholar]
- 81. Bucala R., Model P., Russel M., Cerami A. (1985) Modification of DNA by glucose 6-phosphate induces DNA rearrangements in an Escherichia coli plasmid. Proc. Natl. Acad. Sci. U.S.A. 82, 8439–8442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lee A. T., Cerami A. (1987) Elevated glucose 6-phosphate levels are associated with plasmid mutations in vivo. Proc. Natl. Acad. Sci. U.S.A. 84, 8311–8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kadner R. J., Murphy G. P., Stephens C. M. (1992) Two mechanisms for growth inhibition by elevated transport of sugar phosphates in Escherichia coli. J. Gen. Microbiol. 138, 2007–2014 [DOI] [PubMed] [Google Scholar]
- 84. Irani M. H., Maitra P. K. (1977) Properties of Escherichia coli mutants deficient in enzymes of glycolysis. J. Bacteriol. 132, 398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Papenfort K., Sun Y., Miyakoshi M., Vanderpool C. K., Vogel J. (2013) Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153, 426–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Breitkreutz A., Choi H., Sharom J. R., Boucher L., Neduva V., Larsen B., Lin Z. Y., Breitkreutz B. J., Stark C., Liu G., Ahn J., Dewar-Darch D., Reguly T., Tang X., Almeida R., Qin Z. S., Pawson T., Gingras A. C., Nesvizhskii A. I., Tyers M. (2010) A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Moorhead G. B., Trinkle-Mulcahy L., Ulke-Lemée A. (2007) Emerging roles of nuclear protein phosphatases. Nat. Rev. Mol. Cell Biol. 8, 234–244 [DOI] [PubMed] [Google Scholar]
- 88. Jiang L., Whiteway M., Ramos C., Rodriguez-Medina J. R., Shen S. H. (2002) The YHR076w gene encodes a type 2C protein phosphatase and represents the seventh PP2C gene in budding yeast. FEBS Lett. 527, 323–325 [DOI] [PubMed] [Google Scholar]
- 89. Lu G., Ren S., Korge P., Choi J., Dong Y., Weiss J., Koehler C., Chen J. N., Wang Y. (2007) A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev. 21, 784–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Pagliarini D. J., Wiley S. E., Kimple M. E., Dixon J. R., Kelly P., Worby C. A., Casey P. J., Dixon J. E. (2005) Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic beta cells. Mol. Cell 19, 197–207 [DOI] [PubMed] [Google Scholar]
- 91. Takeda K., Komuro Y., Hayakawa T., Oguchi H., Ishida Y., Murakami S., Noguchi T., Kinoshita H., Sekine Y., Iemura S., Natsume T., Ichijo H. (2009) Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc. Natl. Acad. Sci. U.S.A. 106, 12301–12305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Xu Y. F., Létisse F., Absalan F., Lu W., Kuznetsova E., Brown G., Caudy A. A., Yakunin A. F., Broach J. R., Rabinowitz J. D. (2013) Nucleotide degradation and ribose salvage in yeast. Mol. Syst. Biol. 9, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Carlile T. M., Rojas-Duran M. F., Zinshteyn B., Shin H., Bartoli K. M., Gilbert W. V. (2014) Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515, 143–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cantara W. A., Crain P. F., Rozenski J., McCloskey J. A., Harris K. A., Zhang X., Vendeix F. A., Fabris D., Agris P. F. (2011) The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 39, D195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Charette M., Gray M. W. (2000) Pseudouridine in RNA: what, where, how, and why. IUBMB Life 49, 341–351 [DOI] [PubMed] [Google Scholar]
- 96. Preumont A., Snoussi K., Stroobant V., Collet J. F., Van Schaftingen E. (2008) Molecular identification of pseudouridine-metabolizing enzymes. J. Biol. Chem. 283, 25238–25246 [DOI] [PubMed] [Google Scholar]
- 97. Goldberg I. H., Rabinowitz M. (1963) Comparative utilization of pseudouridine triphosphate and uridine triphosphate by ribonucleic acid polymerase. J. Biol. Chem. 238, 1793–1800 [PubMed] [Google Scholar]
- 98. Kahan F. M., Hurwitz J. (1962) The role of deoxyribonucleic acid in ribonucleic acid synthesis: IV. The incorporation of pyrimidine and purine analogues into ribonucleic acid. J. Biol. Chem. 237, 3778–3785 [PubMed] [Google Scholar]
- 99. Preumont A., Rzem R., Vertommen D., Van Schaftingen E. (2010) HDHD1, which is often deleted in X-linked ichthyosis, encodes a pseudouridine-5′-phosphatase. Biochem. J. 431, 237–244 [DOI] [PubMed] [Google Scholar]
- 100. Tuleva B., Vasileva-Tonkova E., Galabova D. (1998) A specific alkaline phosphatase from Saccharomyces cerevisiae with protein phosphatase activity. FEMS Microbiol. Lett. 161, 139–144 [DOI] [PubMed] [Google Scholar]
- 101. Boiteux S., Guillet M. (2004) Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 3, 1–12 [DOI] [PubMed] [Google Scholar]
- 102. Otterlei M., Kavli B., Standal R., Skjelbred C., Bharati S., Krokan H. E. (2000) Repair of chromosomal abasic sites in vivo involves at least three different repair pathways. EMBO J. 19, 5542–5551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Teresa Pellicer M., Felisa Nuñez M., Aguilar J., Badia J., Baldoma L. (2003) Role of 2-phosphoglycolate phosphatase of Escherichia coli in metabolism of the 2-phosphoglycolate formed in DNA repair. J. Bacteriol. 185, 5815–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Van Vleet J. H., Jeffries T. W., Olsson L. (2008) Deleting the para-nitrophenyl phosphatase (pNPPase), PHO13, in recombinant Saccharomyces cerevisiae improves growth and ethanol production on d-xylose. Metab. Eng. 10, 360–369 [DOI] [PubMed] [Google Scholar]
- 105. Fujitomi K., Sanda T., Hasunuma T., Kondo A. (2012) Deletion of the PHO13 gene in Saccharomyces cerevisiae improves ethanol production from lignocellulosic hydrolysate in the presence of acetic and formic acids, and furfural. Bioresour. Technol. 111, 161–166 [DOI] [PubMed] [Google Scholar]
- 106. Kim S. R., Skerker J. M., Kang W., Lesmana A., Wei N., Arkin A. P., Jin Y. S. (2013) Rational and evolutionary engineering approaches uncover a small set of genetic changes efficient for rapid xylose fermentation in Saccharomyces cerevisiae. PLoS One 8, e57048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schryvers A., Lohmeier E., Weiner J. H. (1978) Chemical and functional properties of the native and reconstituted forms of the membrane-bound, aerobic glycerol-3-phosphate dehydrogenase of Escherichia coli. J. Biol. Chem. 253, 783–788 [PubMed] [Google Scholar]
- 108. Nosaka K. (2006) Recent progress in understanding thiamin biosynthesis and its genetic regulation in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 72, 30–40 [DOI] [PubMed] [Google Scholar]
- 109. Jurgenson C. T., Begley T. P., Ealick S. E. (2009) The structural and biochemical foundations of thiamin biosynthesis. Annu. Rev. Biochem. 78, 569–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Percudani R., Peracchi A. (2003) A genomic overview of pyridoxal-phosphate-dependent enzymes. EMBO Rep. 4, 850–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Lumeng L., Li T. K. (1975) Characterization of the pyridoxal 5′-phosphate and pyridoxamine 5′-phosphate hydrolase activity in rat liver. Identity with alkaline phosphatase. J. Biol. Chem. 250, 8126–8131 [PubMed] [Google Scholar]
- 112. Mukherjee T., Hanes J., Tews I., Ealick S. E., Begley T. P. (2011) Pyridoxal phosphate: biosynthesis and catabolism. Biochim. Biophys. Acta 1814, 1585–1596 [DOI] [PubMed] [Google Scholar]
- 113. Fonda M. L. (1992) Purification and characterization of vitamin B6-phosphate phosphatase from human erythrocytes. J. Biol. Chem. 267, 15978–15983 [PubMed] [Google Scholar]
- 114. Jang Y. M., Kim D. W., Kang T. C., Won M. H., Baek N. I., Moon B. J., Choi S. Y., Kwon O. S. (2003) Human pyridoxal phosphatase: molecular cloning, functional expression, and tissue distribution. J. Biol. Chem. 278, 50040–50046 [DOI] [PubMed] [Google Scholar]
- 115. Moriyama K., Iida K., Yahara I. (1996) Phosphorylation of Ser-3 of cofilin regulates its essential function on actin. Genes Cells 1, 73–86 [DOI] [PubMed] [Google Scholar]
- 116. Theriot J. A. (1997) Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J. Cell Biol. 136, 1165–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Lappalainen P., Fedorov E. V., Fedorov A. A., Almo S. C., Drubin D. G. (1997) Essential functions and actin-binding surfaces of yeast cofilin revealed by systematic mutagenesis. EMBO J. 16, 5520–5530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Giaever G., Chu A. M., Ni L., Connelly C., Riles L., Véronneau S., Dow S., Lucau-Danila A., Anderson K., André B., Arkin A. P., Astromoff A., El-Bakkoury M., Bangham R., Benito R., Brachat S., Campanaro S., Curtiss M., Davis K., Deutschbauer A., Entian K. D., Flaherty P., Foury F., Garfinkel D. J., Gerstein M., Gotte D., Güldener U., Hegemann J. H., Hempel S., Herman Z., Jaramillo D. F., Kelly D. E., Kelly S. L., Kötter P., LaBonte D., Lamb D. C., Lan N., Liang H., Liao H., Liu L., Luo C., Lussier M., Mao R., Menard P., Ooi S. L., Revuelta J. L., Roberts C. J., Rose M., Ross-Macdonald P., Scherens B., Schimmack G., Shafer B., Shoemaker D. D., Sookhai-Mahadeo S., Storms R. K., Strathern J. N., Valle G., Voet M., Volckaert G., Wang C. Y., Ward T. R., Wilhelmy J., Winzeler E. A., Yang Y., Yen G., Youngman E., Yu K., Bussey H., Boeke J. D., Snyder M., Philippsen P., Davis R. W., Johnston M. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418, 387–391 [DOI] [PubMed] [Google Scholar]
- 119. Daiyasu H., Hiroike T., Koga Y., Toh H. (2002) Analysis of membrane stereochemistry with homology modeling of sn-glycerol-1-phosphate dehydrogenase. Protein Eng. 15, 987–995 [DOI] [PubMed] [Google Scholar]
- 120. Guldan H., Sterner R., Babinger P. (2008) Identification and characterization of a bacterial glycerol-1-phosphate dehydrogenase: Ni2+-dependent AraM from Bacillus subtilis. Biochemistry 47, 7376–7384 [DOI] [PubMed] [Google Scholar]
- 121. Nevoigt E., Stahl U. (1997) Osmoregulation and glycerol metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 21, 231–241 [DOI] [PubMed] [Google Scholar]
- 122. Pahlman A. K., Granath K., Ansell R., Hohmann S., Adler L. (2001) The yeast glycerol 3-phosphatases Gpp1p and Gpp2p are required for glycerol biosynthesis and differentially involved in the cellular responses to osmotic, anaerobic, and oxidative stress. J. Biol. Chem. 276, 3555–3563 [DOI] [PubMed] [Google Scholar]
- 123. Wilson W. A., Wang Z., Roach P. J. (2002) Systematic identification of the genes affecting glycogen storage in the yeast Saccharomyces cerevisiae: implication of the vacuole as a determinant of glycogen level. Mol. Cell Proteomics 1, 232–242 [DOI] [PubMed] [Google Scholar]
- 124. Kestler C., Knobloch G., Tessmer I., Jeanclos E., Schindelin H., Gohla A. (2014) Chronophin dimerization is required for proper positioning of its substrate specificity loop. J. Biol. Chem. 289, 3094–3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shi N., Zhang Y. J., Chen H. K., Gao Y., Teng M., Niu L. (2011) Crystal structure of the pyrimidine 5′-nucleotidase SDT1 from Saccharomyces cerevisiae complexed with uridine 5′-monophosphate provides further insight into ligand binding. Proteins 79, 1358–1362 [DOI] [PubMed] [Google Scholar]
- 126. Binkowski T. A., Naghibzadeh S., Liang J. (2003) CASTp: Computed Atlas of Surface Topography of proteins. Nucleic Acids Res. 31, 3352–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Khersonsky O., Tawfik D. S. (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 79, 471–505 [DOI] [PubMed] [Google Scholar]
- 128. Lynch M., Force A. (2000) The probability of duplicate gene preservation by subfunctionalization. Genetics 154, 459–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ward R., Durrett R. (2004) Subfunctionalization: how often does it occur? how long does it take? Theor. Popul. Biol. 66, 93–100 [DOI] [PubMed] [Google Scholar]
- 130. Wu J., Woodard R. W. (2003) Escherichia coli YrbI is 3-deoxy-d-manno-octulosonate 8-phosphate phosphatase. J. Biol. Chem. 278, 18117–18123 [DOI] [PubMed] [Google Scholar]
- 131. Kneidinger B., Marolda C., Graninger M., Zamyatina A., McArthur F., Kosma P., Valvano M. A., Messner P. (2002) Biosynthesis pathway of ADP-L-glycero-β-d-manno-heptose in Escherichia coli. J. Bacteriol. 184, 363–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Tatusov R. L., Fedorova N. D., Jackson J. D., Jacobs A. R., Kiryutin B., Koonin E. V., Krylov D. M., Mazumder R., Mekhedov S. L., Nikolskaya A. N., Rao B. S., Smirnov S., Sverdlov A. V., Vasudevan S., Wolf Y. I., Yin J. J., Natale D. A. (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Li L., Stoeckert C. J., Jr., Roos D. S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.