

Background: It is unclear whether microtubule depolymerization by kinesin-13s is coupled with the hydrolysis step of ATP turnover.
Results: Mutation of the kinesin-13-specific KVD motif blocks ATPase and microtubule depolymerization, whereas another ATPase-deficient mutant depolymerizes microtubules.
Conclusion: Binding of kinesin-13-ATP to microtubule ends is sufficient for depolymerization; ATP hydrolysis is not required.
Significance: Microtubule depolymerization is directly driven by the binding energy of kinesin-13s.
Keywords: ATPase, conformational change, kinesin, microtubule, molecular motor
Abstract
Kinesin-13 proteins depolymerize microtubules in an ATP hydrolysis-dependent manner. The coupling between these two activities remains unclear. Here, we first studied the role of the kinesin-13 subfamily-specific loop 2 and of the KVD motif at the tip of this loop. Shortening the loop, the lysine/glutamate interchange and the additional Val to Ser substitution all led to Kif2C mutants with decreased microtubule-stimulated ATPase and impaired depolymerization capability. We rationalized these results based on a structural model of the Kif2C-ATP-tubulin complex derived from the recently determined structures of kinesin-1 bound to tubulin. In this model, upon microtubule binding Kif2C undergoes a conformational change governed in part by the interaction of the KVD motif with the tubulin interdimer interface. Second, we mutated to an alanine the conserved glutamate residue of the switch 2 nucleotide binding motif. This mutation blocks motile kinesins in a post-conformational change state and inhibits ATP hydrolysis. This Kif2C mutant still depolymerized microtubules and yielded complexes of one Kif2C with two tubulin heterodimers. These results demonstrate that the structural change of Kif2C-ATP upon binding to microtubule ends is sufficient for tubulin release, whereas ATP hydrolysis is not required. Overall, our data suggest that the conformation reached by kinesin-13s upon tubulin binding is similar to that of tubulin-bound, ATP-bound, motile kinesins but that this conformation is adapted to microtubule depolymerization.
Introduction
Although most kinesins translocate along microtubules to transport various cargoes (1), kinesin-13 subfamily members function by depolymerizing microtubules (2) and therefore are involved in many microtubule-dependent events including mitosis (3), neuronal development (4), and ciliogenesis (5). In particular, kinesin-13 proteins play a pivotal role in the correction of erroneous microtubule-kinetochore attachments. As a result, they are important for the faithful segregation of chromosomes during mitosis. Their up-regulated expression in some solid tumors makes them a potential anti-cancer drug target (6). However, despite their potential practical importance, the molecular mechanism of microtubule depolymerization by kinesin-13s is still far from clear (7).
Kinesin-13s have a distinct working cycle that is adapted to microtubule depolymerization (Fig. 1). A previous study of the monomeric minimal functional domain of human kinesin-13 protein Kif2C has established that Kif2C undergoes autonomous nucleotide exchange in solution and that Kif2C-ATP has remarkably high affinity for curved tubulin. These unique properties lead to a proposal that Kif2C starts the microtubule depolymerization process by binding directly to the end of microtubules in the ATP binding state (8). Following this binding is the tubulin release from microtubule end, together with ATP hydrolysis, which produces dissociated Kif2C-ADP for its next round of activity (Fig. 1). Therefore, formation of the Kif2C-ATP-tubulin ternary complex at microtubule ends seems the critical step for microtubule depolymerization, in analogy to the formation of the kinesin-ATP-tubulin complex, which is critical for conventional kinesin motility. What remains to be determined is how strong binding of Kif2C-ATP to tubulin is achieved and how this leads to the release of tubulin from the microtubular lattice.
FIGURE 1.
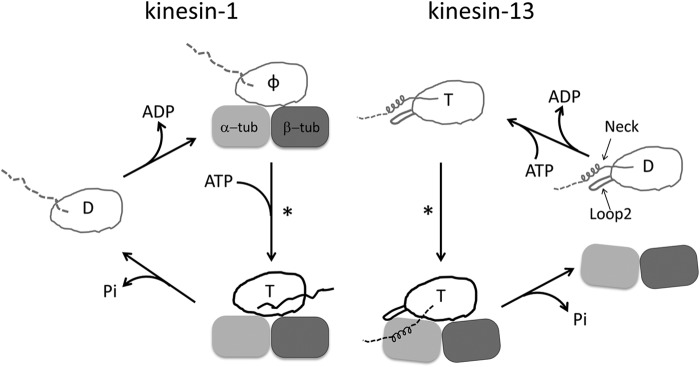
Comparison of the working cycles of motile kinesin-1 and depolymerizing kinesin-13. One motor domain is shown in both cases. The letter inside the motor domain indicates the nucleotide state of kinesin: D, ADP; T, ATP; φ, no nucleotide. In the working cycle of kinesin-1, the microtubule is represented by one tubulin heterodimer. In the ADP binding and nucleotide-free states of kinesin-1, the undocked neck linker is shown as a dashed line, whereas in microtubule-bound kinesin-ATP, the docked neck linker is shown as a solid line pointing toward the microtubule (+) end. In the case of kinesin-13, the subfamily-specific long loop 2 and neck are shown with the motor domain. The microtubule end is shown as a curved tubulin heterodimer. Upon binding to microtubule ends, Kif2C-ATP undergoes a conformational change to form the Kif2C-ATP-tubulin complex, a process to which the neck region contributes significantly (8). However, the binding site of the neck region on tubulin remains unknown, so it is shown in an arbitrary position as a dashed line. In both working cycles, an asterisk indicates the step in which the competent complex forms.
Another unsettled question, albeit central to the microtubule depolymerization mechanism, is how tubulin release by Kif2C-ATP is coupled with ATP hydrolysis in Kif2C. The experimental evidence available so far has been interpreted to mean that ATP turnover of kinesin-13s is required for productive microtubule depolymerization. One such piece of evidence is that in the presence of a nonhydrolysable ATP analogue AMPPNP,5 kinesin-13s failed to release tubulin from microtubule ends (2). Instead, the curved tubulins are longitudinally linked into helical or ring assemblies by the kinesin-13 motor domain, in particular through the subfamily-specific long loop 2 (see Refs. 9 and 10 for a nomenclature of kinesin-13 motor domain secondary structure elements (11, 12). These curved assemblies are proposed to be an intermediate in microtubule depolymerization (2, 12, 13). Additional evidence comes from the study of an ATPase-deficient mutation of kinesin-13s. When the Gly residue of the conserved switch 2 nucleotide-binding motif (DXXGXE) is mutated to Ala, kinesin-13s lose both their ATPase and microtubule depolymerization activities (8, 14). However, to demonstrate coupling between the microtubule depolymerization and ATP hydrolysis activities of kinesin-13s, one needs to ensure the formation of the fully competent ternary complex while preventing ATP hydrolysis in tubulin-bound kinesin-13.
All the kinesin superfamily members share a homologous motor domain that harbors the microtubule-binding site and ATP hydrolysis activity (2). Kinesin-13s are no exception, as demonstrated by the motor domain structures that have been determined (9, 10). In the case of (+) end-directed motile kinesins, the mechanochemical cycle is relatively well established, both biochemically (15) and structurally. In particular, the crystal structures of the motor domain of conventional motile kinesin-1 bound to tubulin were recently determined in two main states of their nucleotide cycle: ATP-like and apo (16, 17). The comparison of these two structures revealed that ATP binding causes motile kinesins to step by inducing rigid body motions of two subdomains of the motor domain with respect to a fixed microtubule-binding subdomain. Given the homology of motor domains of kinesin-13s and motile kinesins (18) and that they share the same binding site on tubulin (11), conformational changes that include motor subdomains movements are probably involved in their working cycle as well. Given the functional importance of the subfamily-specific loop 2 (9–11), this structural element may also contribute to the critical conformational changes of kinesin-13s. Lastly, the crystal structures of kinesin-1 in complex with tubulin could serve as an ideal template to understand the structural mechanism of kinesin-13s.
In this work, we performed mutagenesis studies of the monomeric minimal functional domain of Kif2C, based on a structural model of Kif2C-ATP bound to tubulin. These results revealed that the KVD motif at the tip of Kif2C loop 2 plays an essential role in the conformational change of Kif2C-ATP upon tubulin binding. This conformational change of Kif2C-ATP further determines both the microtubule depolymerization and ATP hydrolysis activities of Kif2C. Furthermore, results on the mutation of the Glu residue in the switch 2 motif (DXXGXE) leading to the Kif2C E497A mutant revealed that the chemical step of ATP hydrolysis in Kif2C is not required for tubulin release from microtubule ends but that the complete conformational change in the Kif2C-ATP-tubulin ternary complex, which is ordinarily coupled to the ATP hydrolysis step in Kif2C, is sufficient. Taken together, our data provide a clearer view of the microtubule depolymerization mechanism of Kif2C, which is probably universal for all the kinesin-13 proteins.
Experimental Procedures
Protein Purification
The minimal functional domain of human Kif2C, a fragment comprising the proximal part of the neck region (named the “short neck”) and the motor domain (Kif2C-(sN+M)), and its mutants were expressed and purified as previously described (8). Mutations were introduced using standard molecular biology methods. Tubulin was purified from pig brain by two cycles of temperature-dependent assembly/disassembly (19). Taxotere-stabilized microtubules were used in this study.
Crystallization and Structure Determination
The Kif2C-(sN+M)-sL2 variant in complex with ADP was crystallized at 293 K by vapor diffusion with a crystallization buffer consisting of 0.1 m Tris-HCl, pH 8.0, 1.5 m ammonium sulfate, and 10% ethylene glycol. Crystals were harvested in 80% saturated lithium sulfate and then flash-frozen in liquid nitrogen. A 2.6 Å data set was collected at 100 K at the Proxima1 beam line (Soleil Synchrotron, Saint Aubin, France). It was processed with XDS (20) and SCALA (21). The structure was solved by molecular replacement with PHASER (22) using HsKif2C motor domain-ADP (Protein Data Bank entry 2HEH) as a starting model. The structure was refined with BUSTER (23) with iterative model building in Coot (24). The final model comprises 330 residues; 57 residues were not included in the model because of weak or absent electron density. Three strong peaks in the Fobs-Fcal electron density map were tentatively attributed to sulfate ions. Statistics for data processing and refinement are reported in Table 1. Coordinates and structure factors have been deposited in the Protein Data Bank with accession code 4Y05.
TABLE 1.
Data collection and refinement statistics of the structure of Kif2C-(sN+M)-sL2
| Kif2C-(sN+M)-sL2 | |
|---|---|
| Data collectiona | |
| Space group | P43212 |
| Cell dimensions | |
| a ( = b), c (Å) | 83.9, 147.9 |
| Resolution (Å) | 42.5-2.59 (2.73-2.59) |
| Rsym | 0.05 (1.22) |
| I/σI | 27.6 (2.4) |
| Completeness (%) | 99.7 (98.0) |
| Multiplicity | 13.6 (11.4) |
| Refinement | |
| Resolution (Å) | 24.7-2.59 |
| No. reflections | 17,069 |
| Rwork/Rfree | 0.192/0.204 |
| No. atoms | |
| Protein | 2532 |
| ADP/Mg2+ | 28 |
| Waters | 52 |
| Ions (SO42−) | 15 |
| B factors | |
| Wilson | 95.3 |
| Protein | 103 |
| ADP/Mg2+ | 104 |
| Waters | 82 |
| Ions (SO42−) | 179 |
| Coordinate error (Å)b | 0.496 |
| Root mean square deviation | |
| Bond lengths (Å) | 0.010 |
| Bond angles (°) | 1.15 |
| Ramachandran | |
| Favored region (%) | 96.92 |
| Allowed region (%) | 2.46 |
| Outliers (%) | 0.62 |
a The data were collected on a single crystal. There is one molecule per asymmetric unit. The values in parentheses are for the highest resolution shell.
b Estimated from a Luzzati plot.
ATPase Measurement
The ATPase activities of Kif2C-(sN+M) mutants were measured at 25 °C using an enzyme-coupled assay, as previously described (8). The buffer used for ATPase measurement is 40 mm PIPES-K, pH 6.8, 75 mm KCl, 1 mm DTT, 2 mm MgCl2, and 1 mm EGTA. When microtubule-stimulated ATPase was measured, 20 μm Taxotere was supplemented in the buffer to prevent the depolymerization of microtubules by Kif2C. For the measurement of Kif2C ATPase stimulated by tubulin-vinblastine assemblies, 100 μm vinblastine was supplemented in the buffer, which is sufficient to fully assemble tubulin up to 10 μm, the maximum concentration used in these experiments.
Microtubule Depolymerization
The microtubule depolymerization activities of Kif2C and its mutants were measured both by turbidity and centrifugation assays, as previously described (8). For turbidity assay, microtubule depolymerization by Kif2C proteins was studied by measuring at 25 °C the turbidity of a Taxotere-stabilized microtubule solution in 40 mm PIPES-K, pH 6.8, 75 mm KCl, 1 mm EGTA, 2 mm MgCl2, 1 mm DTT, and 1 mm ATP. Absorbance at 350 nm was monitored following addition of Kif2C protein, using a Cary 50 spectrophotometer (Varian). Microtubule depolymerization was also monitored in a centrifugation assay by incubating at 25 °C for 15 min Taxotere-stabilized microtubules with Kif2C proteins in the same buffer. Soluble tubulin and microtubules were separated by centrifuging the reaction product for 15 min at 85,000 rpm in a TLA120.1 rotor (Beckman). Equal volumes of supernatant and resuspended pellet were analyzed on SDS-PAGE gels and stained with Coomassie Blue for comparison of tubulin quantities. The relative intensity of the tubulin band was quantified with the ImageJ software.
Tubulin Pelleting
To check the ability of Kif2C proteins to link tubulin longitudinally, typically 1 μm tubulin and 1 μm Kif2C were incubated in the presence of 1 mm AMPPNP for 15 min at room temperature. The mixture was then ultracentrifuged for 15 min at 85,000 rpm in a TLA120.1 rotor (Beckman). The supernatant and pellet were analyzed on SDS-PAGE gels. For comparison of the tubulin pelleting activities between wild type Kif2C-(sN+M), G495A and E497A mutants, 0.5 μm Kif2C variants was incubated with 0.5 μm tubulin, after which ultracentrifugation was at 75,000 rpm.
Structural Modeling
A structural model of the Kif2C motor domain in its ATP state in complex with tubulin (noted Kif2C-ATP-tubulin) was generated using as templates the structures of the ADP-bound Kif2C motor domain (noted Kif2C-ADP; Protein Data Bank entry 2HEH) and of kinesin-1 in the ADP state (noted Kin1-ADP; Protein Data Bank entry 1BG2) and in the ADP-Mg-AlFx state in complex with tubulin (noted Kin1-ATP-tubulin; Protein Data Bank entry 4HNA). Kif2C shares 35% sequence identity with kinesin-1 so that a first model of Kif2C-ATP-tubulin (model 1) could be obtained by comparative modeling using Kin1-ATP-tubulin as template and the SWISS-MODEL server (25). However, regions structurally divergent between Kif2C and kinesin-1 were not properly modeled. They were identified by superimposing the Kif2C-ADP and Kin1-ADP structures. Segments with Cα-Cα distance below 1 Å were considered as common core, whereas the rest was defined as specific to Kif2C. To model these specific segments in Kif2C-ATP, Kif2C-ADP was used as a template. However, given significant rigid body moves occurring between kinesin-1 subdomains in ATP and ADP states, Kif2C-ADP was first superimposed on Kif2C-ATP using rigid subdomains. The subdomains were defined based on the comparison between the Kin1-ADP and Kin1-ATP structures, using the structural alignment algorithm in SwissPDBViewer, available in the SWISS-MODEL server (25). For each of the subdomains, Kif2C-ATP-tubulin model was morphed to match Kif2C-ADP conformations in the regions specific to Kif2C using the Rosetta v3.5 software (26) with the fast relax protocol under positions constraints derived from the Kif2C-ADP coordinates. 1000 models were generated, and the lowest energy model was selected for further optimization. The score of the Kif2C-ATP-tubulin model was further minimized following three relaxation steps without homology restraints and increasing gradually the degrees of freedom in the complex (300 structures generated at each step, keeping the lowest energy model (score 12) for the following steps): step 1: tubulin structure held fixed, whereas both side chains and backbone of Kif2C-ATP within 8 Å of the interface are free to move; step 2: tubulin side chains at interface were mobile, whereas Kif2C-ATP was the same as in step 1; and step 3: tubulin side chains at interface mobile, the whole structure of Kif2C-ATP is flexible. The quality of the resulting model is high as estimated by a Qmean4 score of −1.83 (Z score between 1 and 2) (27). It superimposes with kinesin-1 bound to ATP and tubulin with a root mean square deviation of 1.13 Å over 285 residues without making any steric clash at the interface with tubulin.
Electron Microscopy
Taxotere-stabilized microtubules and the depolymerization product by Kif2C proteins were visualized with EM after negative staining. Briefly, microtubules were incubated with Kif2C proteins, and then a 10-μl aliquot of the product was applied to a glow-discharged carbon-coated 400-mesh grid for 30 s, and stained with 0.75% (w/v) uranyl formate for 30 s. The grids were observed in a JEM-1230 electron microscope (JEOL) operated at 80 kV.
Results
The Interaction of the Kif2C KVD Motif with Tubulin Is Essential
Kinesin-13 proteins were identified based on a motor domain conserved in the kinesin superfamily (28). However, kinesin-13s have a clearly longer loop 2 that folds as a β-hairpin with a conserved KVD motif at its tip (9, 10). This KVD motif is necessary for microtubule depolymerization by kinesin-13s (9–11) and was shown to point toward the part of tubulin that is at a longitudinal intermolecular interface in microtubules (9, 11). Unfortunately, the detailed interaction of the KVD motif and the mechanism of its function are still unknown. To examine the role of this interaction, we first prevented it by producing a short loop 2 mutant of a monomeric construct, noted as Kif2C-(sN+M), which we have utilized for biochemical studies (8). In this Kif2C-(sN+M)-sL2 mutant, three residues flanking the KVD motif on each side were deleted, whereas the KVD motif was kept intact (Fig. 2A). To eliminate the concern of any overall structural change induced by the deletion of in total six residues, we first determined the crystal structure of Kif2C-(sN+M)-sL2 in the ADP state (Table 1). The overall structure is highly similar to the structure of the wild type protein (Protein Data Bank entry 2HEH; root mean square deviation, 0.56 Å over 314 superimposed Cαs, of 324) (Fig. 2B). The β-hairpin structure of loop 2 is conserved but shortened. In the mutant, as in the wild type protein, the hairpin that displays the KVD motif interacts similarly with the neck helix, but the KVD motif contacts neither the rest of the motor domain nor the neck helix.
FIGURE 2.

The functional role of the interaction of the KVD motif with tubulin. A, the sequences of loop 2 in Kif2C and in the Kif2C-(sN+M)-sL2 mutant. Three residues before the KVD motif and three residues after it were deleted in Kif2C-(sN+M)-sL2. B, the crystal structure of Kif2C-(sN+M)-sL2 (orange) was superimposed onto the structure of Kif2C-(sN+M) (Protein Data Bank entry 2HEH, shown in gray). This figure, as well as A–C in Fig. 3, was generated using PyMOL. C, the binding of AMPPNP-bound Kif2C-(sN+M)-sL2 to 1 μm microtubules was analyzed by comparing the supernatant and pellet fractions after ultracentrifugation. S, supernatant; P, pellet. D, turbidity traces of microtubule depolymerization process by 0.1 μm Kif2C-(sN+M) and its Kif2C-(sN+M)-sL2 mutant. The arrow indicates the time point of Kif2C addition. A.U., absorbance unit. E, microtubule (1.5 μm) depolymerization by Kif2C or its short loop 2 mutant. Following incubation and centrifugation, the result was analyzed by SDS-PAGE. F, tubulin (1 μm) aggregation by 1 μm Kif2C-(sN+M) or Kif2C-(sN+M)-sL2. After incubation with the Kif2C proteins in different nucleotide conditions and ultracentrifugation, the result was analyzed by SDS-PAGE.
Consistently, shortening loop 2 does not notably affect the basal ATPase of Kif2C. Neither does this affect the tubulin-stimulated ATPase or the strong microtubule binding property of this protein (Table 2 and Fig. 2C). In contrast, the microtubule-stimulated ATPase of Kif2C-(sN+M)-sL2 is decreased by 17-fold (to 0.21 s−1; Table 2), getting close to the level of tubulin-stimulated ATPase activity of Kif2C-(sN+M). Similarly, whereas the vinblastine-induced curved tubulin assemblies (29) stimulate the ATPase activity of Kif2C-(sN+M) notably more efficiently than unassembled tubulin, the same tubulin-vinblastine assemblies do not stimulate Kif2C-(sN+M)-sL2 ATPase strongly (Table 2). This clearly indicated that the ATPase stimulation by unassembled tubulin is mediated by the interaction through the core structure of the motor domain of Kif2C, whereas the stronger ATPase stimulation by microtubules (8) or tubulin-vinblastine requires an interaction of the KVD motif at the tubulin interdimer interface; this interaction is not fully established with a single tubulin.
TABLE 2.
ATPase activities of Loop2 mutants of Kif2C-(sN+M)
| ATPasea | Kif2C-(sN+M) |
Kif2C-(sN+M)-sL2 |
Kif2C-(sN+M)-DVK |
Kif2C-(sN+M)-DSK |
||||
|---|---|---|---|---|---|---|---|---|
| kcat | Km | kcat | Km | kcat | Km | kcat | Km | |
| s−1 | μm | s−1 | μm | s−1 | μm | s−1 | μm | |
| Microtubule-stimulated | 3.53 ± 0.09b | 1.13 ± 0.09b | 0.21 ± 0.01 | 1.30 ± 0.38 | 0.97 ± 0.02 | 1.05 ± 0.07 | 0.18 ± 0.01 | 0.60 ± 0.06 |
| Tubulin-vinblastine-stimulated | 0.62 ± 0.02 | 3.88 ± 0.28 | 0.069 ± 0.004 | 0.062 ± 0.016 | c | c | c | c |
| Tubulin-stimulatedd | 0.152 ± 0.007b | 0.08 ± 0.01b | 0.117 ± 0.002 | 0.18 ± 0.01 | 0.100 ± 0.002 | 0.058 ± 0.005 | 0.079 ± 0.003 | 0.047 ± 0.006 |
| Basal activity (s−1) | 0.008 ± 0.006a | 0.008 ± 0.002 | 0.010 ± 0.003 | 0.005 ± 0.004 | ||||
Furthermore, this KVD interaction is also required for the depolymerization activity, because the Kif2C-(sN+M)-sL2 mutant is unable to disassemble microtubules (Fig. 2, D and E). This mutant does not even form the pelletable curved tubulin assembly induced by Kif2C in the presence of AMPPNP (8, 11–13) (Fig. 2F). Therefore, the interaction of the KVD motif with tubulin seems also essential both for longitudinally linking and for bending tubulin before it is released from microtubule ends.
The Interaction of the KVD Motif Quantitatively Determines the Activities of Kif2C
To define more precisely the interaction of the KVD motif with tubulin, we built a model of Kif2C-ATP-tubulin based on the structure of human Kif2C (Protein Data Bank entry 2HEH) and on the recent structure of ATP-like state kinesin-1 bound to tubulin (16), taking advantage of the substantial Kif2C-kinesin-1 sequence similarity (Fig. 3A). In the working cycle of the monomeric Kif2C, the Kif2C-ATP-tubulin complex is formed from Kif2C-ATP upon tubulin binding (Fig. 1). Because the structure of isolated Kif2C is not changed between ADP and ATP states (9), the structural model of Kif2C-ATP-tubulin complex was compared with the structure of Kif2C-ADP (Protein Data Bank entry 2HEH) to identify the structural changes of Kif2C upon tubulin binding (Fig. 3B). As expected from the structural changes of kinesin-1 upon tubulin binding, the conformation of Kif2C in this model is reached first through an extension and repositioning of the tubulin-interacting helix α4 (Fig. 3A). When the tubulin-binding subdomains are superposed, it is clear that the P-loop and switch 1/2 subdomains of Kif2C motor domain undergo a substantial rotation upon tubulin binding (Fig. 3B). A direct consequence of this conformational change is the closure of the ATP binding site, in clear contrast to the situation in isolated Kif2C, where the nucleotide-binding switch 1 and switch 2 motifs are disordered (9). The structural model we made suggests that the ATP binding pocket of Kif2C is better formed upon binding to tubulin, which is probably related with the stimulation of Kif2C ATPase by tubulin and microtubules (8).
FIGURE 3.

Structural basis of the KVD interaction. A, structural model of Kif2C-ATP bound to tubulin. Kif2C is colored according to its three subdomains: tubulin-binding subdomain (green), P-loop subdomain (orange), and switch 1/2 subdomain (blue), defined as in kinesin-1 (17). The position of the neighboring β-tubulin (β-tubulin (2)) was modeled as in the curved assembly of two tubulins with a stathmin-like domain protein (Protein Data Bank entry 3RYC). Loop 2 of Kif2C points to and interacts with the tubulin interdimer interface. B, conformational change of Kif2C upon binding to tubulin. The tubulin-binding subdomain of Kif2C alone (Protein Data Bank entry 2HEH, shown in gray) was superimposed with the tubulin-binding subdomain of Kif2C in the structural model of Kif2C-ATP-tubulin (colored as in A), showing that the conformational change is reached through substantial rotation of P-loop subdomain (17°) and switch 1/2 subdomain (14°) of Kif2C. As a consequence, the KVD motif at the tip of loop 2 of Kif2C is well positioned to interact with the tubulin interdimer interface. C, close-up of the binding site of the Kif2C KVD motif on the tubulin interdimer interface. Color code is as in A. Black dashed lines indicate hydrogen bonds or salt bridges; red dashed lines indicate van der Waals contacts. D, SDS-PAGE analysis of the supernatant (S) and pellet (P) following ultracentrifugation of 2 μm Taxotere-stabilized microtubules after incubation with wild type Kif2C-(sN+M) or its DVK or DSK mutants. Note that the Kif2C mutants co-pellet with microtubules, indicating that their binding to microtubules is not impaired.
In this model of the Kif2C-ATP-tubulin complex, the KVD motif of Kif2C interacts directly with tubulin, as suggested by EM modeling (11) and originally in a Kif2C structure analysis (9). We found that the interaction site of the KVD motif on tubulin is comprised of three main components that each interacts with one of the three amino acids of the motif. The lysine is close to the side chain of Glu-434 in an acidic patch on α-tubulin surface; the valine points to a hydrophobic patch composed of the side chains of Tyr-262 and Val-435 and of the Cγ of Glu-434, in α-tubulin too. The aspartate points toward the solvent, but interestingly, when another tubulin is modeled into this complex as in a curved assembly (30), two basic residues Arg-401 and Lys-402 in the neighboring β-tubulin are close to the Asp residue of the KVD motif (Fig. 3, A and C).
To test this model, we interchanged the charged residues in the KVD motif of Kif2C-(sN+M) into DVK and then further mutated the valine into serine, a hydrophilic residue (leading to the DSK mutant). As expected, the microtubule depolymerization activity of the DVK mutant is remarkably decreased, a higher concentration being needed to depolymerize microtubules than with wild type Kif2C-(sN+M). The DSK mutant does not show microtubule depolymerization activity even at high concentrations (1 μm Kif2C mutant for 2 μm microtubules; Fig. 3D). Similarly, in comparison with wild type Kif2C-(sN+M), the DVK and DSK mutants showed stepwise decrease of their microtubule-stimulated ATPase (Table 2). These results suggest that the strength of the interaction of KVD with tubulin quantitatively determines the microtubule depolymerization activity and gates ATP hydrolysis in Kif2C. Therefore, these results support our model of the Kif2C-ATP-tubulin complex and the notion that the structural change in Kif2C-ATP upon tubulin binding is similar to that in a motile kinesin (17). Because the interaction modeled involves curved tubulin, it is therefore most relevant to what occurs at microtubule ends, which have been proposed to stimulate kinesin-13 ATPase most efficiently (31, 32).
ATP Hydrolysis in Kif2C Is Not Required for Tubulin Release
In the working cycle of Kif2C (Fig. 1), the formation of the competent Kif2C-ATP-tubulin complex is followed by both tubulin release from microtubule ends and ATP hydrolysis in Kif2C. We then sought to clarify whether these two events are necessarily coupled or can be uncoupled. To do so, we mutated Kif2C-(sN+M) to dissect the sequential events that precede the ATP hydrolysis step. Recent structural studies of conventional kinesins have revealed that the subdomain movements required to initiate a step are triggered by multiple interactions of ATP with the universally conserved nucleotide-binding motifs, in particular with switch 2 (16, 17). In kinesin-1, mutations of the Gly and Glu residues in the highly conserved switch 2 motif (DXXGXE) trap the ATP-binding kinesin in pre-conformational change and post-conformational change states, respectively (33). We have previously found that the G495A mutant of Kif2C-(sN+M), which is the pre-conformational change-mimicking mutant, does not hydrolyze ATP nor depolymerize microtubules (8). Here, we mutated the switch 2 Glu residue of Kif2C-(sN+M), Glu-497, to Ala and compared the activity of this post-conformational change-mimicking mutant with that of the G495A one.
As expected, Kif2C-(sN+M)-E497A is nearly completely ATPase-deficient, this activity being detectable only on the time scale of days (data not shown). However, in contrast to the fully inactive G495A mutant (8), the E497A mutant definitely depolymerizes microtubules, an activity that gets more obvious at higher E497A concentrations (Fig. 4A). When the depolymerization of 2 μm microtubules was monitored by measuring the turbidity, 1 μm E497A mutant yielded a clear turbidity decrease (Fig. 4B), suggesting a fast disassembly of microtubules. Interestingly, this activity is not shown even by AMPPNP-bound wild type Kif2C-(sN+M), in either turbidity or centrifugation assays (Fig. 4, B and C). Therefore, the postconformational change-mimicking E497A mutant of Kif2C seems more competent than AMPPNP-bound wild type Kif2C-(sN+M) and than the G495A mutant. The results with the E497A mutant demonstrate that the ATPase and microtubule depolymerization activities of Kif2C are dissociable, and that the ATP hydrolysis step is not directly required for microtubule depolymerization.
FIGURE 4.
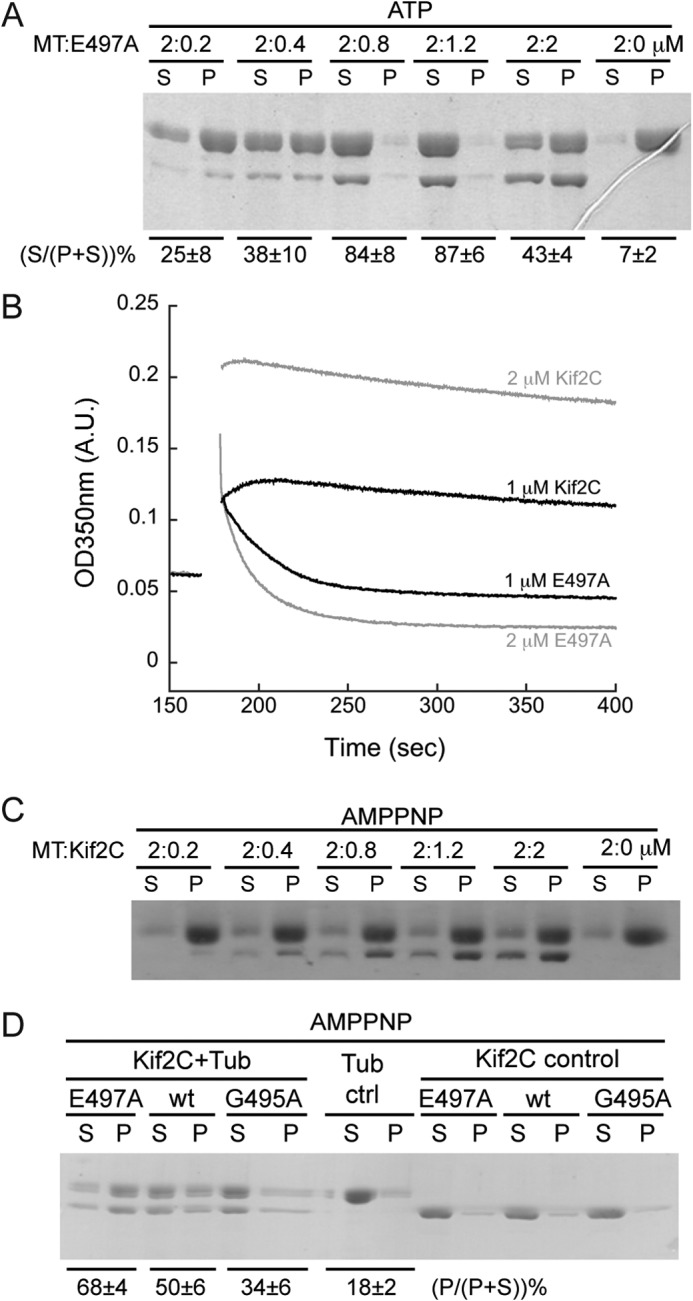
The microtubule depolymerization activity of the ATPase-deficient post-conformational change-mimicking Kif2C-(sN+M)-E497A mutant. A, SDS-PAGE analysis of the supernatant (S) and pellet (P) following ultracentrifugation of 2 μm Taxotere-stabilized microtubules after incubation with various concentrations of Kif2C-(sN+M)-E497A mutant. The relative amount of tubulin band in each lane was quantified with ImageJ, and the percentage of tubulin in the supernatant in each group is given below the gel (average ± S.D., n = 2). B, turbidity traces of 2 μm microtubules depolymerized by 1 μm (black) or 2 μm (gray) Kif2C-(sN+M) or its Kif2C-(sN+M)-E497A mutant in AMPPNP conditions. After adding such high concentrations of Kif2C protein, the turbidity of microtubules had a remarkable, Kif2C concentration-dependent, increase, probably because of the binding of Kif2C onto the microtubule surface. In the case of the E497A mutant, the depolymerization is demonstrated by the quick decrease of turbidity, in clear contrast to the rather stable turbidity in the presence of wild type Kif2C. C, SDS-PAGE analysis of the supernatant (S) and pellet (P) following ultracentrifugation of 2 μm microtubules after incubation with various concentrations of Kif2C-(sN+M) in the presence of 1 mm AMPPNP. D, aggregation of tubulin (0.5 μm) by Kif2C-(sN+M) or its E497A or G495A mutants (0.5 μm) in AMPPNP condition. Following incubation and ultracentrifugation (75,000 rpm), the result was checked by SDS-PAGE. The percentage of tubulin in the pellet in each group is given below the gel (average ± S.D., n = 3).
Based on findings on kinesin-1 (33), the expected main structural difference between the G495A and E497A mutants of Kif2C-(sN+M) is that they undergo different extents of conformational change upon tubulin binding. We looked for further evidence to support this proposal. A necessary consequence of the Kif2C structural change is that the KVD motif is in a position to contact a tubulin longitudinally associated to the tubulin heterodimer to which the kinesin is bound (Fig. 3A). This is similar to what has been seen at medium resolution (11 Å) by electron microscopy of complexes of tubulin with another kinesin-13 (11). This would favor tubulin-tubulin associations in the presence of Kif2C-(sN+M) and lead to the formation of pelletable tubulin assemblies. To check for this, we compared the tubulin pelleting ability of the E497A mutant with those of the wild type Kif2C-(sN+M) and of the G495A mutant in the AMPPNP state. As expected, the post-conformational change-mimicking E497A mutant exhibited a stronger tubulin pelleting ability than wild type Kif2C-(sN+M), whereas the G495A mutant showed a weaker ability (Fig. 4D). Taken together, these results clarified that tubulin release from microtubule ends does not require ATP hydrolysis in Kif2C but does require the proper conformational state of tubulin-bound Kif2C-ATP, which is reached in the E497A mutant but not fully reached in AMPPNP-bound wild type Kif2C nor in the G495A mutant.
The Unexpected 1:2 Stoichiometry of Microtubule Depolymerization by Kif2C
We also noted from the centrifugation result of microtubule depolymerization that, as the Kif2C-(sN+M)-E497A concentration became higher than half the concentration of microtubular tubulin, tubulin remained in the pellet rather than in the supernatant (Fig. 4A). Intriguingly, the quick decrease of the turbidity of 2 μm microtubules depolymerized by 2 μm Kif2C-(sN+M)-E497A argues for the occurrence of depolymerization in this stoichiometric condition (Fig. 4B). The apparent inconsistency between the results from these two methods may come from the property of kinesin-13s to stabilize curved tubulin assemblies that stay in the pellet upon centrifugation (Fig. 4D). Through direct observation of the reaction product by using negative staining electron microscopy, we confirmed that microtubules were depolymerized by Kif2C-(sN+M)-E497A in both cases, but into different tubulin assemblies (Fig. 5, A–C). When 2 μm microtubules were depolymerized by 0.5 μm Kif2C-(sN+M)-E497A, a lot of short tubulin oligomers of ∼16-nm length were observed, implying that each one comprises two tubulin heterodimers (34) (Fig. 5F). However, when a 2 μm Kif2C-(sN+M)-E497A concentration was used, more numerous longer tubulin oligomers or tubulin rings were obtained (Fig. 5, C and F). These observations may be rationalized in terms of our structural model of the tubulin-Kif2C interaction. In that model, Kif2C interacts with two tubulin heterodimers (Fig. 3, A and B). As a consequence, when the concentration of the E497A mutant is limiting, the most likely microtubule depolymerization product is the ternary 1:2 E497A-tubulin complex. In that complex, the two tubulin heterodimers associate through their longitudinal interaction, as usual, but they are also linked through the interaction of the KVD motif of Kif2C molecule. This ternary complex is soluble, but when the amount of the E497A mutant increases, tubulin gets saturated by E497A and longitudinally linked into a pelletable assembly (Fig. 4A).
FIGURE 5.

The 1:2 binding stoichiometry between the E497A Kif2C-(sN+M) and tubulin. A–C, negative staining EM images of microtubules (A), 2 μm microtubules depolymerized by 0.5 μm Kif2C-(sN+M)-E497A (B), and 2 μm microtubules depolymerized by 2 μm Kif2C-(sN+M)-E497A (C) in the presence of ATP for 15 min. D and E, negative staining EM images of 2 μm microtubules treated with 1 μm wild type Kif2C-(sN+M) in the presence of 1 mm AMPPNP for 1 min (D) and 15 min (E). F, length distribution of tubulin oligomers observed in negative staining EM images. For each condition, images of two different fields were used for statistics. In the case of wild type Kif2C-(sN+M), statistics are from the 15-min incubation time. G, aggregation of tubulin:Kif2C-(sN+M)-E497A as a function of the concentration ratio. The result was analyzed by SDS-PAGE following ultracentrifugation. H, same experiment as in G, but with wild type Kif2C-(sN+M).
By using negative staining electron microscopy, we also confirmed that microtubules are not depolymerized into individual tubulin or short oligomers of two tubulin heterodimers by AMPPNP-bound wild type Kif2C-(sN+M) (Fig. 5D), in agreement with the results of turbidity and centrifugation assays (Fig. 4, B and C). Tubulin rings and curls are only formed following a longer incubation (Fig. 5E), as previously observed with other kinesin-13s (11–13, 35, 36). These EM results, together with the clearly different kinetics of the E497A mutant and AMPPNP-bound wild type Kif2C (Fig. 4B), demonstrate that these two proteins disassemble microtubules through different mechanisms: fast release of two-tubulin assemblies from microtubule ends by the E497A mutant or slow peeling of protofilaments by AMPPNP-bound wild type Kif2C. The rapid depolymerization by the E497A mutant suggests that its mechanism is more similar to that of tubulin removal from microtubules during catalytic depolymerization by kinesin-13s than to the mechanism of tubulin removal by Kif2C-AMPPNP. The less efficient disassembly by AMPPNP-bound wild type Kif2C is likely because of its incomplete conformational change upon binding to tubulin. Fully establishing this will require the crystal structures of Kif2C and its E497A mutant in complex with tubulin.
Furthermore, the property of releasing two tubulins each time by the E497A mutant of Kif2C suggests that, when given sufficient tubulin, the E497A mutant preferably binds to two tubulins. This is indeed what we observed when Kif2C-(sN+M)-E497A was incubated with nonmicrotubular tubulin. Although the E497A mutant heavily pellets tubulin at a 1:1 Kif2C:tubulin ratio, one additional molar equivalent of tubulin largely prevents pelleting (Fig. 5G). Because this ratio-dependent tubulin-pelleting property is also applicable to wild type Kif2C-(sN+M) (Fig. 5H), the 1:2 stoichiometry is likely to be an intrinsic property of Kif2C and not caused by the E497A mutation.
Discussion
Since the discovery of kinesin-13 proteins (28), substantial efforts have been devoted to decipher their biological function and working mechanisms (7). The minimal functional domain of kinesin-13s has been narrowed down to the conserved motor domain together with its N-terminal short neck region (8), and the nucleotide cycle of kinesin-13s has been largely clarified (8, 32). Here, we further investigated the coupling between the ATP hydrolysis in Kif2C and tubulin release from microtubule ends both from biochemical and structural points of view, taking advantage of recent progress in structural studies of conventional kinesins (16, 17). Our results provide new insights into the microtubule depolymerization mechanism of Kif2C.
Our structural model of the Kif2C-ATP-tubulin complex, supported by mutagenesis studies (Figs. 2 and 3), suggests that Kif2C undergoes conformational changes upon microtubule binding and that the interaction of the Kif2C KVD motif with tubulin plays an essential role. Because both the ATPase and microtubule depolymerizing activities of Kif2C have parallel sensitivities to the order of the amino acids in the KVD motif (Fig. 3) and to the length of loop 2 at the tip of which the KVD motif is located (Fig. 2), this conformational change is most likely involved in both activities. However, these two activities of Kif2C are not necessarily interdependent and can be decoupled. The biochemical study of the depolymerization competent Kif2C-(sN+M)-E497A mutant (Fig. 4) demonstrates that the conformational change in Kif2C-ATP upon binding to microtubule ends is sufficient to drive tubulin release from microtubules but that the ATP cleavage step, which is ordinarily coupled to this conformational change, is not required for microtubule depolymerization. Rather, because the affinity of Kif2C for tubulin is remarkably decreased in ADP-Pi or ADP state (8), ATP hydrolysis is required to dissociate Kif2C from tubulin and perform additional reaction cycles (Fig. 6).
FIGURE 6.
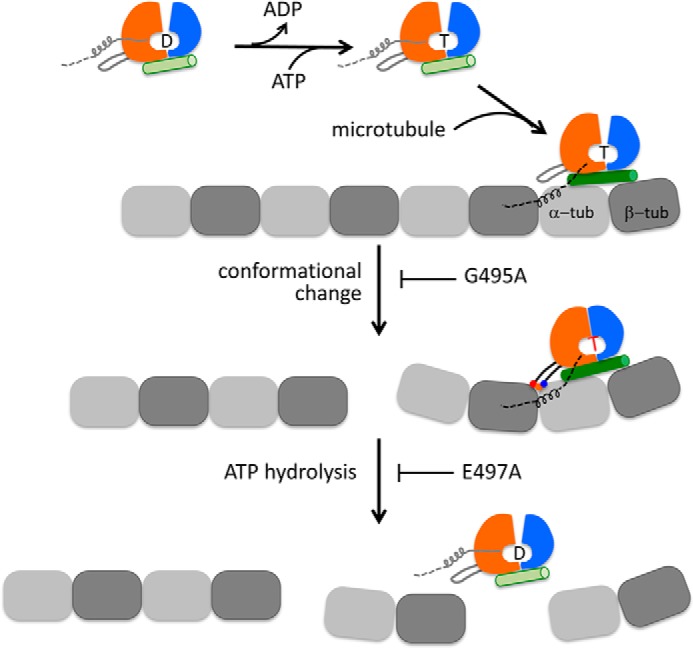
Model of microtubule depolymerization by Kif2C and other kinesin-13s. A microtubule is represented as a single protofilament; the motor domain of Kif2C is illustrated as three subdomains that are colored as in Fig. 3A. The letter between the P loop subdomain and switch 1/2 subdomain indicates the nucleotide state of Kif2C: D, ADP; T, ATP. Loop 2 is highlighted as an appendage of the P loop subdomain. In Kif2C molecules in solution, the neck region, which is indispensable for microtubule depolymerization, is shown as a four-turn helix attached to the P loop subdomain, oriented as in the crystal structure of human Kif2C (in Fig. 3B; Protein Data Bank entry 2HEH). In solution, Kif2C releases ADP autonomously and then binds ATP (8). Kif2C-ATP binds directly to the end of microtubule, which induces the elongation of tubulin-interacting helix α4 (shown as a green cylinder) in the tubulin-binding subdomain of Kif2C. The neck region may contribute to Kif2C binding at microtubule ends by interacting with tubulin. Because the precise binding site of neck is still unknown, it is arbitrarily positioned and shown as a dashed line. Kif2C-ATP bound at a microtubule end undergoes conformational changes, for which the interaction of the KVD motif (shown in red, orange, and blue triple dots) with the tubulin interdimer longitudinal interface is essential. The G495A mutation of Kif2C prevents the conformational change from occurring. After the conformational change of Kif2C-ATP bound at a microtubule end, Kif2C reaches the depolymerization competent state and releases the terminal two tubulins in a ternary complex. In addition, ATP hydrolysis in Kif2C takes place following the conformational change. This is blocked by the E497A mutation. Kif2C dissociates from tubulin only after ATP hydrolysis; it is then available for the next round of depolymerization.
The essential role of the KVD interaction for Kif2C ATPase activity explains the much stronger ATPase stimulation by microtubules than by tubulin (20-fold difference) (8). The ratio of the stimulation factors by microtubules and tubulin is substantially lowered in the Kif2C variant with a shortened loop 2 that we designed (a 2-fold difference in this case). Because shortening loop 2 does not change the structure of the nucleotide binding site and because the effect of loop 2 length is mostly apparent upon interaction of Kif2C with microtubules, this effect is most likely due to a specific interaction of the KVD motif with microtubules. Microtubules differ from isolated tubulin by the structure of tubulin (straight versus curved) (37) and by the contacts, in particular longitudinal, that each tubulin establishes with other heterodimers. To distinguish between these two contributions to the different KVD interactions with tubulin and microtubules, we also measured Kif2C ATPase stimulated by a second type of tubulin assembly, induced by vinblastine. This assembly combines tubulin structure as in solution with one of the microtubule features: longitudinal contacts between heterodimers (29). Kif2C ATPase stimulated by tubulin-vinblastine assemblies is intermediate between ATPase stimulated by tubulin and by microtubules (Table 2). The difference between tubulin and tubulin-vinblastine stimulations means that tubulins longitudinally contacting the one bound to the Kif2C motor core modulate ATPase stimulation. The difference between tubulin-vinblastine and microtubule stimulations means that curvature is also important. We also note that the Kif2C-(sN+M)-sL2 ATPase stimulation is moderate whatever tubulin assembly it is bound to (Table 2). Therefore, the Kif2C structural change upon binding to a tubulin assembly, as reflected by its ATPase and by its microtubule depolymerization activity (see below), depends on the interaction of the KVD motif with longitudinally associated tubulins, as in the model we built.
The essential role of the KVD interaction for the microtubule depolymerization activity of Kif2C explains the microtubule depolymerization properties of Kif2C. Electron microscopy of negatively stained samples demonstrates that numerous individual complexes of the Kif2C-(sN+M)-E497A mutant with tubulin released from microtubules have a 1:2 stoichiometry, at least for a low Kif2C:tubulin ratio (Fig. 5B). It has also been observed in an EM study that when a kinesin-13 construct comprising the neck and the motor domain is added to dolastatin-stabilized tubulin rings, most rings are decorated with the kinesin at a 1:2 kinesin:tubulin stoichiometry (38). The question that arises then is how these 1:2 complexes form. Because of its three-pronged interaction (Fig. 3A) (11), Kif2C stabilizes a curved conformation of tubulin that is not accommodated in a microtubule and therefore released as a Kif2C-tubulin complex. Because of the interactions mediated by the KVD motif, the first tubulin drags a second, longitudinally interacting, heterodimer (Fig. 6). Another possibility is that the released 1:1 Kif2C-tubulin complex may associate in solution with tubulin at the critical concentration to form a curved ternary complex. Importantly, because the KVD binding site is at the α-tubulin longitudinal interface contacting β-tubulin from another heterodimer, it is not completely formed when Kif2C binds to the tubulin at the (−) end of a microtubule. Because Kif2C depolymerizes microtubules from both ends (31), it likely does so by binding to the penultimate tubulin at the (−) end, which allows the KVD interaction to be fully established and leads to the release of the terminal two tubulins (Fig. 6).
It is also worth pointing out that the microtubule depolymerization activity of most kinesin-13 proteins requires the presence of the subfamily-specific neck region N-terminal to the motor domain (8, 39). Nevertheless, we have previously found that the neck-less motor domain of Kif2C has the full microtubule-stimulated ATPase of the depolymerization-competent Kif2C-(sN+M) (8), implying that Kif2C-Motor undergoes a complete conformational change as Kif2C-(sN+M), but that this is not utilized to release tubulin from microtubule ends. Considering that Kif2C-(sN+M)-E497A releases tubulin from microtubule ends without hydrolyzing ATP (Fig. 4), it appears that the two events following the conformational change of Kif2C upon binding to microtubule ends, i.e. tubulin release and ATP hydrolysis, are mutually independent of each other. These two events just occur coincidentally after forming the competent Kif2C-ATP-tubulin complex, to finish a round of microtubule depolymerization (tubulin release) and to make Kif2C available for a next round of work (ATP hydrolysis).
To sum up, by combining a structural model and a mutagenesis study of the minimal functional domain of Kif2C, we demonstrated that the KVD motif, a structural element unique to the kinesin-13 subfamily, plays a central role in mediating the conformational change of Kif2C-ATP upon binding to microtubule ends, which in turn determines the microtubule depolymerization and ATPase activities of Kif2C. Furthermore, we have shown that tubulin release from microtubule ends does not require ATP hydrolysis in Kif2C but does require the Kif2C-ATP conformational change upon binding to microtubule ends. Although the crystal structure of tubulin-bound Kif2C is needed to further our understanding of microtubule depolymerization, Kif2C and probably all the kinesin-13 subfamily members most likely share a conformational change similar to that of motile kinesins. This conformational change, either because of its detailed nature or because of the sequence elements that are specific to kinesin-13s, is adapted to microtubule depolymerization.
Author Contributions
C. W., M. K., and B. G. designed the study and wrote the paper. W. W. and T. S. performed the biochemical characterization of Kif2C mutants. W. W. crystallized and determined the structure of Kif2C-(sN+M)-sL2. R. G. built the structural model of Kif2C-ATP-tubulin complex. F. Z. and H. K. carried out the negative staining EM observations. Y. L. studied the depolymerization activity of wild type Kif2C-AMPPNP. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank Prof. Jian Zhu and all the staff at the EM facility of Tongji University and Mi Cao from National Center for Protein Science Shanghai for help with negative staining experiments. We thank the staff at the IMAGIF crystallization platforms (CNRS, Gif-sur-Yvette, France). Diffraction data were collected at the SOLEIL synchrotron (PX1 and PX2 beam lines). We thank the machine and beam line groups for making these experiments possible.
This work was supported by National Science Foundation of China Grants 30700125 and 31370771 and Specialized Research Fund for the Doctoral Program of Ministry of Education of China Grant 20130072110031 (to C. W.) and by Agence Nationale de la Recherche Grant ANR-12-BSV8-0002-01 (to B. G.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 4Y05) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AMPPNP
- adenosine 5′-(β,γ-imido)triphosphate.
References
- 1. Vale R. D. (2003) The molecular motor toolbox for intracellular transport. Cell 112, 467–480 [DOI] [PubMed] [Google Scholar]
- 2. Desai A., Verma S., Mitchison T. J., Walczak C. E. (1999) Kin I kinesins are microtubule-destabilizing enzymes. Cell 96, 69–78 [DOI] [PubMed] [Google Scholar]
- 3. Kline-Smith S. L., Khodjakov A., Hergert P., Walczak C. E. (2004) Depletion of centromeric MCAK leads to chromosome congression and segregation defects due to improper kinetochore attachments. Mol. Biol. Cell 15, 1146–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Homma N., Takei Y., Tanaka Y., Nakata T., Terada S., Kikkawa M., Noda Y., Hirokawa N. (2003) Kinesin superfamily protein 2A (KIF2A) functions in suppression of collateral branch extension. Cell 114, 229–239 [DOI] [PubMed] [Google Scholar]
- 5. Kobayashi T., Tsang W. Y., Li J., Lane W., Dynlacht B. D. (2011) Centriolar kinesin Kif24 interacts with CP110 to remodel microtubules and regulate ciliogenesis. Cell 145, 914–925 [DOI] [PubMed] [Google Scholar]
- 6. Sanhaji M., Friel C. T., Wordeman L., Louwen F., Yuan J. (2011) Mitotic centromere-associated kinesin (MCAK): a potential cancer drug target. Oncotarget 2, 935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Walczak C. E., Gayek S., Ohi R. (2013) Microtubule-depolymerizing kinesins. Annu. Rev. Cell Dev. Biol. 29, 417–441 [DOI] [PubMed] [Google Scholar]
- 8. Wang W., Jiang Q., Argentini M., Cornu D., Gigant B., Knossow M., Wang C. (2012) Kif2C minimal functional domain has unusual nucleotide binding properties that are adapted to microtubule depolymerization. J. Biol. Chem. 287, 15143–15153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ogawa T., Nitta R., Okada Y., Hirokawa N. (2004) A common mechanism for microtubule destabilizers-M type kinesins stabilize curling of the protofilament using the class-specific neck and loops. Cell 116, 591–602 [DOI] [PubMed] [Google Scholar]
- 10. Shipley K., Hekmat-Nejad M., Turner J., Moores C., Anderson R., Milligan R., Sakowicz R., Fletterick R. (2004) Structure of a kinesin microtubule depolymerization machine. EMBO J. 23, 1422–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Asenjo A. B., Chatterjee C., Tan D., DePaoli V., Rice W. J., Diaz-Avalos R., Silvestry M., Sosa H. (2013) Structural model for tubulin recognition and deformation by kinesin-13 microtubule depolymerases. Cell Rep. 3, 759–768 [DOI] [PubMed] [Google Scholar]
- 12. Moores C. A., Yu M., Guo J., Beraud C., Sakowicz R., Milligan R. A. (2002) A mechanism for microtubule depolymerization by KinI kinesins. Mol. Cell 9, 903–909 [DOI] [PubMed] [Google Scholar]
- 13. Tan D., Rice W. J., Sosa H. (2008) Structure of the kinesin 13-microtubule ring complex. Structure 16, 1732–1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagenbach M., Domnitz S., Wordeman L., Cooper J. (2008) A kinesin-13 mutant catalytically depolymerizes microtubules in ADP. J. Cell Biol. 183, 617–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cross R. A. (2004) The kinetic mechanism of kinesin. Trends Biochem. Sci 29, 301–309 [DOI] [PubMed] [Google Scholar]
- 16. Gigant B., Wang W., Dreier B., Jiang Q., Pecqueur L., Plückthun A., Wang C., Knossow M. (2013) Structure of a kinesin-tubulin complex and implications for kinesin motility. Nat. Struct. Mol. Biol. 20, 1001–1007 [DOI] [PubMed] [Google Scholar]
- 17. Cao L., Wang W., Jiang Q., Wang C., Knossow M., Gigant B. (2014) The structure of apo-kinesin bound to tubulin links the nucleotide cycle to movement. Nat. Commun. 5, 5364. [DOI] [PubMed] [Google Scholar]
- 18. Dagenbach E. M., Endow S. A. (2004) A new kinesin tree. J. Cell Sci. 117, 3–7 [DOI] [PubMed] [Google Scholar]
- 19. Castoldi M., Popov A. V. (2003) Purification of brain tubulin through two cycles of polymerization-depolymerization in a high-molarity buffer. Protein Expr. Purif. 32, 83–88 [DOI] [PubMed] [Google Scholar]
- 20. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Smart O., Vonrhein C., Womack T. O. (2011) BUSTER. 2.10.0 Ed., Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Biasini M., Bienert S., Waterhouse A., Arnold K., Studer G., Schmidt T., Kiefer F., Cassarino T. G., Bertoni M., Bordoli L., Schwede T. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252-W258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leaver-Fay A., Tyka M., Lewis S. M., Lange O. F., Thompson J., Jacak R., Kaufman K., Renfrew P. D., Smith C. A., Sheffler W., Davis I. W., Cooper S., Treuille A., Mandell D. J., Richter F., Ban Y. E., Fleishman S. J., Corn J. E., Kim D. E., Lyskov S., Berrondo M., Mentzer S., Popović Z., Havranek J. J., Karanicolas J., Das R., Meiler J., Kortemme T., Gray J. J., Kuhlman B., Baker D., Bradley P. (2011) ROSETTA3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 487, 545–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Benkert P., Biasini M., Schwede T. (2011) Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aizawa H., Sekine Y., Takemura R., Zhang Z., Nangaku M., Hirokawa N. (1992) Kinesin family in murine central nervous system. J. Cell Biol. 119, 1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gigant B., Wang C., Ravelli R. B., Roussi F., Steinmetz M. O., Curmi P. A., Sobel A., Knossow M. (2005) Structural basis for the regulation of tubulin by vinblastine. Nature 435, 519–522 [DOI] [PubMed] [Google Scholar]
- 30. Nawrotek A., Knossow M., Gigant B. (2011) The determinants that govern microtubule assembly from the atomic structure of GTP-tubulin. J. Mol. Biol. 412, 35–42 [DOI] [PubMed] [Google Scholar]
- 31. Hunter A. W., Caplow M., Coy D. L., Hancock W. O., Diez S., Wordeman L., Howard J. (2003) The kinesin-related protein MCAK is a microtubule depolymerase that forms an ATP-hydrolyzing complex at microtubule ends. Mol Cell 11, 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Friel C. T., Howard J. (2011) The kinesin-13 MCAK has an unconventional ATPase cycle adapted for microtubule depolymerization. EMBO J. 30, 3928–3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rice S., Lin A. W., Safer D., Hart C. L., Naber N., Carragher B. O., Cain S. M., Pechatnikova E., Wilson-Kubalek E. M., Whittaker M., Pate E., Cooke R., Taylor E. W., Milligan R. A., Vale R. D. (1999) A structural change in the kinesin motor protein that drives motility. Nature 402, 778–784 [DOI] [PubMed] [Google Scholar]
- 34. Steinmetz M. O., Kammerer R. A., Jahnke W., Goldie K. N., Lustig A., van Oostrum J. (2000) Op18/stathmin caps a kinked protofilament-like tubulin tetramer. EMBO J. 19, 572–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moores C. A., Cooper J., Wagenbach M., Ovechkina Y., Wordeman L., Milligan R. A. (2006) The role of the kinesin-13 neck in microtubule depolymerization. Cell Cycle 5, 1812–1815 [DOI] [PubMed] [Google Scholar]
- 36. Tan D., Asenjo A. B., Mennella V., Sharp D. J., Sosa H. (2006) Kinesin-13s form rings around microtubules. J. Cell Biol. 175, 25–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ravelli R. B., Gigant B., Curmi P. A., Jourdain I., Lachkar S., Sobel A., Knossow M. (2004) Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 428, 198–202 [DOI] [PubMed] [Google Scholar]
- 38. Mulder A. M., Glavis-Bloom A., Moores C. A., Wagenbach M., Carragher B., Wordeman L., Milligan R. A. (2009) A new model for binding of kinesin 13 to curved microtubule protofilaments. J. Cell Biol. 185, 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maney T., Wagenbach M., Wordeman L. (2001) Molecular dissection of the microtubule depolymerizing activity of mitotic centromere-associated kinesin. J. Biol. Chem. 276, 34753–34758 [DOI] [PubMed] [Google Scholar]