

Background: Endothelial cells line blood vessels, forming a monolayer that acts as a regulated barrier. Actin helps create and maintain the junctions between endothelial cells.
Results: Endothelial N-WASP organizes VE-cadherin and actin at cellular junctions, which alters barrier integrity.
Conclusion: N-WASP regulates monolayer integrity by remodeling cell-cell contacts.
Significance: Our results show how endothelial cells use one actin regulator to modulate the endothelial barrier.
Keywords: actin, Arp2/3 complex, cytoskeleton, endothelial cell, endothelial dysfunction, N-WASP, barrier, monolayer
Abstract
Endothelial cells (ECs) form a monolayer that serves as a barrier between the blood and the underlying tissue. ECs tightly regulate their cell-cell junctions, controlling the passage of soluble materials and immune cells across the monolayer barrier. We studied the role of N-WASP, a key regulator of Arp2/3 complex and actin assembly, in EC monolayers. We report that N-WASP regulates endothelial monolayer integrity by affecting the organization of cell junctions. Depletion of N-WASP resulted in an increase in transendothelial electrical resistance, a measure of monolayer integrity. N-WASP depletion increased the width of cell-cell junctions and altered the organization of F-actin and VE-cadherin at junctions. N-WASP was not present at cell-cell junctions in monolayers under resting conditions, but it was recruited following treatment with sphingosine-1-phosphate. Taken together, our results reveal a novel role for N-WASP in remodeling EC junctions, which is critical for monolayer integrity and function.
Introduction
Endothelial cells (ECs)2 line the vasculature, and they regulate the trafficking of fluid, metabolites, and cells between the blood and tissue. Endothelial cell-cell junctions are highly dynamic and tightly regulated (1–3); dysfunctional junctions can be associated with human disease (4–7). Actin filaments are organized as a loose circumferential band near endothelial cell junctions. The actin network and cell junctions are organized in a coordinated manner to regulate a variety of physiological and pathological processes, including transendothelial migration by immune and cancer cells.
Endothelial cells are highly dynamic at their junctions, frequently sending out lamellipodial protrusions to establish and maintain the cell-cell contacts that hold the monolayer together (8, 9). Endothelial junctions consist of adherens junctions and tight junctions, each with their associated molecules. The junctions between endothelial cells are not spatially segregated into zones as found in epithelial monolayers (10); the junctional molecules of ECs are dispersed throughout the junctional area (11). A large body of work has documented several molecules involved in endothelial junctional integrity and how these molecules contribute to overall monolayer barrier function (1, 11–13).
How actin regulatory proteins contribute to the organization and integrity of endothelial junctions is not well understood. Arp2/3 complex is a major nucleator for actin filament assembly in cells. Arp2/3 complex alone has low actin nucleation activity, and cells utilize several regulator proteins to target and activate the complex. N-WASP, one of these proteins, is localized to cell membranes and functions there to promote actin assembly and actin-based cellular functions, such as endocytosis (14, 15). The adherens junction component, p120-catenin, interacts with N-WASP to provide a link between the actin cytoskeleton and cell-cell contacts (16).
In this study, we report that N-WASP is involved in endothelial monolayer integrity and in proper organization of actin and VE-cadherin at cell-cell junctions. Overall, we propose that N-WASP coordinates actin with endothelial junctions to regulate endothelial monolayer integrity.
Experimental Procedures
Recombinant DNA and siRNAs
Two pools of N-WASP-targeting siRNA oligonucleotide duplexes were obtained from Dharmacon, GE Life Sciences (Lafayette, CO). The pools were transfected into ECs using Lipofectamine RNAiMAX transfection reagent (Invitrogen). The first pool (ON-TARGETplus WASL siRNA, catalog no. L-006444-00-0005) contained the following sequences: 07, CAGCAGAUCGGAACUGUAU; 08, UAGAGAGGGUGCUCAGCUA; 09, GGUGUUGCUUGUCUUGUUA; and 10, CCAGAAAUCACAACAAAUA. At 48, 72, and 96 h, N-WASP protein levels were not decreased by immunoblot. However, the second pool of oligonucleotides (ON-TARGETplus WASL siRNA, catalog no. L-006444-01-0005) was effective at decreasing N-WASP protein levels. The second pool was not a standard Dharmacon product; Dharmacon created it for us because the first pool was not effective. The second pool contained the following siRNA sequences: 11, UCUGAAAGCCUUCCGUAUA; 12, GGCAAAGGUGUGCGCUAAA, 13 - UGUAAAGGUUUGUCGUAAU; and 14, AGGAAUUGUGGGUGCAUUA. As a control, we used a nontargeting siRNA (catalog no. D-001810-01-05) with the following sequence: UGGUUUACAUGUCGACUAA.
To test for off target effects, we expressed full-length N-WASP from a plasmid to attempt to rescue the effects of siRNA-induced knockdown. The N-WASP coding region from a cDNA, BC052955 from Origene (Rockville, MD) was cloned into pAcGFP, producing GFP fused to the N terminus of N-WASP. The GFP-N-WASP region from this plasmid was subcloned into the lentiviral expression vector pBOB-GFP, a gift of Dr. George Bloom (University of Virginia), which removed the GFP-encoding portion of the vector. Five codon-neutral point mutations were created in the N-WASP coding region to prevent targeting by siRNA 14, as follows: AGGtATaGTcGGaGCcTTA (changes in lowercase). siRNAs 11, 12, and 13 targeted the 3′-untranslated region of N-WASP, which was not present in the expression rescue construct. Lentivirus carrying the GFP-N-WASP-pBOB plasmid was generated as described (8).
Cells
Human dermal microvascular endothelial cells from a neonate were purchased from Lonza (Basel, Switzerland). Cells were maintained in EGM-2MV medium (Lonza) and used between passages 2 and 12.
Reagents and Antibodies
Sphingosine-1-phosphate (S1P; Sigma-Aldrich) was incubated with cells at 0.5 μm for 5 min. Latrunculin A (ENZO Life Sciences, Farmingdale, NY) was incubated with cells at 1 μm for 30 min.
Anti-VE-cadherin antibodies included a mouse monoclonal antibody (clone 55-7H1; BD Biosciences, San Jose, CA) and a rabbit polyclonal antibody (antibody 2158; Cell Signaling, Danvers, MA). N-WASP antibodies included two rabbit polyclonal antibodies: 30D10 from Cell Signaling and H-100 from Santa Cruz Biotechnology (Dallas, TX). As controls for loading, we probed for GAPDH with mouse monoclonal 6C5 (Abcam, Cambridge, MA) and for actin with mouse monoclonal C4 (MAB1501; EMD Millipore, Billerica, MA). Fluorescent secondary antibodies and phalloidin, conjugated with Alexa dyes, were obtained from Molecular Probes (Life Technologies).
Immunofluorescence
Cells were fixed using 2× fixation buffer (5% paraformaldehyde in PIPES buffer) (8) for 10 min at 37 °C. Cells were permeabilized using 0.1% Triton X-100 in PIPES buffer for 10 min at room temperature. Cells were treated with blocking buffer (3% BSA in PBS) for >1 h, followed by primary antibody for 2 h and secondary antibody for 1 h. Antibodies were diluted in blocking buffer and incubated at room temperature. Phalloidin staining was performed by adding fluorescent phalloidin to the secondary antibody solution.
Light Microscopy
For immunofluorescence, a set of z-stack images were collected using either a laser scanning confocal microscope (Zeiss 510 LSM) with a 40× 1.2 NA water immersion objective or a Yokogawa spinning-disk confocal inverted microscope (Olympus IX-81) with a 40× 0.75 NA objective and a Hamamatsu ORCA-Flash 4.0 CMOS camera. Wide field fluorescence images were collected using an inverted microscope (Olympus IX-81) with a 40× 0.75 NA objective, a mercury lamp light source and a Hamamatsu EM-CCD video camera. Image analysis was performed with ImageJ (17).
Transendothelial Resistance Measurements
Human dermal microvascular endothelial cells were plated on fibronectin-coated Transwells (8) and grown to confluence, confirmed by phase contrast microscopy. ECs were treated with siRNAs or lentivirus prior to plating on Transwells. Transwell electrical resistance measurements were made with an Endohm-6 electrode adapter or chopstick electrodes with an EVOM-2 meter (World Precision Instruments, Sarasota, FL). Transendothelial resistance (TER) values were calculated by subtracting a measurement with no endothelial cells from the sample measurement and then multiplying by the surface area of the Transwell. All TER values were normalized to control TER readings for each day and then averaged.
Results
Depletion of N-WASP Enhances the Integrity of the Endothelial Barrier
To determine whether N-WASP plays a role in endothelial monolayer formation and barrier integrity, we depleted N-WASP from human dermal microvascular endothelial cells in monolayers and measured TER. We pooled three siRNAs: 11, 12, and 13, to produce a large (∼90%) depletion of N-WASP by immunoblots (Fig. 1A). The pool of three siRNAs caused TER values of endothelial monolayers to increase (Fig. 1B); this effect was consistent and reproducible. Moreover, the increase in TER was suppressed by expression of siRNA-resistant N-WASP (Fig. 1, A and B), documenting that the phenotype was due specifically to the loss of N-WASP. N-WASP depletion also caused TER values to increase for endothelial monolayers treated with TNF-α as a mimic of inflammatory conditions; this effect was also rescued by N-WASP expression (Fig. 1C).
FIGURE 1.

Depletion of N-WASP increases the electrical resistance of the endothelial monolayer. A, immunoblots of endothelial cells, probed for N-WASP and GAPDH. On the left, cells were treated with control or knockdown (N-WASP targeting) siRNAs. On the right, cells treated with control siRNA are compared with cells treated with knockdown siRNAs. The N-WASP-depleted cells expressed either GFP (knockdown) or GFP-N-WASP (rescue). B, TER values for control, knockdown, and rescue endothelial monolayers, normalized within 1 day of experiments. The difference between control and knockdown is statistically significant (p < 0.01), and the difference between control and rescue is not (p = 0.71). For control and knockdown data, n = 15 values, with measurements over 4 days. For rescue data, n = 6 values, with measurements on 2 days. C, the effect of addition of TNFα. For both untreated and TNFα-treated monolayers, the differences between control and knockdown are statistically significant (p ≤ 0.01), and the differences between control and rescue are not (p ≥ 0.3). n = 9 measurements on 1 day.
Our observation of increased TER following N-WASP depletion conflicts with a previous study, which found the opposite effect: decreased TER following N-WASP depletion in endothelial cells (16). Our studies reveal that the previous result was due to an off target effect of one siRNA in the pool that was used. Indeed, we observed decreased TER in our initial experiments when using a pool of four siRNAs (siRNAs 11–14) from Dharmacon. This pool depleted N-WASP from ECs (Fig. 2A), as shown by immunoblot, and this pool of four siRNAs caused a decrease in TER (Fig. 2B), with and without treatment by TNF-α (Fig. 2B). In positive control experiments, latrunculin A and EGTA, which disrupt F-actin and cadherin-based cell junctions, respectively, lowered TER values to even greater extents (Fig. 2B).
FIGURE 2.

Effects of four siRNA oligonucleotides on N-WASP levels and TER in EC monolayers. A, immunoblot showing depletion of N-WASP by pools of four (siRNAs 11–14) or three (siRNAs 11–13) siRNAs and by each of the individual siRNAs, 11–14. The graph below shows the quantitated levels of protein based on densitometry of the immunoblot, normalized to control. n = 2 immunoblots. B, effect of monolayer disrupting agents and N-WASP knockdown by the pool of four siRNA oligonucleotides, 11–14, on TER of EC monolayers. TER values are normalized and plotted as an index, with the value for latrunculin A (Lat A) defined as 0 and the untreated control value defined as 1. The decrease caused by the siRNA pool of four occurs in the absence or presence of TNFα treatment. C, effect of the four siRNAs on TER. TER is decreased by the pool of four siRNAs and by siRNA 14 alone. The other three siRNAs (siRNAs 11, 12, and 13) cause modest increases in TER. Asterisks denote statistical significance with p < 0.001. The values for n are as follows: control, 20; pool with siRNAs 11–14, 9; siRNAs 11 and 12, 15; siRNA 13, 13; and siRNA 14, 16. The values are derived from experiments on 3–5 different days. D, expression of siRNA-resistant full-length N-WASP does not rescue the TER phenotype caused by the pool of four siRNAs or by the individual siRNA 14. n = 9 values, collected on 3 days. The values for samples expressing N-WASP did not increase toward the values for controls.
We tested each individual siRNA oligonucleotide from the pool of four (Fig. 2A). Oligonucleotides 11, 12, and 13 decreased the level of N-WASP, but oligonucleotide 14 had only a small effect, based on immunoblots (Fig. 2A). Oligonucleotides 11, 12, and 13 caused the TER value to increase or not change (Fig. 2C), and only oligonucleotide 14 caused TER values to decrease (Fig. 2C). Thus, decreased TER did not correlate with decreased N-WASP, comparing the effects of the four siRNAs.
To test for off target effects, we expressed siRNA-resistant N-WASP in cells treated with each of the individual siRNAs and with the pool of four siRNAs. For siRNA 14, TER levels did not return to normal when N-WASP was expressed (Fig. 2D). A similar result was seen for the pool of four oligonucleotides (Fig. 2D). This failure to rescue was in striking contrast to the rescue we observed for the other three oligonucleotides, 11, 12 and 13, without oligonucleotide 14, described above. Therefore, we conclude that the decrease in TER caused by the pool of four oligonucleotides was caused by siRNA 14 alone and that this phenotype is due to an off target effect, not to depletion of N-WASP.
We conclude that depletion of N-WASP from endothelial monolayers enhances the integrity of the monolayer barrier, as measured by electrical resistance. Subsequent experiments utilized the pool of three siRNAs, which were specific for N-WASP.
N-WASP Controls Endothelial Junction Width and Organization
We asked whether the increase in TER caused by depletion of N-WASP was associated with changes in the junctions between ECs in the monolayer. Endothelial monolayers express the tight junctional protein, ZO-1, which contributes to the barrier function of the monolayer. We first examined the localization and organization of ZO-1 in control and N-WASP-depleted endothelial monolayers by immunofluorescence. In control cells, ZO-1 localized to cell-cell contacts (Fig. 3, upper panels), F-actin co-localized with ZO-1 in junctional structures that were oriented perpendicular to the line of cell-cell contact (Fig. 3, arrows). In contrast, F-actin did not co-localize with ZO-1 along junctions running parallel to the line of cell-cell contact (Fig. 3, arrowheads). We did not observe any changes in ZO-1 localization at cell-cell contacts in N-WASP-depleted monolayers (Fig. 3, lower panels). Therefore, the increase in TER resulting from N-WASP depletion does not appear to result from changes in the organization of the tight junctional protein ZO-1.
FIGURE 3.

ZO-1 recruitment and organization is not altered by depletion of N-WASP. Control and N-WASP-depleted monolayers were fixed and stained for ZO-1 and F-actin. Scale bar, 50 μm. Merge shows co-localization of ZO-1 (red) and F-actin (green) at junctional structures aligned perpendicular to the line of cell-cell contact (arrows) and separation of ZO-1 and F-actin at junctional structures aligned parallel to cell-cell contacts (arrowheads). Scale bar for merge, 25 μm. high mag, high magnification.
Next we examined adherens junctions between the endothelial cells, using immunofluorescence localization of VE-cadherin. N-WASP depletion increased the apparent width of the adherens junctions of the endothelial monolayers, based on anti-VE-cadherin staining. In views of the monolayer from above, in x-y projection planes, the average width of N-WASP-depleted adherens junctions was 4.35 ± 0.24 μm (median ± S.E., n = 126; Fig. 4B) compared with 2.93 ± 0.14 μm (n = 109) for control cell adherens junctions (Fig. 4A). This difference was statistically significant (p < 0.001). Expression of siRNA-resistant N-WASP in N-WASP-depleted cells restored the width of the junctions to nearly normal (2.87 μm ± 0.16 μm, n = 105; Fig. 4C).
FIGURE 4.

Top row, depletion of N-WASP increases the apparent width of cell-cell junctions in x-y projection images in the upper row of images, based on anti-VE-cadherin staining. Middle row, orthogonal views of x-z and y-z image planes. Bottom row, representative line scans from the vertical (y-z) lines. A, control endothelial monolayers have thin bands of VE-cadherin at cell-cell junctions. Scale bar, 50 μm. B, N-WASP-depleted monolayers show thick bands of VE-cadherin at junctions in the x-y plane image. However, the band is thin in the x-z and y-z image planes. Scale bar, 10 μm. C, expression of full-length N-WASP restores VE-cadherin junctional bandwidth to that of control monolayers.
To assess the height of the cell junctions in the z axis perpendicular to the monolayer, we collected a set of images from different focal planes and prepared orthogonal views (x-z and y-z) along lines that were drawn parallel and perpendicular to the adherens junctions in the x-y plane. These images reveal that the thickness of the junctional band in the z axis did not increase when N-WASP was depleted (Fig. 4). To account for this result, we envision that membrane protrusions from adjacent cells overlap each other by a substantial distance, with VE-cadherin at the contact surface, which causes the junction to appear wider in the x-y projection. Therefore, we propose that the role of N-WASP is to control the overlapping protrusions of adjacent cells and thereby promote the ultimate organization of VE-cadherin into a thin band at cell-cell junctions, characteristic of mature endothelial junctions. We also propose that this increased overlap in N-WASP-depleted monolayers results in a higher TER value.
To investigate the molecular role of N-WASP in more detail, we examined the organization of VE-cadherin and F-actin at cell-cell junctions. In control endothelial monolayers, VE-cadherin and F-actin co-localized at many places along contacts between cells (Fig. 5, arrows). In N-WASP-depleted monolayers, with thickened cell junctions, actin and VE-cadherin were largely not co-localized (Fig. 5, arrows); VE-cadherin was organized as thick networks devoid of actin. Expression of siRNA-resistant N-WASP restored the co-localization and organization of VE-cadherin and actin to patterns similar to those of control monolayers (Fig. 5, arrows). Therefore, we conclude that N-WASP contributes to the organization of VE-cadherin and actin networks at endothelial cell-cell junctions.
FIGURE 5.

N-WASP controls the organization of F-actin and VE-cadherin at endothelial cell junctions. In views of x-y projection planes, control endothelial monolayers show co-localization of F-actin (red) and VE-cadherin (green) at cell-cell junctions (arrows). N-WASP depletion results in thickened bands of VE-cadherin at cell junctions, which do not contain F-actin (arrows). Expression of full-length N-WASP restores the width of the cell junctions, defined by anti-VE-cadherin, along with the co-localization of F-actin (arrows). Scale bar, 10 μm.
Depletion of N-WASP Does Not Alter Endothelial Actin Superassemblies
Actin filaments in ECs are organized into two major sets of fibers composed of many overlapping actin filaments. Circumferential fibers are loosely packed, and they lie close to cell-cell junctions. Longitudinal fibers are more densely packed, and they are located in the interior of the cell, with their long axis often oriented parallel to the long axis of the cell. The number and staining intensity of longitudinal fibers increases in the presence of inflammatory mediators, such as TNF-α (8, 18–20).
First, we used the pool of four siRNAs, which included the off-target siRNA 14. We observed an increase in the number and staining intensity of longitudinal actin fibers (Fig. 6, upper panels), similar to the previous report (16). In contrast, when we used the pool of three siRNAs (11, 12, and 13, excluding 14), we did not observe this change in actin longitudinal fibers (Fig. 6, lower panels). In addition, the increase in longitudinal stress fibers caused by the pool of four siRNAs was not rescued by expression of N-WASP (data not shown), confirming that this phenotype is also an off target effect caused by siRNA 14. Depletion of N-WASP by the pool of three specific siRNAs also did not affect circumferential actin fibers. We conclude that N-WASP does not influence the organization of actin fiber superassemblies in ECs.
FIGURE 6.

Actin fiber phenotypes caused by the pool of four siRNAs are off-target effects not due to loss of N-WASP. In the upper panels, ECs treated with the pool of four siRNAs show increases in the number of longitudinal actin fibers, which are found in the cell interior and are oriented parallel to the long axis of the cells. In control cells, in contrast, actin is predominantly organized in a circumferential pattern at the periphery. In the lower panels, this change in actin organization is not seen when using only the three siRNAs (siRNAs 11, 12, and 13) that specifically target N-WASP. Scale bar, 20 μm.
N-WASP Localization to Cell-Cell Junctions
To investigate how N-WASP might be controlling cell-cell junctions, we localized N-WASP in endothelial monolayers by immunofluorescence. In resting monolayers fixed and stained with anti-N-WASP and anti-VE-cadherin, we did not observe accumulation of N-WASP at cell-cell junctions (Fig. 7A).
FIGURE 7.

Localization of N-WASP in endothelial monolayers by immunofluorescence. Endothelial monolayers were fixed and stained with anti-N-WASP, anti-VE-cadherin, and fluorescent phalloidin. A, in resting endothelium, N-WASP does not localize to cell-cell junctions. Scale bar, 100 μm. B, treatment with S1P (0.1 μm, 5 min) rapidly recruits N-WASP to cell junctions. Merge shows N-WASP (green) and VE-cadherin (red) localization in untreated and S1P-treated monolayers. Scale bar, 25 μm.
Endothelial cells treated with S1P, a biologically active lysophospholipid that regulates the immune and vascular systems (21), display rapid recruitment of a number of regulators of actin and Arp2/3 complex to the cell periphery, in association with increases in dynamic F-actin and lamellipodia-based ruffling of cell edges. In addition, S1P treatment causes recruitment of adherens junction components VE-cadherin, α-catenin, and β-catenin (22, 23). Together, these molecular recruitments lead to enhanced assembly of cell-cell junctions, and they strengthen the monolayer barrier (23).
We treated our endothelial monolayers with S1P, and we observed rapid recruitment of N-WASP to cell-cell junctions, in association with VE-cadherin at many junctions (Fig. 7B). N-WASP recruitment was transient, peaking at ∼5 min and dissipating by ∼10 min after S1P addition (data not shown).
Because N-WASP was recruited to junctions by S1P treatment, we asked whether N-WASP depletion had an effect on S1P-generated ruffling and recruitment of the Arp2/3 activator cortactin. We found no effect of N-WASP depletion on the response of the endothelial cell monolayer to S1P (Fig. 8). We conclude that N-WASP is recruited to endothelial cell-cell junctions to assist in remodeling cell junctions following S1P treatment, but that N-WASP is not involved in producing the initial response to S1P.
FIGURE 8.
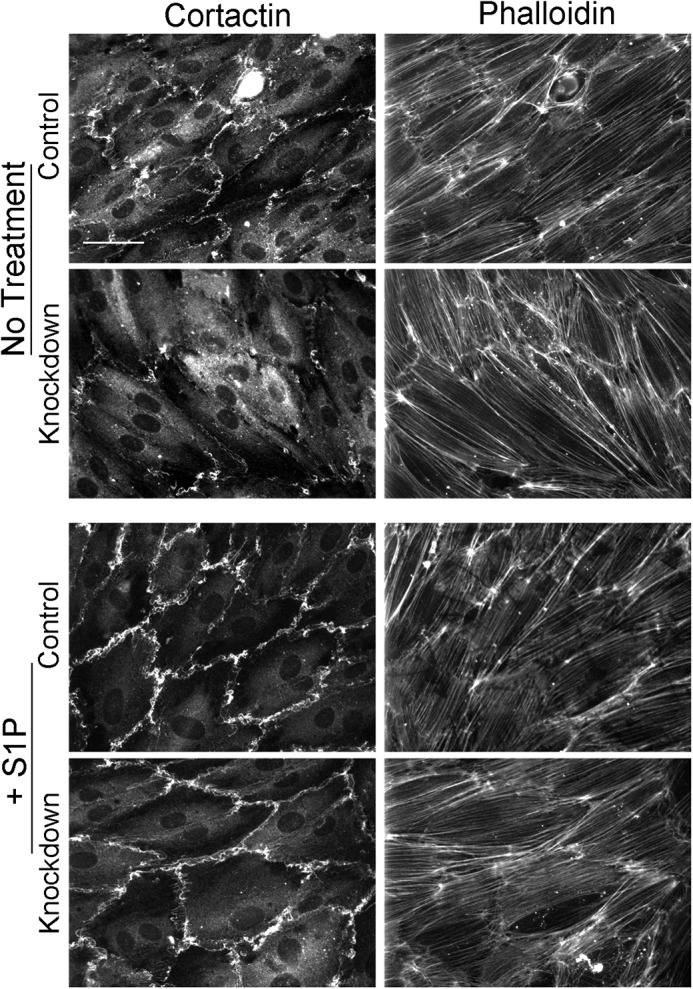
N-WASP depletion does not affect the response of endothelial cell monolayer to S1P. Monolayers were treated with 0.1 μm S1P for 5 min, followed by fixation and staining for cortactin and F-actin. S1P induced the formation of junctional ruffles and caused cortactin recruitment to ruffles in similar ways in control and N-WASP-depleted monolayers. Scale bar, 100 μm.
Discussion
The most important finding of our study is that N-WASP depletion enhances endothelial monolayer integrity, as assessed by electrical resistance. This increase in monolayer integrity is associated with increased VE-cadherin at cell-cell contacts and altered organization of F-actin and VE-cadherin networks at adherens junctions.
A previous report reached the opposite conclusion, that depletion of N-WASP leads to loss of endothelial monolayer integrity as measured by transendothelial electrical resistance, consistent with the finding of gaps between endothelial cells in the monolayer (16). We initially replicated these results and observed similar phenotypes using a pool of four siRNA oligonucleotides. However, we found that these phenotypes were off target effects of one of the four siRNAs (siRNA 14) in the pool, because the phenotypes were not rescued by expression of siRNA-resistant N-WASP.
In striking contrast, when siRNA 14 was removed from the pool, we observed the opposite phenotype: N-WASP depletion caused an increase in monolayer integrity as measured by transendothelial electrical resistance. Consistent with that finding, and in contrast to the previous report, we also did not observe gaps between cells in the monolayer. The phenotype of increased electrical resistance was specific for N-WASP, revealed by rescue with expression of N-WASP. Furthermore, the pool of three specific siRNA oligonucleotides (lacking siRNA 14) produced a substantial decrease in the level of N-WASP protein, whereas siRNA 14 alone produced little to no effect on the level of N-WASP. Thus, we conclude that the phenotypes of gaps between cells and decreased electrical resistance was an off target effect of siRNA 14, one that is not caused by depletion of N-WASP. BLAST searches with the sequence of siRNA 14 against the human genome database revealed a 17- of 18-base pair match to an uncharacterized long noncoding RNA, LOC101927657, which we speculate may be the target responsible for the cellular phenotypes caused by siRNA 14.
Upon close inspection, we observed that endothelial junctions were altered in N-WASP-depleted monolayers, with loss of coordination of actin and VE-cadherin at cell-cell junctions. The altered junctions likely account for the increase in TER we measured. Endothelial junctions are dynamic, with neighboring cells constantly ruffling and sending out lamellipodia to create and maintain cell contacts (8). We envision that these lamellipodia create regions of overlap between the plasma membrane surfaces of neighboring cells. When cell surface junctional molecules contact one another, they bind and create adhesions. The adhesions eventually coalesce into mature junctions, where overlap between cells is minimal. We propose that N-WASP contributes to the last step of creating mature junctions, perhaps by promoting turnover of junctional molecules or retracting the lamellipodial protrusions.
Our results with N-WASP depletion are important and striking because loss of function phenotypes for many actin regulatory proteins, including regulators of Arp2/3 complex, feature defects in endothelial monolayer integrity (8, 13, 24). However, biochemical and cell biological studies reveal that different Arp2/3 regulators, present at the same time and location in the cell, have different functions (25, 26). Arp2/3 complex has two binding sites for acidic/DDW motif regulators, with distinct affinities (27, 28). Activation of Arp2/3 complex has been proposed to involve the replacement of one regulator by another (29).
A role for N-WASP in junctional organization of F-actin and VE-cadherin has also been reported in epithelial cells (30); the effects of N-WASP depletion in that study were similar to the ones we observed here in endothelial cells. In epithelial monolayers, depletion of N-WASP produced distorted junctions, with E-cadherin networks that had little co-localization with F-actin, compared with control cells (30). Therefore, we conclude that N-WASP promotes co-localization and coordinate organization of F-actin and VE-cadherin at cell-cell junctions.
Endothelial-cell Arp2/3 complex is important for the formation of lamellipodia at the cell periphery, which promote junction formation and maintenance (8, 9). Arp2/3 complex regulators, such as N-WASP, may be recruited to junctions to recruit and/or activate Arp2/3 complex, which would promote the assembly of branched networks of actin filaments. WAVE2, a different regulator of Arp2/3 complex, does not co-localize with VE-cadherin at mature endothelial cell-cell junctions (8), which suggests that it has an early role in junction formation. In this report, we observed that N-WASP is not recruited to any large extent to endothelial junctions under basal resting conditions, unlike what is observed in epithelial monolayers (31). Therefore, we propose that N-WASP is recruited transiently to endothelial junctions when they are undergoing remodeling.
Additional support for the idea that N-WASP is recruited to actively remodeling junctions was provided by our result that S1P treatment caused recruitment of N-WASP to cell-cell contacts. Recruitment and activation of N-WASP may be downstream of FAK (32), which is recruited to S1P-induced lamellipodia (33). In addition, N-WASP is known to interact with p120-catenin at endothelial adherens junctions (16), providing a molecular link between adherens junctions and actin assembly. In turn, p120-catenin controls VE-cadherin levels at endothelial cell junctions by effects on endocytosis (34–36). We speculate that N-WASP is activated at sites of junctional remodeling to regulate the dynamic assembly of actin and junction components.
Author Contributions
O. L. M. designed and performed the experiments described in this manuscript. J. K. assisted in setting up and performing portions of the experiments (Figs. 1, 3, and 8). J. L. cared for the cells and assisted in setting up the experiments carried out in this manuscript. J. A. C. oversaw the experiments and was a mentor for the project. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM 38542. The authors declare that they have no conflicts of interest with the contents of this article.
- EC
- endothelial cell
- S1P
- sphingosine-1-phosphate
- TER
- transendothelial resistance.
References
- 1. Dejana E., Orsenigo F., Lampugnani M. G. (2008) The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122 [DOI] [PubMed] [Google Scholar]
- 2. Rigor R. R., Shen Q., Pivetti C. D., Wu M. H., Yuan S. Y. (2013) Myosin light chain kinase signaling in endothelial barrier dysfunction. Med. Res. Rev. 33, 911–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wojciak-Stothard B., Ridley A. J. (2002) Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 39, 187–199 [DOI] [PubMed] [Google Scholar]
- 4. Weber C., Fraemohs L., Dejana E. (2007) The role of junctional adhesion molecules in vascular inflammation. Nat. Rev. Immunol. 7, 467–477 [DOI] [PubMed] [Google Scholar]
- 5. Bakker W., Eringa E. C., Sipkema P., van Hinsbergh V. W. (2009) Endothelial dysfunction and diabetes: roles of hyperglycemia, impaired insulin signaling and obesity. Cell Tissue Res. 335, 165–189 [DOI] [PubMed] [Google Scholar]
- 6. Kumar P., Shen Q., Pivetti C. D., Lee E. S., Wu M. H., Yuan S. Y. (2009) Molecular mechanisms of endothelial hyperpermeability: implications in inflammation. Expert Rev. Mol. Med. 11, e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sima A. V., Stancu C. S., Simionescu M. (2009) Vascular endothelium in atherosclerosis. Cell Tissue Res. 335, 191–203 [DOI] [PubMed] [Google Scholar]
- 8. Mooren O. L., Li J., Nawas J., Cooper J. A. (2014) Endothelial cells use dynamic actin to facilitate lymphocyte transendothelial migration and maintain the monolayer barrier. Mol. Biol. Cell 25, 4115–4129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Abu Taha A., Taha M., Seebach J., Schnittler H. J. (2014) Arp2/3-mediated junction-associated lamellipodia control VE-cadherin-based cell junction dynamics and maintain monolayer integrity. Mol. Biol. Cell 25, 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Citalán-Madrid A. F., García-Ponce A., Vargas-Robles H., Betanzos A., Schnoor M. (2013) Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 1, e26938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Aghajanian A., Wittchen E. S., Allingham M. J., Garrett T. A., Burridge K. (2008) Endothelial cell junctions and the regulation of vascular permeability and leukocyte transmigration. J. Thromb. Haemost. 6, 1453–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dudek S. M., Garcia J. G. (2001) Cytoskeletal regulation of pulmonary vascular permeability. J. Appl. Physiol. 91, 1487–1500 [DOI] [PubMed] [Google Scholar]
- 13. Spindler V., Schlegel N., Waschke J. (2010) Role of GTPases in control of microvascular permeability. Cardiovasc. Res. 87, 243–253 [DOI] [PubMed] [Google Scholar]
- 14. Padrick S. B., Rosen M. K. (2010) Physical mechanisms of signal integration by WASP family proteins. Annu. Rev. Biochem. 79, 707–735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Derivery E., Gautreau A. (2010) Generation of branched actin networks: assembly and regulation of the N-WASP and WAVE molecular machines. Bioessays 32, 119–131 [DOI] [PubMed] [Google Scholar]
- 16. Rajput C., Kini V., Smith M., Yazbeck P., Chavez A., Schmidt T., Zhang W., Knezevic N., Komarova Y., Mehta D. (2013) Neural wiskott-aldrich syndrome protein (N-WASP)-mediated p120-catenin interaction with Arp2-actin complex stabilizes endothelial adherens junctions. J. Biol. Chem. 288, 4241–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stolpen A. H., Guinan E. C., Fiers W., Pober J. S. (1986) Recombinant tumor necrosis factor and immune interferon act singly and in combination to reorganize human vascular endothelial cell monolayers. Am. J. Pathol. 123, 16–24 [PMC free article] [PubMed] [Google Scholar]
- 19. Blum M. S., Toninelli E., Anderson J. M., Balda M. S., Zhou J., O'Donnell L., Pardi R., Bender J. R. (1997) Cytoskeletal rearrangement mediates human microvascular endothelial tight junction modulation by cytokines. Am. J. Physiol. 273, H286–H294 [DOI] [PubMed] [Google Scholar]
- 20. Clark P. R., Manes T. D., Pober J. S., Kluger M. S. (2007) Increased ICAM-1 expression causes endothelial cell leakiness, cytoskeletal reorganization and junctional alterations. J. Invest Dermatol. 127, 762–774 [DOI] [PubMed] [Google Scholar]
- 21. Rosen H., Goetzl E. J. (2005) Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat. Rev. Immunol. 5, 560–570 [DOI] [PubMed] [Google Scholar]
- 22. Lee M. J., Thangada S., Claffey K. P., Ancellin N., Liu C. H., Kluk M., Volpi M., Sha'afi R. I., Hla T. (1999) Vascular endothelial cell adherens junction assembly and morphogenesis induced by sphingosine-1-phosphate. Cell 99, 301–312 [DOI] [PubMed] [Google Scholar]
- 23. Sun X., Shikata Y., Wang L., Ohmori K., Watanabe N., Wada J., Shikata K., Birukov K. G., Makino H., Jacobson J. R., Dudek S. M., Garcia J. G. (2009) Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc. Res. 77, 304–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schnoor M., Lai F. P., Zarbock A., Kläver R., Polaschegg C., Schulte D., Weich H. A., Oelkers J. M., Rottner K., Vestweber D. (2011) Cortactin deficiency is associated with reduced neutrophil recruitment but increased vascular permeability in vivo. J. Exp. Med. 208, 1721–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Campellone K. G., Welch M. D. (2010) A nucleator arms race: cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pollard T. D., Cooper J. A. (2009) Actin, a central player in cell shape and movement. Science 326, 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ti S. C., Jurgenson C. T., Nolen B. J., Pollard T. D. (2011) Structural and biochemical characterization of two binding sites for nucleation-promoting factor WASP-VCA on Arp2/3 complex. Proc. Natl. Acad. Sci. U.S.A. 108, E463–E471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Padrick S. B., Doolittle L. K., Brautigam C. A., King D. S., Rosen M. K. (2011) Arp2/3 complex is bound and activated by two WASP proteins. Proc. Natl. Acad. Sci. U.S.A. 108, E472–E479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helgeson L. A., Nolen B. J. (2013) Mechanism of synergistic activation of Arp2/3 complex by cortactin and N-WASP. Elife 2, e00884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Otani T., Ichii T., Aono S., Takeichi M. (2006) Cdc42 GET Tuba regulates the junctional configuration of simple epithelial cells. J. Cell Biol. 175, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kovacs E. M., Verma S., Ali R. G., Ratheesh A., Hamilton N. A., Akhmanova A., Yap A. S. (2011) N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat. Cell Biol. 13, 934–943 [DOI] [PubMed] [Google Scholar]
- 32. Wu X., Suetsugu S., Cooper L. A., Takenawa T., Guan J. L. (2004) Focal adhesion kinase regulation of N-WASP subcellular localization and function. J. Biol. Chem. 279, 9565–9576 [DOI] [PubMed] [Google Scholar]
- 33. Belvitch P., Dudek S. M. (2012) Role of FAK in S1P-regulated endothelial permeability. Microvasc. Res. 83, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xiao K., Allison D. F., Buckley K. M., Kottke M. D., Vincent P. A., Faundez V., Kowalczyk A. P. (2003) Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J. Cell Biol. 163, 535–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xiao K., Garner J., Buckley K. M., Vincent P. A., Chiasson C. M., Dejana E., Faundez V., Kowalczyk A. P. (2005) p120-catenin regulates clathrin-dependent endocytosis of VE-cadherin. Mol. Biol. Cell 16, 5141–5151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Alcaide P., Newton G., Auerbach S., Sehrawat S., Mayadas T. N., Golan D. E., Yacono P., Vincent P., Kowalczyk A., Luscinskas F. W. (2008) p120-catenin regulates leukocyte transmigration through an effect on VE-cadherin phosphorylation. Blood 112, 2770–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]