

Abstract
Background and Objective
Circadian rhythms may influence the pharmacokinetics of drugs. This study aimed to elucidate whether the pharmacokinetics of the orally administered drug sunitinib are subject to circadian variation.
Methods
We performed studies in male FVB-mice aged 8–12 weeks, treated with single-dose sunitinib at six dosing times. Plasma and tissue samples were obtained for pharmacokinetic analysis and to monitor messenger RNA (mRNA) expression of metabolizing enzymes and drug transporters. A prospective randomized crossover study was performed in which patients took sunitinib once daily at 8 a.m., 1 p.m., and 6 p.m at three subsequent courses. Patients were blindly randomized into two groups, which determined the sequence of the sunitinib dosing time. The primary endpoint in both studies was the difference in plasma area under the concentration–time curve (AUC) of sunitinib and its active metabolite SU12662 between dosing times.
Results
Sunitinib and SU12662 plasma AUC in mice followed an ~12-h rhythm as a function of administration time (p ≤ 0.04). The combined AUC from time zero to 10 h (AUC10) was 14–27 % higher when sunitinib was administered at 4 a.m. and 4 p.m. than at 8 a.m. and 8 p.m. Twenty-four-hour rhythms were seen in the mRNA levels of drug transporters and metabolizing enzymes. In 12 patients, sunitinib trough concentrations (C trough) were higher when the drug was taken at 1 p.m. or 6 p.m. than when taken at 8 a.m. (C trough-1 p.m. 66.0 ng/mL; C trough-6 p.m. 58.9 ng/mL; C trough-8 a.m. 50.7 ng/mL; p = 0.006). The AUC was not significantly different between dosing times.
Conclusions
Our results indicate that sunitinib pharmacokinetics follow an ~12-h rhythm in mice. In humans, morning dosing resulted in lower C trough values, probably resulting from differences in elimination. This can have implications for therapeutic drug monitoring.
Electronic supplementary material
The online version of this article (doi:10.1007/s40262-015-0239-5) contains supplementary material, which is available to authorized users.
Key Points
| Sunitinib is known for its narrow therapeutic window and wide inter-patient variability in drug exposure, which in part may be explained by a within-patient variability in drug exposure, possibly due to daily variations in absorption, distribution, metabolism, and excretion of sunitinib. |
| In this study, we showed that the area under the concentration–time curve of sunitinib and its active metabolite SU12662 follows a 12-h rhythm in mice. Patients had lower trough concentrations (C trough) of both sunitinib and SU12662 when the drug was administered in the morning, rather than intake at later times of the day. |
| This is particularly interesting, since therapeutic drug monitoring (TDM) is currently being suggested as a step forward in the individualization of sunitinib treatment. In TDM, the drug dose may be increased or decreased based on C trough values to aim for improved survival. Therefore, it is crucial to take the administration time into account to prevent erroneous dose escalations or reductions. |
Introduction
Sunitinib (Sutent®; Pfizer Labs, Division of Pfizer Inc, New York, NY, USA) is a multi-targeted tyrosine kinase inhibitor (TKI) that is registered for the treatment of advanced clear cell renal cell carcinoma (RCC), imatinib-resistant or intolerant gastrointestinal stromal tumors (GIST), and pancreatic neuro-endocrine tumors (pNET) [1–5]. There is a large inter-patient variability in plasma concentrations of sunitinib, which may be due to patient non-compliance, drug–drug interactions [cytochrome P450 (CYP) 3A4-modulating drugs], and inter-patient variability in pharmacokinetics [6]. Besides inter-patient variability in drug exposure, variability within a patient due to intra-patient variations in pharmacokinetic processes may also exist.
After oral intake, sunitinib is absorbed from the gastrointestinal tract, resulting in peak plasma concentrations 6–12 h after administration [1]. Food has no significant effect on the exposure to sunitinib [1, 7]. In the liver, sunitinib is mainly metabolized by the CYP3A4 enzyme to its primary active metabolite SU12662, which is further converted by CYP3A4 into several inactive compounds. Both sunitinib and SU12662 are eliminated from the body via the bile in the feces through efflux transporters in the liver, namely the adenosine triphosphate (ATP)-binding cassette sub-family B member 1 (ABCB1) and sub-family G member 2 (ABCG2) [8].
Enzymes and transporters involved in the pharmacokinetics of drugs, such as CYP3A4 and ABCB1, have time-dependent variations in expression which may have a profound effect on the exposure to several drugs [9, 10]. These variations may change the efficacy and/or toxicity profile of drugs. This was previously shown in patients treated with tacrolimus, a drug frequently used to prevent organ rejection after transplantation, where morning administration resulted in higher area under the concentration–time curve (AUC) levels than evening administration [11].
Two previous studies have compared the toxicity levels of patients treated with sunitinib in the morning with those of patients treated with sunitinib in the evening. No differences in toxicity levels between morning and evening dosing of sunitinib were found in either study. Although the authors concluded that it is safe to take the drug either in the morning or at night, sunitinib pharmacokinetics were only examined during morning sunitinib administration and these studies were not designed to study the chrono-efficacy and chrono-tolerance of sunitinib exposure [12, 13].
Several studies have shown an association between higher exposure to sunitinib and improved survival [14, 15]. In order to improve the efficacy of sunitinib treatment, we wanted to gain insight into possible time-dependent changes in sunitinib pharmacokinetics, for which we designed a translational study. The primary aim of this study was to examine whether the administration time of sunitinib influences its pharmacokinetics. We studied the effect of administration time on pharmacokinetic parameters of sunitinib by measuring sunitinib and its active metabolite in plasma and organ tissue of mice as well as in plasma of cancer patients treated with sunitinib monotherapy. As a secondary aim, we studied the daily variation in the activity of sunitinib-metabolizing enzymes and transporters in murine hepatocytes and bowel tissue and through the midazolam clearance test in patients, which is a well-known marker of CYP3A activity [16].
Methods
Mice
Seven groups of 18 male FVB mice 8–12 weeks of age were used in this study. For logistic reasons, mice were housed under normal and reversed 12 h light/12 h dark cycles (lights on/off or off/on at 8 a.m. and 8 p.m.) for at least 1 week before the start of the experiment. Mice were fed a standard diet ad libitum. The mice were fasted from 3 h before pharmaceutical treatment to 1 h after administration. Water was available ad libitum throughout the entire experiment. This study was approved by an independent Animal Ethical Committee under protocol number EMC139-12-19.
Study Protocol
Sunitinib was dissolved in acidified water with a maximum pH of 6.0, at a concentration of 5 mg/mL. All mice were administered a fixed single dose of 1.06 mg, which was based on a dose of 42.4 mg/kg and a mean weight of mice of 25 g. Every 4 h, one group consisting of 18 mice was administered a single dose of sunitinib through gavage, starting at 8 a.m.
At six different times after the administration of sunitinib (t = 1, 2, 3, 5, 10, and 20 h), three mice with an identical sunitinib dosing time were administered isoflurane as anesthetic followed by 1 mL blood withdrawal by eye extraction and euthanasia by cervical dislocation. Directly after euthanasia, one liver lobe and the small bowel were removed. Blood from each single mouse was separately processed to plasma. Both plasma and tissue samples were snap-frozen in liquid nitrogen and stored at −80 °C until further analysis.
The seventh group of mice served as a control group and was administered solvent. Mice in this group were sacrificed at 4-h intervals. Blood and organs of the mice in this group were collected and stored under the same conditions as those of the treated mice. Quantitative real-time polymerase chain reaction (PCR) was applied to tissue samples to reveal potential time-dependent changes in the circadian expression of murine orthologs of genes encoding drug transporters and metabolizing enzymes involved in sunitinib pharmacokinetics, using known core clock genes as a control (see the Electronic Supplementary Material).
Patients
A prospective randomized crossover trial was performed in patients treated with sunitinib at the Department of Medical Oncology of the Erasmus MC-Cancer Institute, Rotterdam, The Netherlands. Patients were allowed to participate in the study at any time during sunitinib treatment, with both 4 weeks on/2 weeks off treatment and with continuous dosing regimen. Patients could participate in the study if (1) they were aged ≥18 years; (2) they had a histologically or cytologically confirmed diagnosis of advanced RCC, GIST, or pNET for which treatment consisted of sunitinib monotherapy; (3) they were using any stable dose of sunitinib at study entry (defined as at least 2 weeks’ continuous dosing without dose modifications); (4) hematologic, renal, and liver functions were adequate; and (5) written informed consent was given. Exclusion criteria consisted of (1) pregnancy or breastfeeding; (2) medical unstable condition requiring treatment; (3) symptomatic central nervous system metastases or a history of a psychiatric disorder that would prohibit the understanding and giving of informed consent; (4) major surgery within 2 weeks before start of the protocol; (5) use of CYP3A4-inhibiting or -inducing drugs; (6) problematic blood withdrawal; and (7) a known allergy to sunitinib and/or midazolam.
Study Protocol
Patients were randomized to start intake of sunitinib either at 8 a.m. (group A) or 6 p.m. (group B) in the next treatment course after inclusion. In the course thereafter, a crossover was performed to 6 p.m. for group A and 8 a.m. for group B. Accordingly, the sequence of the time of dosing of sunitinib in group A was morning–evening and in group B was evening–morning. During both courses, patients were hospitalized for 24 h in the third or fourth week of the treatment cycle, when steady-state plasma concentrations were achieved [8]. Blood samples for pharmacokinetic measurements were taken at timepoints t = 0, 1, 2, 4, 6, 8, 12, and 24 h after oral sunitinib intake.
During both hospitalization days, patients were subjected to a midazolam clearance test as a marker of hepatic CYP3A activity 2 h after administration of sunitinib. Midazolam (2.5 mg) was administered intravenously through slow infusion, and blood withdrawals to determine the pharmacokinetics of midazolam and its metabolite 1′-OH-midazolam were taken at timepoints t = 0.5, 2, and 6 h after administration, as previously described by Lee et al. [17].
After amendment of the study protocol, based on the outcome of the animal experiments, a subset of patients was asked to participate in the study for a third treatment course in which sunitinib was taken at 1 p.m.
This study was approved by the local medical ethical board (MEC2012-138), and registered at the Dutch trial registry (http://www.trialregister.nl, number NTR3526). According to the instructions stated in the Codes for Proper Use and Proper Conduct in the Self-Regulatory Codes of Conduct (http://www.federa.org), all samples for sunitinib and midazolam pharmacokinetic assessment were coded and anonymized. The analytical methods for sunitinib and midazolam quantification are described in the Electronic Supplementary Material. Parameters for sunitinib and SU12662 pharmacokinetics were combined AUC, trough concentrations (C trough), and clearance of sunitinib.
Statistics
For an 83 % probability that the study would detect a treatment difference at a two-sided 0.05 significance level and a 20 % true mean difference in AUC and/or clearance between treatment times, 18 patients were required. This is based on the assumption that the within-patient standard deviation of the pharmacokinetic parameters is 0.2. Patients were considered eligible after completing pharmacokinetic blood withdrawal during two treatment courses. To compare sunitinib pharmacokinetic parameters between morning, afternoon, and evening dosing times, the Wilcoxon signed-rank test was used. CircWave Batch (Dr. Roelof Hut, University of Groningen, Groningen, The Netherlands) v5.0 with cosinor analysis was used for harmonic regression analysis of circadian oscillation using a 24-h wave in the expression of genes and a 12-h wave in the pharmacokinetics of sunitinib, with forward linear harmonic regression using an F test. User defined α was chosen at 0.05.
Results
Sunitinib Pharmacokinetics in Mice
A total of 108 mice were treated with a single dose of sunitinib at six different timepoints over 24 h. Cosinor analysis of the AUC time zero to 10 h (AUC10) plot showed a clear 12-h rhythm in the exposure to sunitinib and SU12662 (p = 0.0342 and p = 0.0027, respectively; Fig. 1a) and the combined exposure (p = 0.0174) as a function of administration time. The combined AUC of sunitinib and SU12662 was 14–27 % higher when the drug was administered at 4 a.m. and 4 p.m., rather than at 8 a.m. and 8 p.m.
Fig. 1.
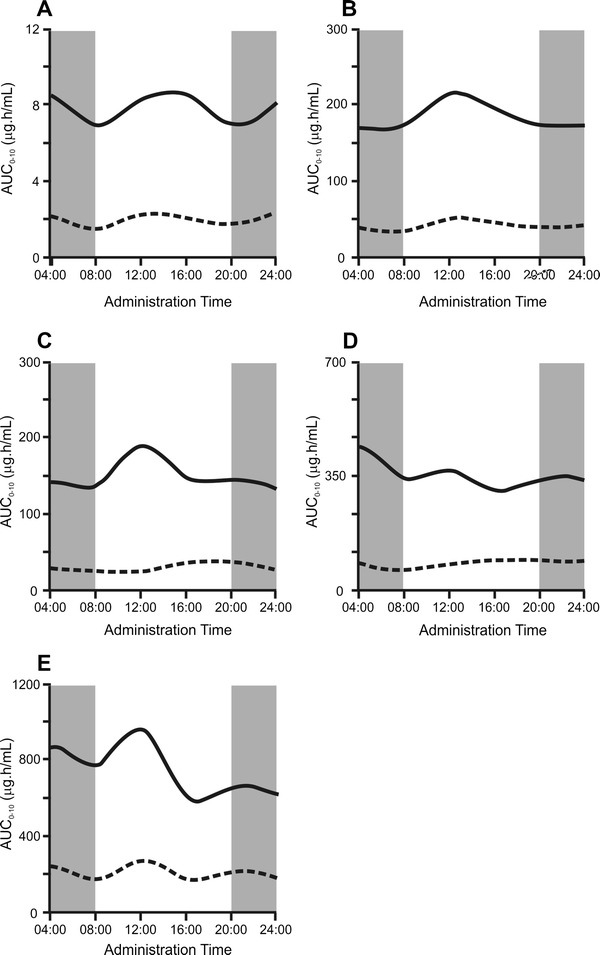
Daily variations in the area under the concentration–time curve (AUC) for sunitinib (solid lines) and SU12662 (dotted lines) in plasma (a), liver (b), duodenum (c), jejunum (d), and ileum (e) of FVB mice treated with a single dose of sunitinib. The x axis indicates the time of sunitinib administration, the y axis represents the mean AUC corresponding to the six consecutive administration times. Gray areas indicate it was dark in the cages of the mice, white areas indicate it was light. AUC 0–10 area under the concentration–time curve from time zero to 10 h
Figure 1b–e shows the AUCs of sunitinib and SU12662 for liver and three consecutive sections of the intestine, corresponding with the duodenum, jejunum, and ileum, respectively. As shown, oscillations also occur in the accumulation of sunitinib and its metabolite in these tissues. However, the 12-h rhythm was only statistically significant for SU12662 accumulation in the duodenum (p = 0.0179; Fig. 1c) but not for sunitinib or SU12662 accumulation in other tissues. Sunitinib and SU12662 concentrations in both plasma and tissue samples showed a broad inter-mouse variability.
Quantitative real-time PCR on liver tissue samples taken from mice in various stages of their circadian rhythm revealed daily fluctuations with a 24-h period in the activity of the clock genes Bmal1, Cryptochrome 1 (Cry1), Dbp, Period 2 (Per2), and Rev-erbα. This was confirmed by the cosinor analysis (p ≤ 0.0214), showing that the mice in a normal and reversed light–dark cycle were properly entrained at the time of the experiment (Electronic Supplementary Material Fig. 1).
In the liver, cosinor analysis showed circadian fluctuations in the messenger RNA (mRNA) levels of Abcb1a, Abcb1b, and Cyp3a11 (p ≤ 0.047), but not in the expression of Abcg2 (p = 0.254; Electronic Supplementary Material Fig. 2A). In duodenum and jejunum, the expression of Abcb1a, Abcg2, and Cyp3a11 followed a circadian rhythm (p ≤ 0.0162; Electronic Supplementary Material Fig. 2B and 2C). In the ileum, only a circadian variation in Abcb1a expression was seen (p < 0.001; Electronic Supplementary Material Fig. 2D). This implicates circadian expression of the genes involved in the uptake and metabolism of sunitinib as a cause for daily variations in sunitinib pharmacokinetics. The peak activities, as judged by their mRNA expression levels, for these genes were all at different times of the day.
Patient Demographics
Twenty-seven patients were included in this study, of whom 16 patients completed at least two courses with pharmacokinetic blood withdrawals (group A, n = 7; group B, n = 9). One patient was excluded from the study due to inability to withdraw blood, and one patient retracted informed consent before the end of the study protocol. Nine patients stopped sunitinib treatment during the study due to toxicity or progressive disease. A subgroup of 12 patients had pharmacokinetic measurements at three different times of the day during three courses of sunitinib. All patients used sunitinib as first-line anti-cancer treatment.
Group A and group B were comparable in sex, age, number of courses within the study protocol, and amount of dose reductions before and within the study protocol. Of the 16 patients who completed at least two courses with pharmacokinetic blood withdrawals, three patients underwent dose reductions in between the study courses. Six patients had already undergone dose reductions before the start of the study protocol. There were no dose escalations prior to or during the study protocol in any of the patients. Demographic characteristics are presented in Table 1.
Table 1.
Characteristics of the 27 individuals who participated in the clinical study
| Characteristic | Cohort A | Cohort B |
|---|---|---|
| No. of patients | 13 (48.1) | 14 (51.9) |
| No. of courses within study | ||
| 0–1 | 6 (46.2) | 5 (35.7) |
| 2 | 7 (53.8) | 9 (64.3) |
| 3 | 5 (38.5) | 7 (50.0) |
| Sex | ||
| Male | 10 (76.9) | 12 (85.7) |
| Female | 3 (23.1) | 2 (14.3) |
| Age (years) | 63.1 (10.9) | 61.3 (7.4) |
| WHO-PS | ||
| 0–1 | 13 (100) | 13 (92.9) |
| 2 | 0 (0) | 1 (7.1) |
| Tumor type | ||
| RCC | 13 (100) | 12 (85.7) |
| pNET | 0 (0) | 2 (14.3) |
| Dose reductions during study | 2 (22.2) | 1 (11.1) |
| Duration of sunitinib treatment (months) | 16.7 (26.8) | 19.3 (22.2) |
Values are expressed as n (%), except for age and duration of treatment, which are mean (SD)
pNET pancreatic neuro-endocrine tumor, RCC renal cell carcinoma, SD standard deviation, WHO-PS World Health Organization Performance Score
Sunitinib Pharmacokinetic Parameters
The mean AUC from time zero to the end of dosing period (AUCτ) in 16 patients was 1,599 ± 592 ng·h/mL when sunitinib was administered at 8 a.m. (AUC8 a.m.) and 1,444 ± 536 ng·h/mL when sunitinib was administered at 6 p.m. (AUC6 p.m.). In the 12 patients who underwent pharmacokinetic blood sampling during three courses, no difference was seen in AUC1 p.m. when compared with dosing times at 8 a.m. (p = 0.21) or 6 p.m. (p = 0.24), as shown in Fig. 2. However, a relevant difference in combined C trough values was seen between morning and evening dosing (n = 16, C trough-8 a.m. 50.7 ± 17.6 ng/mL, C trough-6 p.m. 58.9 ± 21.6 ng/mL; p = 0.006). This was also seen when comparing morning dosing and dosing at noon (n = 12, C trough-8 a.m. 56.0 ± 19.6 ng/mL; C trough-1 p.m. 66.0 ± 25.2 ng/mL; p = 0.003). The clearance (CL) of sunitinib was not significantly different between the three dosing times (CL8 a.m. 42.0 ± 14.7, CL1 p.m. 40.2 ± 13.5, CL6 p.m. 39.8 ± 9.8 L/h; p ≥ 0.5). Patients in group A had slightly higher AUC8 a.m. than patients in group B. For all other pharmacokinetic parameters, there were no significant differences between the both treatment groups (Table 2).
Fig. 2.
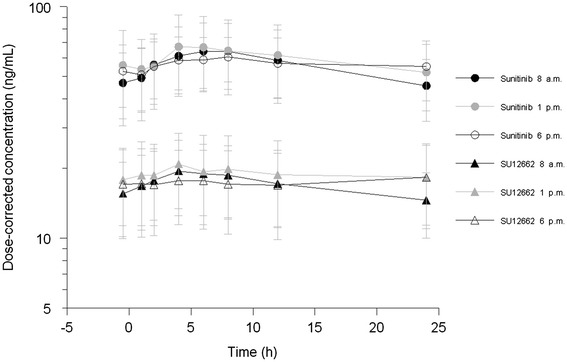
24-h concentration curves of sunitinib and SU12662 (mean ± SD) for patients treated with sunitinib at 8 a.m., 1 p.m., and 6 p.m
Table 2.
Pharmacokinetic parameters in group A and group B
| Pharmacokinetic measurementa | Group A [mean (SD)] | Group B [mean (SD)] | p Value |
|---|---|---|---|
| AUC8 a.m. | 1,832 (623) | 1,314 (426) | 0.048 |
| AUC1 p.m. | 1,965 (679) | 1,572 (665) | 0.340 |
| AUC6 p.m. | 1,711 (663) | 1,274 (377) | 0.092 |
| C trough-8 a.m. | 64.1 (22.6) | 48.1 (16.4) | 0.094 |
| C trough-1 p.m. | 76.3 (26.2) | 58.6 (23.6) | 0.249 |
| C trough-6 p.m. | 69.0 (27.8) | 52.4 (15.4) | 0.122 |
| CL8 a.m. | 35.1 (14.0) | 45.5 (12.0) | 0.145 |
| CL1 p.m. | 34.5 (16.5) | 44.3 (25.4) | 0.435 |
| CL6 p.m. | 36.0 (12.9) | 42.7 (5.8) | 0.235 |
| 1′-OH-MD/MD10 a.m. | 0.081 (0.014) | 0.095 (0.027) | 0.251 |
| 1′-OH-MD/MD8 p.m. | 0.087 (0.027) | 0.121 (0.084) | 0.311 |
Bold value indicates that the difference is statistically significant
1′-OH-MD/MD x ratio between 1′-OH-midazolam and midazolam at time x, AUC x combined area under the concentration–time curve of sunitinib and SU12662 at sunitinib dosing time x, C trough-x combined trough concentration of sunitinib and SU12662 at sunitinib dosing time x, CL x sunitinib clearance at sunitinib dosing time x
Units of AUC ng·h/mL, C trough-x ng/mL, CL L/h
Midazolam Clearance Test
Three out of 16 patients did not undergo midazolam clearance tests at both hospitalization days, due to patient request. In the remaining 13 patients, there was no significant difference in the ratio between midazolam and 1′-OH-midazolam between the administration times 10 a.m. and 8 p.m. (mean difference in 1′-OH-midazolam/midazolam ratio = 0.0184; p = 0.256). Group A was not different from group B in outcome of the 1′-OH-midazolam/midazolam ratio in the morning or in the evening (Table 2).
Discussion
In this translational study, sunitinib pharmacokinetics in mice showed daily variations depending on the time of administration. Sunitinib and SU12662 AUCs follow a similar oscillation pattern as a function of administration time, with a 14–27 % higher combined plasma AUC when sunitinib was administered at 4 a.m. or 4 p.m., which is in the middle of the active or inactive phase of the mice respectively, rather than at 8 a.m. or 8 p.m. Although a recent study in rabbits revealed significant differences in the exposure to sunitinib between dosing at 8 a.m. or 8 p.m., this was not seen in our study in mice [18].
Both the sunitinib and the SU12662 AUCs in plasma follow a similar pattern in an apparent 12-h rhythm. The oscillation pattern is probably due to circadian rhythms in both Cyp3a11 enzyme activity and expression of the efflux transporters Abcb1a, Abcb1b, and Abcg2. The 12-h rhythm in sunitinib and SU12662 pharmacokinetics may be due to counteracting activities of these drug transporters and metabolizing enzyme, since we observed variation in mRNA expression with peak concentrations at different times of the day.
In the 16 patients treated with sunitinib, the plasma AUC was equal at three dosing times, which is in contrast to our pre-clinical results. Despite the equality in daily exposure to sunitinib, the C trough values of sunitinib were significantly lower when patients were administered sunitinib in the morning than with noon or evening dosing. C trough values were sampled just before the intake of a new dose of sunitinib. In this phase of the pharmacokinetic process, metabolism and elimination are the most important determinants of drug concentrations.
This study showed that the midazolam clearance test in patients was similar at 10 a.m. and 8 p.m., suggesting similarity in hepatic CYP3A activity between these timepoints. Since CYP3A4 has a key role in sunitinib metabolism [8], this suggests that sunitinib metabolism was similar at morning and evening dosing. Based on the midazolam clearance test, it can be assumed that metabolism did not vary between these timepoints. Therefore, it is hypothesised that the difference in sunitinib C trough values may be attributed to daily changes in elimination.
The daily variation in sunitinib C trough values is relevant for future research. For sunitinib and other TKIs, a threshold concentration for efficacy is known [15]. Therapeutic drug monitoring (TDM) is currently being investigated as a potential improvement of sunitinib therapy, using C trough values of sunitinib and SU12662 as an indicator for daily exposure. In these studies, dose levels of sunitinib are increased if C trough values are below the threshold. A recently published study revealed that it is feasible to administer sunitinib based on C trough values, with threshold concentrations between 50 and 100 ng/mL [19]. Patients in our study had fewer fluctuations in the daily sunitinib concentration when sunitinib was administered at noon or in the evening. Therefore, the chance of dropping below the threshold of 50 ng/mL, where sunitinib is thought to be ineffective, is smaller when the drug is administered at these times of the day, although the daily exposure is the same. Patients dosed in the morning may therefore potentially undergo erroneous dose escalations and suffer from more toxicity [20].
Interestingly, the daily variation in sunitinib AUCs observed in mice was not confirmed in patients. The discrepancy between pre-clinical models and patients may be due to various reasons. First, the pharmacokinetics of sunitinib may be different between mice and humans due to species specificity in the orthologous CYP enzymes and drug transporters involved in the pharmacokinetics of sunitinib. Second, the mice in this study only received a single dose of sunitinib and therefore steady-state plasma concentrations were not reached at the time of pharmacokinetic sampling. This is in contrast with the patients in our study who were administered sunitinib for at least 2 weeks before pharmacokinetic sampling took place. A third explanation can be that while mice were kept under strict light/dark cycles resulting in synchronized behavior, the circadian rhythm in the patients in this study may be disturbed by lifestyle or during the hospital stay for pharmacokinetic sampling. Pharmacokinetic samples were taken both during day times and during night hours, which resulted in waking of the patient. Fourth, the mice in this study were genetically homogeneous, while in the human population there is genetic heterogeneity.
Of note, there were some limitations in the pre-clinical study. Only one blood withdrawal for pharmacokinetic determination was possible from each mouse. Therefore, the AUC is calculated from sunitinib concentrations that are measured in different mice, which showed a large inter-individual difference in sunitinib and SU12662 concentrations. These large variations in pharmacokinetics may have been due to several causes. For instance, sunitinib was administered through gavage. Possibly, the solvent was partially left in the gavage tube and it was not noticed whether mice vomited after gavage. Also, mice ranged in age from 8 to 12 weeks and therefore may differ in weight. In addition, mice were fasted from 3 h before gavage until 1 h after oral gavage to make sure the stomachs of all mice were empty at the time of sunitinib administration. However, this may have caused stress to the animals and may have affected the phase of peripheral circadian rhythms, although analysis of mRNA levels of the core clock genes excluded the latter possibility.
This study was not designed to detect differences in pharmacodynamics (efficacy of treatment and toxicity) between different dosing times of sunitinib. Patients were allowed to participate at any time during sunitinib treatment, and there was a broad variation in the on-treatment time between patients. Six out of 16 (38 %) patients had previously undergone dose reductions due to severe toxicity, and at the time of participation in the study these patients were treated at a dose level with an acceptable toxicity profile. Therefore, differences in toxicity levels between dosing times were not an endpoint in this study. However, previous studies have reported that toxicities from sunitinib treatment are similar in morning and evening dosing [12, 13].
Conclusion
Altogether, we conclude that daily variation in the pharmacokinetics of sunitinib exists, most likely resulting from differences in sunitinib elimination. Patient’s plasma sunitinib C trough values are higher when administered in the afternoon or evening, and during these dosing times more stable drug concentrations are achieved than when administered in the morning. It is therefore advised that sunitinib should be dosed in the afternoon or evening in daily clinical practice, and if TDM is implemented in clinical practice, the administration time of sunitinib should be taken into account.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary material 2 (TIFF 23851 kb)
Supplementary material 3 (TIFF 17231 kb)
Acknowledgments
We would like to thank Ton Boersma, Inge Ghobadi Moghaddam-Helmantel, Annemieke Nieuweboer, Ellen de Morrée, Cindy Bolder, Anna Pagani, Stefanie Vester, Xander den Dekker, Anne-Joy de Graan, Els Moltzer, Dennis de Meulder, and Dominique Kuiper for their specific contributions to this study.
This research received no grant from any funding agency in the public, commercial, or not-for-profit sectors.
The authors declare that there is no conflict of interest.
Contributor Information
Jacqueline S. L. Kloth, Phone: +31 10 704 1757, Email: j.kloth@erasmusmc.nl
Gijsbertus T. J. van der Horst, Phone: +31 10 7043199, Email: g.vanderhorst@erasmusmc.nl
References
- 1.Sunitinib prescribing information, 2009. http://labeling.pfizer.com/ShowLabeling.aspx?id=607. Accessed 21 July 2009.
- 2.Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368(9544):1329–1338. doi: 10.1016/S0140-6736(06)69446-4. [DOI] [PubMed] [Google Scholar]
- 3.Faivre S, Demetri G, Sargent W, Raymond E. Molecular basis for sunitinib efficacy and future clinical development. Nat Rev Drug Discov. 2007;6(9):734–745. doi: 10.1038/nrd2380. [DOI] [PubMed] [Google Scholar]
- 4.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356(2):115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 5.Raymond E, Dahan L, Raoul JL, Bang YJ, Borbath I, Lombard-Bohas C, et al. Sunitinib malate for the treatment of pancreatic neuroendocrine tumors. N Engl J Med. 2011;364(6):501–513. doi: 10.1056/NEJMoa1003825. [DOI] [PubMed] [Google Scholar]
- 6.Klumpen HJ, Samer CF, Mathijssen RH, Schellens JH, Gurney H. Moving towards dose individualization of tyrosine kinase inhibitors. Cancer Treat Rev. 2011;37(4):251–260. doi: 10.1016/j.ctrv.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 7.Bello CL, Sherman L, Zhou J, Verkh L, Smeraglia J, Mount J, et al. Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs. 2006;17(3):353–358. doi: 10.1097/00001813-200603000-00015. [DOI] [PubMed] [Google Scholar]
- 8.van Erp NP, Gelderblom H, Guchelaar HJ. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat Rev. 2009;35(8):692–706. doi: 10.1016/j.ctrv.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 9.Takiguchi T, Tomita M, Matsunaga N, Nakagawa H, Koyanagi S, Ohdo S. Molecular basis for rhythmic expression of CYP3A4 in serum-shocked HepG2 cells. Pharmacogenet Genomics. 2007;17(12):1047–1056. doi: 10.1097/FPC.0b013e3282f12a61. [DOI] [PubMed] [Google Scholar]
- 10.Okyar A, Dressler C, Hanafy A, Baktir G, Lemmer B, Spahn-Langguth H. Circadian variations in exsorptive transport: in situ intestinal perfusion data and in vivo relevance. Chronobiol Int. 2012;29(4):443–453. doi: 10.3109/07420528.2012.668996. [DOI] [PubMed] [Google Scholar]
- 11.Park SI, Felipe CR, Pinheiro-Machado PG, Garcia R, Tedesco-Silva H, Jr, Medina-Pestana JO. Circadian and time-dependent variability in tacrolimus pharmacokinetics. Fundam Clin Pharmacol. 2007;21(2):191–197. doi: 10.1111/j.1472-8206.2007.00468.x. [DOI] [PubMed] [Google Scholar]
- 12.Escudier B, Roigas J, Gillessen S, Harmenberg U, Srinivas S, Mulder SF, et al. Phase II study of sunitinib administered in a continuous once-daily dosing regimen in patients with cytokine-refractory metastatic renal cell carcinoma. J Clin Oncol. 2009;27(25):4068–4075. doi: 10.1200/JCO.2008.20.5476. [DOI] [PubMed] [Google Scholar]
- 13.George S, Blay JY, Casali PG, Le Cesne A, Stephenson P, Deprimo SE, et al. Clinical evaluation of continuous daily dosing of sunitinib malate in patients with advanced gastrointestinal stromal tumour after imatinib failure. Eur J Cancer. 2009;45(11):1959–1968. doi: 10.1016/j.ejca.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 14.Houk BE, Bello CL, Poland B, Rosen LS, Demetri GD, Motzer RJ. Relationship between exposure to sunitinib and efficacy and tolerability endpoints in patients with cancer: results of a pharmacokinetic/pharmacodynamic meta-analysis. Cancer Chemother Pharmacol. 2010;66(2):357–371. doi: 10.1007/s00280-009-1170-y. [DOI] [PubMed] [Google Scholar]
- 15.Yu H, Steeghs N, Nijenhuis CM, Schellens JH, Beijnen JH, Huitema AD. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin Pharmacokinet. 2014;53(4):305–325. doi: 10.1007/s40262-014-0137-2. [DOI] [PubMed] [Google Scholar]
- 16.Streetman DS, Bertino JS, Jr, Nafziger AN. Phenotyping of drug-metabolizing enzymes in adults: a review of in vivo cytochrome P450 phenotyping probes. Pharmacogenetics. 2000;10(3):187–216. doi: 10.1097/00008571-200004000-00001. [DOI] [PubMed] [Google Scholar]
- 17.Lee LS, Bertino JS, Jr, Nafziger AN. Limited sampling models for oral midazolam: midazolam plasma concentrations, not the ratio of 1-hydroxymidazolam to midazolam plasma concentrations, accurately predicts AUC as a biomarker of CYP3A activity. J Clin Pharmacol. 2006;46(2):229–234. doi: 10.1177/0091270005283466. [DOI] [PubMed] [Google Scholar]
- 18.Szalek E, Karbownik A, Sobanska K, Polom W, Grabowski T, Wolc A, et al. The influence of the time-of-day administration of the drug on the pharmacokinetics of sunitinib in rabbits. Eur Rev Med Pharmacol Sci. 2014;18(16):2393–2399. [PubMed] [Google Scholar]
- 19.Lankheet NA, Kloth JS, Gadellaa-van Hooijdonk CG, Cirkel GA, Mathijssen RH, Lolkema MP et al. Pharmacokinetically guided sunitinib dosing: a feasibility study in patients with advanced solid tumours. Br J Cancer. 2014;110(10):2441–9. doi:10.1038/bjc.2014.194. [DOI] [PMC free article] [PubMed]
- 20.Mathijssen RH, Sparreboom A, Verweij J. Determining the optimal dose in the development of anticancer agents. Nat Rev Clin Oncol. 2014;11(5):272–281. doi: 10.1038/nrclinonc.2014.40. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material 2 (TIFF 23851 kb)
Supplementary material 3 (TIFF 17231 kb)