

Key Points
The geometric orientation of the underlying matrix regulates platelet α-granule secretion.
On geometrically constrained matrices, platelets self-deposit additional matrix, providing more cell membrane to extend spreading.
Abstract
Although the biology of platelet adhesion on subendothelial matrix after vascular injury is well characterized, how the matrix biophysical properties affect platelet physiology is unknown. Here we demonstrate that geometric orientation of the matrix itself regulates platelet α-granule secretion, a key component of platelet activation. Using protein microcontact printing, we show that platelets spread beyond the geometric constraints of fibrinogen or collagen micropatterns with <5-µm features. Interestingly, α-granule exocytosis and deposition of the α-granule contents such as fibrinogen and fibronectin were primarily observed in those areas of platelet extension beyond the matrix protein micropatterns. This enables platelets to “self-deposit” additional matrix, provide more cellular membrane to extend spreading, and reinforce platelet-platelet connections. Mechanistically, this phenomenon is mediated by actin polymerization, Rac1 activation, and αIIbβ3 integrin redistribution and activation, and is attenuated in gray platelet syndrome platelets, which lack α-granules, and Wiskott-Aldrich syndrome platelets, which have cytoskeletal defects. Overall, these studies demonstrate how platelets transduce geometric cues of the underlying matrix geometry into intracellular signals to extend spreading, which endows platelets spatial flexibility when spreading onto small sites of exposed subendothelium.
Introduction
Hemostasis begins with platelet adhesion and activation at the site of vascular injury.1 Platelets then secrete their α- and dense granules, leading to further recruitment of other platelets and the formation of a hemostatic plug.2 Extensive research has characterized the underlying biological signaling pathways that govern these processes at the bulk level via in vivo and in vitro techniques.3 However, how platelets interact with their microenvironment at the single-cell level and how these interactions subsequently influence platelet physiology remain poorly understood. For example, although single platelets have long been observed to fill submicron-sized “gaps” in the endothelium,4 how platelets establish adhesion on such small areas of exposed matrix that is strong enough to withstand the shear forces of the circulation is unknown. Studies of single platelet-matrix interactions may help explain the role of platelets in maintaining vascular integrity and supporting the semipermeable barrier function of the endothelium during homeostatic conditions.5 Furthermore, because platelets within different regions of the same thrombus exhibit significantly different levels of activation,6,7 this concept of physical and microenvironmental regulation of platelet physiology at the single-cell level is also important for our overall understanding of clot formation.
Secretion of platelet α-granules, which are tenfold more abundant than other granule types, plays major roles in hemostasis, thrombosis, and inflammation.8 Components of the secretory machinery interact directly with the platelet cytoskeleton, and actin polymerization especially has a substantial influence on granule secretion.9-11 However, those mechanisms of platelet granule secretion have largely been studied on platelets in suspension,12 obscuring the potential effects adhesion and cytoskeletal dynamics may have on granule exocytosis in physiologic settings. Platelet granules may also represent an internal reservoir of “extra” membrane essential for membrane remodeling via granule exocytosis.13 Indeed, in other cell types, granule membranes mediate remodeling during cell movement,14 neurite outgrowth,15-17 membrane resealing,18 and phagocytic cup formation.19 In addition, α-granules contain matrix proteins, the deposition of which could provide substrate and scaffolding for increased platelet spreading. Collectively, these observations raise the question of whether matrix geometry can control the orientation of granule secretion in spreading platelets, which could directionally direct further adhesion and spreading.
Evaluation of platelet geometry sensing and the spatial regulation of granule secretion and actin polymerization requires the study of single platelets on geometrically defined protein patterns, which can be achieved using microcontact printing as demonstrated previously by our laboratory and others.20-23 In this work, we apply microcontact printing to create micro/nanoscale patterns of matrix proteins that mediate platelet adhesion and activation, to demonstrate that the microenvironmental geometry directly mediates platelet physiology and function. Specifically, we demonstrate that platelet α-granule secretion is spatially regulated at the micro/nanoscale via rearrangement of the actin cytoskeleton. Moreover, this spatially regulated α-granule secretion enables the selective exposure of the adhesion molecule P-selectin, the directed deposition of matrices such as fibrinogen and fibronectin, and provides cell membrane to support platelet filopodial extension and lamellipodial spreading beyond the geometric boundaries of the protein micropattern. This capability, which does not occur in gray platelet syndrome (GPS) patient platelets (ie, platelets that congenitally lack α-granules) or in Wiskott-Aldrich syndrome (WAS) patient platelets (ie, platelets with cytoskeletal defects, impaired αIIbβ3-mediated activation, and decreased and dysfunctional α-granules24), likely enables platelets to increase their total contact area of spreading when adhering to small areas of exposed subendothelial matrix or subclinical sites of vascular injury.
Materials and methods
Microfabrication
The polydimethylsiloxane (PDMS) stamp was fabricated via soft lithography using a positive photoresist (model S1813, Microposit) spin-coated onto a silicon wafer. The patterned soft mold was silanized using hexamethyldisilazane (Sigma-Aldrich) to prevent adhesion to the PDMS, which was poured over the soft mold and cured at 60°C overnight.
Microcontact printing
The PDMS stamp surfaces were incubated with either Alexa Fluor 647 human fibrinogen (Life Technologies) or rat tail Collagen Type 1 (BD) or human fibronectin (Sigma-Aldrich) mixed with Alexa Fluor 647 bovine serum albumin (BSA; Life Technologies) at 37°C. The surfaces were then rinsed with water and dried via nitrogen gas. Protein micropatterns were then printed onto clean coverslips and were blocked with 1% BSA before the platelet adhesion assay. For experiments involving flow, a sheet of PDMS with double-sided tape as a gasket was attached to a patterned glass surface to create a microfluidic channel, and 1% BSA was perfused before the experiment.
Platelet isolation and adhesion assay
Human blood was collected in anticoagulant citrate dextrose solution-A (BD Biosciences) and centrifuged at 150g for 15 minutes at room temperature without brakes. Platelet-rich plasma was collected, then either centrifuged at 900g for 5 minutes or gel-filtered using Sepharose 2B (Sigma-Aldrich). The platelets were then resuspended or collected in Tyrode’s buffer and diluted to a concentration of 5 to 20 million platelets per milliliter. Two mM MgCl2 was added to promote adhesion of platelets on substrates. For select experiments, pharmacologic inhibitors, namely nocodazole (2 µM; Sigma-Aldrich), latrunculin A (2 µM; Life Technologies), eptifibatide (Integrilin, 10 µg/mL; COR Therapeutics), Y27632 (75 µM; Sigma-Aldrich), ML-7 (30 µM; Sigma-Aldrich), and NSC23766 (150 µM; Cayman Chemicals) were added. Platelets were then incubated on micropatterned surfaces and washed and fixed with 1.6% paraformaldehyde. Our results show that as incubation time increases, the number of adherent/spread platelets on protein micropattern increases. More importantly, however, is that the rate-limiting step in this process is for platelets to settle from the suspension onto the surface. Once platelets adhere to the protein micropattern, spreading onto and beyond the micropattern boundaries ensues. Therefore, to optimize the number of detectable platelets interacting with the protein micropatterns, two-hour incubations were used for most experiments. All blood samples were obtained with permission from the institutional review boards of Emory University School of Medicine, Georgia Institute of Technology, and Beth Israel Deaconess Medical Center.
Immunocytochemistry
Immunocytochemistry was performed using antibodies against P-selectin (G-1, AK4; Santa Cruz Biotechnology), fibrinogen (Abcam), and fibronectin (Abcam) for select experiments. For experiments involving permeabilization, fixed platelets were permeabilized with 0.5% Triton-X100. All samples were blocked in 5% goat serum (Rockland) before incubation with primary and secondary antibodies (Alexa Fluor conjugated IgGs, Life Technologies). Platelets were also counterstained with a plasma membrane dye (CellMask Orange, Life Technologies) or phalloidin (Alexa Fluor conjugated, Life Technologies) and then exposed to mounting medium (Dako Cytomation). For detecting activated αIIbβ3, FITC-PAC-1 antibody (BD Biosciences) was applied directly to platelets after spreading was visibly confirmed, then immediately replaced with 1.6% paraformaldehyde for fixation. To induce membrane retraction, fully spread platelets were exposed to 200 µM latrunculin A for 30 minutes before fixation. For experiments involving flow, upon platelet adhesion (<1 hour), P-selectin and its secondary antibody were perfused into the microfluidic device at a shear rate of ∼100 s−1.The shear rate was calculated via Equation (1).
 |
where 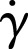 is shear rate, Q is the flow rate, w is the width of the channel, and h is the height of the channel.
is shear rate, Q is the flow rate, w is the width of the channel, and h is the height of the channel.
Images were obtained with a confocal microscope (LSM700, Zeiss) and a total internal reflection fluorescent (TIRF) microscope (IX71, Olympus). For TIRF images, Volocity 6.3 (PerkinElmer) was used for image deconvolution.
Statistical analysis
Line plots of averaged fluorescence signal intensity and total signal intensity were measured by ImageJ (National Institutes of Health). Platelet-platelet connections and filopodial projections were counted in randomly selected fields of arrays of microdots. The Wilcoxon rank-sum test was performed using GraphPad Prism 5.04 (GraphPad Software) and MATLAB (MathWorks). Extended platelet areas were measured using ZEN (Zeiss), and Student t test was used for statistical analysis. P < .05 was considered to be significant.
Results
Platelets extend their spreading upon adhesion to geometrically constrained areas of matrix
First, we investigated how platelet spreading is affected by protein matrix size. Circular fluorescently-labeled fibrinogen or collagen-microstamped dots of various diameters (2-10 µm) were simultaneously patterned on glass surfaces and then incubated with washed human platelets isolated from healthy subjects. These circular matrix protein micropatterns (microdots), although certainly not physiologic in shape, enable the assessment of how the geometry and geometric boundaries of the microenvironment affect platelet physiology. As platelets adhered onto larger protein microdots (>7 µm diameter), platelet spreading and lamellipodial formation ensued but was restricted by the geometric constraints of the microdots, which platelet morphology followed with high fidelity. However, on smaller fibrinogen or collagen microdots, platelets spread beyond the geometric boundaries of the microdot; the extent of this phenomenon inversely correlated with microdot size (Figure 1 and supplemental Figure 1, available on the Blood Web site).
Figure 1.

Platelet spreading is spatially regulated at the microscale. (A) Platelets (red, cell membrane stain) adhere and spread onto fibrinogen microdots (blue) fabricated via protein microcontact printing. On larger fibrinogen microdots (eg, 7 and 10 µm in diameter; “∞” denotes no geometric boundary), platelet spreading conforms to the microenvironmental geometric boundaries of the microdots with high fidelity. Decreasing the fibrinogen microdot size (eg, diameters of 2 and 5 µm), however, leads to platelet spreading beyond the geometric boundaries of the microdots. (B) This phenomenon is quantified by measuring the surface area of spread platelets on fibrinogen (blue) and collagen (green) microdots of different diameters. The gray line denotes the protein microdot area and diameters for reference. The scale bar = 5 µm. Error bars indicate standard error (SE).
Moreover, the area of platelet spreading varied among the different microdot conditions. Platelets optimally spread to 35.67 ± 4.93 µm2 (mean ± standard error [SE]), on 7-µm diameter fibrinogen microdots, which, interestingly, is higher than the average area of platelets spread on larger diameter or non–geometrically constrained areas of protein-printed surfaces (24.56 ± 2.27 µm2) (Figure 1B), suggesting that platelets have a geometry sensing mechanism that triggers increased spreading upon contact with the matrix protein boundary. In addition, as microdot diameters decreased and platelet spreading occurred beyond the geometric constraints, platelets exhibited a minimal average spread area of 7.95 ± 0.47 µm2 on collagen microdots, which is not significantly different than when platelets adhered to nonsubstrate area (7.39 ± 0.32 µm2). These results indicate that although platelet spreading is tightly regulated by the microenvironmental geometry, platelet geometry sensing mediates spreading beyond the geometric boundaries of protein microdots below a specific microdot diameter.
Subcellular spatial regulation of α-granule secretion occurs at the geometric boundaries of protein matrices
To explore how microenvironmental geometry affects platelet physiology, we investigated whether platelet α-granule release is spatially regulated. P-selectin, a transmembrane cell adhesion molecule stored in platelet α-granules has been used as a marker for α-granule release.25 We performed P-selectin immunostaining to evaluate α-granule distribution and secretion by platelets exposed to microdots of different diameters. Along with the expected high expression at the granulomere, the granule-dense platelet center,13 P-selectin expression was most intense in parts of the platelets that spread beyond the protein micropattern boundaries (Figure 2A,E). This phenomenon inversely correlated with microdot size (Figure 2B) and also occurred at different time points (supplemental Figure 2) and under physiologic flow conditions (supplemental Figure 3). Peripheral P-selectin expression beyond the microdot boundaries was observed in both permeabilized and nonpermeabilized samples (supplemental Figure 5B). This high P-selectin expression was not observed in platelets that spread fully without geometric constraint in larger microdots (Figure 2E). Total P-selectin signal showed no statistically significant difference between platelets on larger microdots (5 µm) compared with platelets on matrices without geometric constraint, although a statistically significant difference did exist between platelets on smaller vs larger microdots (3 µm vs 5 µm, Figure 2F), suggesting that α-granule release is not complete when platelets were spread on geometrically constrained matrices. In addition, pairs of platelets on adjacent protein microdots that came into contact during spreading also expressed high levels of P-selectin at the areas where platelets connected beyond the microdot boundaries (Figure 2C).
Figure 2.

Platelet α-granule secretion is spatially regulated at the micro- and nanoscales. (A) Platelet spreading beyond protein microdot (blue) boundaries is associated with increased P-selectin expression (green) in those specific regions of unpermeabilized platelets (red). Individual fluorescence microscopy channels and a merged image of all 3 channels are shown. (B) High levels of anti–P-selectin staining colocalize with regions of platelets that spread beyond the geometric boundaries of protein microdots, and this effect inversely correlates with microdot size. (C) Permeabilized platelets on adjacent protein microdots form bridging connections associated with high levels of P-selectin in those regions. (D) Protein micropatterns with arrays of nanoscale “holes” reveal that P-selectin colocalizes with the patterned holes in permeabilized platelets. (E) Averaged (n = 7-10 platelets) fluorescence intensity line plots of P-selectin expression (green) on platelets (red, membrane dye) that spread beyond 3- and 5-µm diameter fibrinogen microdots (blue) show peak P-selectin expression outside the micropattern boundaries, whereas no clear peak is shown in platelets that spread on evenly coated fibrinogen surfaces. (F) Total P-selectin signal intensity is significantly increased on platelets spread on 5-µm diameter microdots compared with platelets on 3-μm microdots, but was not significantly different compared with platelets on evenly coated surfaces (“∞” µm diameter). The scale bars = 5 µm.
To determine if α-granule release is spatially regulated at the nanoscale and/or the platelet basal surface, we fabricated protein micropatterns with arrays of “holes” with diameters ranging from 400 nm to 2 µm. When platelets adhered to protein micropatterns with holes of diameters <1 µm, they completely spread over those holes. Interestingly, immunostaining revealed strong P-selectin expression localized to the micropatterned holes (Figure 2D and supplemental Figure 4). This spatially regulated P-selectin distribution was only observed in permeabilized samples (supplemental Figure 5C), indicating that this phenomenon occurs either inside and/or at the basal surface rather than the apical surface of the platelet.
To confirm whether nanoscale-mediated α-granule secretion occurs on the basal surface of the platelet, we used TIRF. When the TIRF focal plane was positioned at the glass surface, directly below the basal surface of the platelet membrane, P-selectin staining revealed sharply focused α-granules where the platelet spread beyond the protein microdot boundaries but not the granulomere at the platelet center (Figure 3A). When the focal plane was elevated to just above the basal platelet surface, P-selectin staining beyond the protein pattern defocused, whereas the focus of the overlying platelet membrane and the granulomere sharpened (Figure 3B). Similarly, on protein patterns with arrays of nanoscale holes, P-selectin immunostaining colocalized with the holes was sharply focused only when the TIRF focal plane was positioned below the basal platelet surface and blurred out of focus as the focal plane was elevated (Figure 3C-D). Overall, the TIRF microscopy results confirmed that as platelets spread beyond the micropattern boundaries, α-granule secretion occurred at the basal surface of the platelet membrane. This is consistent with the 3D confocal imaging data of platelets spreading beyond microdot boundaries (Figure 3E), which also strongly suggest that spatially regulated α-granule secretion occurs at the basal platelet membrane.
Figure 3.

Spatially regulated α-granule secretion occurs at the basal platelet surface. (A) Total internal reflection fluorescence (TIRF) microscopy reveals that on platelets adhered and spread onto fibrinogen microdots (traced with dotted circle), anti–P-selectin staining (green) was clearly focused at the basal surface of the platelet membrane (red, cell membrane stain). (B) As the TIRF focal plane is elevated, the focus of the outer edges of the platelet membrane and anti–P-selectin staining at the granulomere center of the platelet sharpens, whereas the anti–P-selectin staining beyond the micropattern boundaries loses focus. (C) P-selectin expression localized to the submicron “holes” of the micropatterned protein matrix is clearly focused at the basal surface of the platelet membrane, whereas this focus is lost as the focal plane is elevated and focused at the outer edges of the platelet membrane (D). (E) The orthogonal sectional view of a platelet (red) on a fibrinogen microdot (blue). Strong expression of P-selectin (green) is detected in areas where platelet spreading extended beyond the geometric constraints of the microdot pattern, as well as at the granulomere center of the platelet. P-selectin expression beyond microdot pattern was also observed at the glass surface (arrows). All samples are permeabilized upon staining.
Deposition of α-granule contents, fibrinogen and fibronectin, supports platelet spreading in an integrin αIIbβ3-dependent manner
Platelet α-granules contain proteins including several matrix proteins such as fibrinogen and fibronectin.26,27 Immunostaining for fibrinogen on collagen microdots and fibronectin on fibrinogen microdots revealed high concentrations localized at the regions of the platelet spread beyond the protein microdot boundaries and at platelet-platelet connections between 2 adjacent protein microdots (Figure 4A-B). In addition, pharmacologic retraction of platelet membranes with 200 µM latrunculin A revealed fibronectin deposition to the surface on which platelet membranes were formerly adhered (Figure 4C). Live-cell immunostaining with PAC-1, an antibody that binds to activated αIIbβ3 integrins, revealed high concentrations of activated αIIbβ3 on regions of platelets at and beyond the geometric borders of fibrinogen micropatterns (Figure 4D). Other membrane proteins, such as nonactivated αIIbβ3 and glycoprotein Ib, did not exhibit this phenomenon of spatial regulation (supplemental Figure 6). Exposure to eptifibatide, an αIIbβ3 antagonist, inhibited platelet spreading beyond the geometric boundaries of collagen microdots completely but still allowed spreading within the collagen micropattern, which is most prominent on small microdots (Figure 4E). These data suggest that when adhering and spreading on limited area of extracellular matrices (≤5-µm diameter), platelets will secrete α-granule contents beyond the border of the matrices. Secretion of α-granule contents will then likely lead platelets to deposit fibrinogen and fibronectin, then adhere and spread onto in an αIIbβ3-dependent manner.
Figure 4.

Matrix self-deposition by spatially regulated α-granule secretion supports platelet spreading beyond the geometric boundaries of the protein micropattern in an αIIbβ3-dependent manner. (A) Immunostaining with antifibronectin reveals that small amounts of fibronectin (green) colocalized with the platelet membrane (red) margins on platelets spread on evenly coated fibrinogen (blue) surfaces (left) but shows increased levels of fibronectin beyond the protein micropattern boundaries (right). Averaged (n = 5-8 platelets) fluorescence intensity line plots of fibronectin expression show that the fibronectin signal is highest at and immediately beyond the micropattern boundaries and not at the edge of platelet membrane. (B) Regions of platelets (red) that spread beyond the collagen microdot (blue) boundaries stain strongly with antifibrinogen (left, green, arrow). Strong expression of fibronectin was also observed at connections between 2 platelets adhered to adjacent protein microdots (middle), and also at the area around the connection (right). (C) Retraction of spread platelets via latrunculin A exposure reveal the deposited fibronectin (green) that remains on the surface after retraction of the platelet membrane. (D). Live cell staining with FITC-PAC-1 (green) shows high concentrations of activated αIIbβ3 integrins distributed at and beyond the geometric boundaries of fibrinogen microdots (blue). (E) When platelets (red) were treated with eptifibatide, an αIIbβ3 antagonist, platelet spreading beyond the geometric constraints of the collagen micropattern (blue) is completely inhibited. Platelets (B,E) were permeabilized upon staining. The scale bars= 5 µm.
The actin cytoskeleton mediates spatial regulation of platelet spreading and α-granule secretion
In platelets, the actin cytoskeleton is essential for filopodia formation and spreading,28 and is therefore a likely mediator of spatially regulated α-granule secretion. To determine the role of those, first we examined the effects of pharmacologic cytoskeletal inhibitors of actin polymerization (latrunculin A) and microtubule assembly (nocodazole). When platelets exposed to 2 µM latrunculin adhered to protein microdots, platelet spreading and P-selectin expression beyond the microdot boundaries were reduced and the overall spread surface area was smaller than those of control platelets. In contrast, exposure to 2 µM nocodazole did not affect platelet morphology or area, and platelets still spread beyond the geometric constraints of the protein microdots (Figure 5A). In addition, on smaller microdots, latrunculin-treated platelets adhered and spread to encompass the entire surface of the micropattern; however, platelet spreading beyond the geometric constraints of the protein micropattern was completely inhibited (supplemental Figure 7). These results indicate that rearrangement of actin, but not microtubules, is essential for platelet extension and α-granule secretion beyond boundaries of protein microdots.
Figure 5.

The actin cytoskeleton mediates spatial regulation of platelet spreading and α-granule secretion in a Rac1- and Rho-dependent manner. (A) Latrunculin A inhibits platelet (red, cell membrane stain) spreading and P-selectin expression (green) beyond the microdot patterns, whereas nocodazole has no effect. (B) Phalloidin binding (red) shows thick bundles of filamentous actin (f-actin) at the geometric boundaries of the micropatterns and at platelet-platelet interconnections. The thick f-actin “rings” are observed on smaller microdots (4 µm), but f-actin is dispersed over the entire microdot region as microdot diameter increases (5 µm). P-selectin binding is mutually excluded from these thick f-actin bundles but colocalize with thinner strands of filopodial f-actin (arrow), where platelets extend beyond the microdot boundaries. (C) Platelets (red) adhere and spread onto 5-µm fibrinogen microdot arrays (blue). Platelets treated with Y27632 exhibit more spreading beyond the microdot boundaries and formed more platelet connections, whereas platelets treated with ML-7 and NSC23766 exhibit less spreading beyond the microdot boundaries and decreased platelet connections. F-actin bundles (green) are observed at platelet-platelet connections in the control condition and Y27632-treated platelets, but not in ML-7– and NSC23766-treated platelets, although actin stress fiber expression within the microdot boundaries appears intact. (D) Platelet-platelet connections on collagen (left) and fibrinogen (right) microdot arrays (2700-dot arrays per condition) were quantified and normalized by vehicle control. All conditions are significantly different compared with the controls (P < .0005) using the Mann-Whitney U test. All samples are permeabilized upon staining. The scale bars = 5 µm; error bars indicate SE.
Next, we stained platelets spread on protein micropatterns with fluorescent phalloidin, which binds to polymerized filamentous actin (f-actin), and we also stained with anti–P-selectin. Platelets spread on the microdots showed thick bundles of f-actin along the geometric boundaries inside the micropatterns (Figure 5B, 4 µm). Thinner f-actin bundles were also observed at peripheries of the filopodia/lamellipodia, and also at platelet connections between 2 adjacent microdots. When the microdot was larger, thick bundles of f-actin were more dispersed within the microdots (Figure 5B, 5 µm). Although P-selectin expression did not colocalize with, but in fact was excluded by, these thick bundles of f-actin, P-selectin did colocalize with thinner strands of filopodial and lamellipodial actin in regions of the platelets that spread beyond the microdot boundaries (Figure 5B, right).
Spatial regulation of platelet spreading and α-granule secretion are mediated by Rac1
We used other pharmacologic cytoskeletal inhibitors to elucidate the underlying molecular mechanisms of how platelets sense cues in matrix geometry and biologically respond in a spatially regulated manner. Because actomyosin activity and rearrangement contribute to platelet granule secretion,29 we exposed platelets on collagen and fibrinogen microdots to ML-7, a myosin light-chain kinase inhibitor, and Y27632, an inhibitor of Rho-kinase. We then counted the number of platelets that extended beyond the microdot boundaries and formed connections between adjacent microdots. Compared with platelets under control conditions, platelets treated with ML-7 formed significantly fewer platelet-platelet interconnections. Interestingly, platelets treated with Y27632 formed significantly more connections compared with both ML-7–treated and control platelets (Figure 5C).
Rho inhibition is known to increase Rac activity and vice versa in nucleated adherent cells.30 Therefore, to evaluate the role of Rac1, which is associated with platelet adhesion, spreading, granule secretion, and aggregation,31,32 we exposed platelets to NSC23766, a Rac1 inhibitor. Platelets exposed to NSC23766 spread over the entire protein microdot area but did not spread beyond the geometric boundaries (Figure 5C and supplemental Figure 8). In addition, platelets exposed to NSC23766 formed fewer than half the number of platelet-platelet connections compared with platelets under control conditions, whereas platelets exposed to Y27632 formed approximately 3 times the number of connections as control platelets (Figure 5D). When platelets were exposed to Y27632 or NSC23766 in a standard platelet adhesion assay (with no geometric patterning of extracellular proteins) and stained with phalloidin, Y27632-exposed platelets exhibited high densities of filamentous actin at the leading edges, suggesting a pro-lamellipodial state, whereas NSC23766-exposed platelets exhibited high densities of actin stress fibers but no filamentous actin at the leading edges, suggesting a pro-adhesion state (supplemental Figure 9). These experiments indicate that Rac1 plays an important role in how platelets transduce the spatial cues of the microenvironment to create a cytoskeletal response that enables platelet spreading beyond the geometric boundaries of extracellular matrices, while also implicating Rho as a negative regulator of this process.
Spatially regulated platelet spreading and α-granule secretions are impaired in gray platelet syndrome and Wiskott-Aldrich syndrome
To further evaluate the role of α-granules in directional spreading, adhesion and spreading of α-granule–deficient platelets from a patient with GPS were examined on collagen micropatterns. We used matrices of “microstrips” to measure filopodia projections away from the platelet body. In control platelets, directed spreading with extended filopodia formation oriented away from the collagen strip was observed, whereas GPS platelets demonstrated very little directional filopodia formation and tended not to spread beyond micropattern boundaries (Figure 6A-B). In addition, healthy control platelets readily formed platelet-platelet interconnections between adjacent microdots, whereas GPS platelets were significantly less likely to form connections (Figure 6C), suggesting that filopodia extension to form the platelet-platelet connection is oriented by the underlying matrix in an α-granule–dependent manner.
Figure 6.

Spatially regulated platelet spreading and α-granule secretion are impaired in gray platelet syndrome and Wiskott-Aldrich syndrome. (A) Gray platelet syndrome (GPS) patient platelets, which are deficient in α-granules, adhere and spread onto collagen microstrip patterns (blue). Healthy donor platelets extend filopodial projections (red) on protein microstrips, whereas GPS platelets remained geometrically constrained within the boundaries of the protein microstrips. (B) The number of filopodia extending beyond the collagen microstrips are significantly reduced in GPS platelets (P < .05). (C) In addition, platelet-platelet interconnections between adjacent protein microdots (of 5- and 10-μm diameters) are significantly reduced in GPS platelets (P < .0001 for both sizes of microdots). (D) Compared with healthy control platelets, Wiskott-Aldrich syndrome platelets exhibit significantly less spreading areas beyond the microdot boundaries. The scale bars = 10 μm; error bars indicate SE.
Finally, we compared healthy control platelets to platelets isolated from a patient with WAS. WAS is caused by mutations of the WAS protein gene, which adversely affects Arp2/3- and Cdc42-mediated actin polymerization.33 WAS platelets are smaller than healthy control platelets and have impaired outside-in αIIbβ3 signaling and abnormal α-granule distributions.24,34 When adhered to protein microdots, WAS platelets exhibited less spreading beyond the microdot boundaries compared with healthy wild-type platelets (Figure 6D). As expected, on non–geometrically constrained areas of collagen, spread areas of WAS and normal platelets were 12.7 ± 1.3 µm2 vs 17.3 ± 1.1 µm2, respectively. However, this 26.5% difference in spread area likely does not account for the vast difference we observed in spatially regulated spreading on protein microdots (Figure 6D; 70.1%, 76.2%, 85.2%, and 91.2% differences on 2-, 3-, 4-, and 5-µm diameter microdots, respectively; P < .01), suggesting that intact cytoskeletal machinery and/or integrin αIIbβ3 activation are essential for spreading beyond the geometric constraints of the matrix.
Discussion
These studies demonstrate that the geometric orientation of the matrix itself can regulate platelet function at the subcellular level. Specifically, our protein micropattern data show that platelets transduce geometric cues of the matrix into intracellular signals to spatially orient granule exocytosis and enable deposition of a matrix that supports filopodial extension and lamellipodial spreading beyond a preexisting matrix. Physiologically, this phenomenon may serve several purposes.
First, this capability may enable platelets to achieve hemostasis in small, subclinical perturbations of the endothelium. Platelets have been shown to fill small “gaps” of exposed subendothelial matrix that are significantly smaller than the platelets themselves.4 In this context, however, because the overall force of adhesion correlates with the contact area, the platelets must maintain sufficient adhesion to resist the dynamic shear forces of the circulation. Secreting α-granule contents, which contain matrix proteins such as fibrinogen and fibronectin, adjacent to those gaps may enable platelets to establish firmer adhesion by increasing their effective spreading area, which our data indicate to be >8 µm2 (Figure 1B). In addition, exposed P-selectin on the platelet surface via α-granule exocytosis may play critical roles in platelet adhesion to endothelial cells at the site of vascular injury. PSGL-1, a ligand for P-selectin, is expressed in microvascular and venular endothelium35,36 and has been shown to directly mediate adhesion of platelet-monocyte complexes.37 As such, when platelets encounter small areas of exposed subendothelium, spatially regulated α-granule exocytosis may enable platelets to establish adhesion beyond the area of exposed subendothelial matrix via self-deposition of adhesive matrix as well as P-selectin-PSGL interactions with the surface of contiguous endothelial cells (Figure 7). This process may also represent an underlying mechanism whereby platelets support and maintain vascular integrity during angiogenesis and inflammation.5
Figure 7.

Proposed role of spatial regulation of platelet α-granule secretion in hemostasis and maintenance of endothelial integrity. As a platelet under flow (left) encounters and initially adheres to a small site of vascular injury (middle), α-granules will redistribute and exocytose their contents primarily in regions beyond the geometric boundaries of the exposed subendothelial matrix. This spatially regulated α-granule secretion results in platelet “self-deposition” of adhesive matrix proteins such as fibrinogen and fibronectin, high expression of P-selectin beyond the matrix boundaries, and increased cell membrane to extend the overall platelet spreading area (right). Adhesion in those regions likely involves integrin αIIbβ3 redistribution and activation as well as P-selectin binding to PSGL-1 on the endothelial cell surface (zoomed in).
In addition, spatial regulation of α-granule secretion may reinforce platelet-platelet connections between platelets adhered on small areas of exposed subendothelial matrix as platelets on adjacent protein microdots form bridging connections with associated high levels of α-granule secretion in those regions (Figure 2C). Because α-granules contain fibronectin and fibrinogen, which serve as the primary bridging molecules between platelets, concentration of α-granule secretion at those connections may increase the number of αIIbβ3-fibrinogen-αIIbβ3 interactions at those sites, thereby enhancing platelet-platelet adhesion.
We discovered that the underlying molecular mechanisms of this process involve the Rho GTPases, specifically Rac1 and Rho. In nucleated mammalian cells, Rac1 expression leads to lamellipodia formation, whereas Rho expression leads to the formation of stress fibers and focal adhesions.38 Expression of Rac1 can result in downregulation of Rho and vice versa, and inhibition of one can lead to upregulation of the other and vice versa, so nucleated cells are often balancing a pro-lamellipodial (Rac1) state or pro-stress fiber (Rho) state. Our findings in platelets are consistent with these observations. Platelets exposed to the Rac1 inhibitor NSC23766 spread within the protein microdot area but remained geometrically constrained (Figure 5C-D), which is consistent with the lamellipodial inhibition in NSC23766-exposed nucleated cells38,39 and platelets.31,32 Platelets exposed to the ROCK inhibitor Y27632 downregulate Rho, which is known to increase pro-spreading Rac1 activity in other cells and, interestingly, caused a marked increase in actin-rich, platelet-platelet connections (Figure 5C-D). As such, the balance between Rac1 and Rho activity dictates the spatial regulation of α-granule–mediated platelet spreading.
The molecular mechanisms that dictate α-granule secretion have been evaluated primarily in suspension platelets, using assays that measure bulk secretion.12 Physiologically relevant granule release, however, occurs after adhesion of platelets to a matrix and in the context of spreading, and would involve remodeling of the actin cytoskeleton. In fact, our studies using protein micropatterns show that actin polymerization controls the spatial regulation of granule secretion by platelets. Specifically, granule release is directed away from sites of actin polymerization to increase the plasma membrane surface area in a lateral dimension. Such membrane may be necessary to cover growing actin structures during spreading. In the absence of functional α-granules, such as in GPS platelets, filopodial formation is markedly decreased (Figure 6A-B). The possibility that α-granule secretion at the periphery of a platelet spreading on a protein microdot may be caused in part by physical exclusion cannot be ruled out. However, even if this were the case, this process would still be spatially regulated because the end result is secretion of α-granular contents that conform to the geometric constraints of the underlying matrix. These studies also support a second function of α-granule secretion during spreading: deposition of matrix proteins to support the outgrowth of platelet filopodia.
Overall, these studies demonstrate a previously unrecognized relationship between platelet actin polymerization, αIIbβ3 redistribution and activation, and α-granule secretion that directs spatially oriented secretion and spreading. On the basis of these studies, we propose the following mechanistic model. First, platelet adhesion onto <20 µm2 areas of exposed matrices results in spatially regulated actin polymerization at adhesion sites. Via Rac1, platelet filopodial extension, αIIbβ3 redistribution, and α-granule release are then oriented away from the sites of adhesion. Directed α-granule release results in the deposition of matrix proteins such as fibrinogen and fibronectin from the α-granule stores. This “self-deposition” of matrix facilitates αIIbβ3-mediated spreading beyond the initial area of adhesion, and α-granule exocytosis provides additional membrane from fused granules to cover growing actin structures. In addition, exposed P-selectin binds to PSGL-1 on endothelial cells to secure the platelet–endothelial cell interaction (Figure 7). Such spatial regulation of platelet spreading could facilitate lateral coverage of denuded endothelium and contribute to vascular hemostasis.
Acknowledgments
The authors thank the entirety of the Lam laboratory, including E. T. Hardy, M. Rollins, H. A. Gole, E. Tyburski, J. Ciciliano, and R. Mannino, for thoughtful discussions.
This study was supported by NSF CAREER Award 1150235 and National Institutes of Health National Heart, Lung, and Blood Institute grants U54 HL112309 and R01 HL121264 (W.A.L.).
Footnotes
The online version of this article contains a data supplement.
There is an Inside Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: W.A.L., R.F., and Y.S. planned all experiments; Y.S., J.L.F.-T., Y.Q., B.A., R.T., D.R.M., and M.E.F. conducted experiments and data analysis; P.W.S. and L.D. assisted with TIRF microscopy experiments; A.D.M. provided vital patient samples; and W.A.L., R.F., and Y.S. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wilbur A. Lam, Emory University School of Medicine, 2015 Uppergate Dr NE #448, Atlanta, GA 30322; e-mail: wilbur.lam@emory.edu.
References
- 1.Brass LF, Wannemacher KM, Ma P, Stalker TJ. Regulating thrombus growth and stability to achieve an optimal response to injury. J Thromb Haemost. 2011;9(Suppl 1):66–75. doi: 10.1111/j.1538-7836.2011.04364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koseoglu S, Flaumenhaft R. Advances in platelet granule biology. Curr Opin Hematol. 2013;20(5):464–471. doi: 10.1097/MOH.0b013e3283632e6b. [DOI] [PubMed] [Google Scholar]
- 3.Coller BS. Historical perspective and future directions in platelet research. J Thromb Haemost. 2011;9(Suppl 1):374–395. doi: 10.1111/j.1538-7836.2011.04356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tranzer JP, Baumgartner HR. Filling gaps in the vascular endothelium with blood platelets. Nature. 1967;216(5120):1126–1128. doi: 10.1038/2161126a0. [DOI] [PubMed] [Google Scholar]
- 5.Ho-Tin-Noé B, Demers M, Wagner DD. How platelets safeguard vascular integrity. J Thromb Haemost. 2011;9(Suppl 1):56–65. doi: 10.1111/j.1538-7836.2011.04317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stalker TJ, Traxler EA, Wu J, et al. Hierarchical organization in the hemostatic response and its relationship to the platelet-signaling network. Blood. 2013;121(10):1875–1885. doi: 10.1182/blood-2012-09-457739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Welsh JD, Stalker TJ, Voronov R, et al. A systems approach to hemostasis: 1. The interdependence of thrombus architecture and agonist movements in the gaps between platelets. Blood. 2014;124(11):1808–1815. doi: 10.1182/blood-2014-01-550335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blair P, Flaumenhaft R. Platelet alpha-granules: basic biology and clinical correlates. Blood Rev. 2009;23(4):177–189. doi: 10.1016/j.blre.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woronowicz K, Dilks JR, Rozenvayn N, et al. The platelet actin cytoskeleton associates with SNAREs and participates in alpha-granule secretion. Biochemistry. 2010;49(21):4533–4542. doi: 10.1021/bi100541t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge S, White JG, Haynes CL. Cytoskeletal F-actin, not the circumferential coil of microtubules, regulates platelet dense-body granule secretion. Platelets. 2012;23(4):259–263. doi: 10.3109/09537104.2011.620657. [DOI] [PubMed] [Google Scholar]
- 11.Flaumenhaft R, Dilks JR, Rozenvayn N, Monahan-Earley RA, Feng D, Dvorak AM. The actin cytoskeleton differentially regulates platelet alpha-granule and dense-granule secretion. Blood. 2005;105(10):3879–3887. doi: 10.1182/blood-2004-04-1392. [DOI] [PubMed] [Google Scholar]
- 12.Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apheresis Sci. 2003;28(3):307–317. doi: 10.1016/S1473-0502(03)00050-8. [DOI] [PubMed] [Google Scholar]
- 13.Peters CG, Michelson AD, Flaumenhaft R. Granule exocytosis is required for platelet spreading: differential sorting of α-granules expressing VAMP-7. Blood. 2012;120(1):199–206. doi: 10.1182/blood-2011-10-389247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bretscher MS. Moving membrane up to the front of migrating cells. Cell. 1996;85(4):465–467. doi: 10.1016/s0092-8674(00)81246-5. [DOI] [PubMed] [Google Scholar]
- 15.Martinez-Arca S, Alberts P, Zahraoui A, Louvard D, Galli T. Role of tetanus neurotoxin insensitive vesicle-associated membrane protein (TI-VAMP) in vesicular transport mediating neurite outgrowth. J Cell Biol. 2000;149(4):889–900. doi: 10.1083/jcb.149.4.889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Arca S, Coco S, Mainguy G, et al. A common exocytotic mechanism mediates axonal and dendritic outgrowth. J Neurosci. 2001;21(11):3830–3838. doi: 10.1523/JNEUROSCI.21-11-03830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alberts P, Rudge R, Irinopoulou T, Danglot L, Gauthier-Rouvière C, Galli T. Cdc42 and actin control polarized expression of TI-VAMP vesicles to neuronal growth cones and their fusion with the plasma membrane. Mol Biol Cell. 2006;17(3):1194–1203. doi: 10.1091/mbc.E05-07-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bi GQ, Alderton JM, Steinhardt RA. Calcium-regulated exocytosis is required for cell membrane resealing. J Cell Biol. 1995;131(6 Pt 2):1747–1758. doi: 10.1083/jcb.131.6.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Braun V, Fraisier V, Raposo G, et al. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J. 2004;23(21):4166–4176. doi: 10.1038/sj.emboj.7600427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kita A, Sakurai Y, Myers DR, et al. Microenvironmental geometry guides platelet adhesion and spreading: a quantitative analysis at the single cell level. PLoS ONE. 2011;6(10):e26437. doi: 10.1371/journal.pone.0026437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corum LE, Eichinger CD, Hsiao TW, Hlady V. Using microcontact printing of fibrinogen to control surface-induced platelet adhesion and activation. Langmuir. 2011;27(13):8316–8322. doi: 10.1021/la201064d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basabe-Desmonts L, Ramstrom S, Meade G, et al. Single-step separation of platelets from whole blood coupled with digital quantification by interfacial platelet cytometry (iPC). Langmuir. 2010;26(18):14700–14706. doi: 10.1021/la9039682. [DOI] [PubMed] [Google Scholar]
- 23.Van de Walle AB, Fontenot J, Spain TG, et al. The role of fibrinogen spacing and patch size on platelet adhesion under flow. Acta Biomater. 2012;8(11):4080–4091. doi: 10.1016/j.actbio.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kajiwara M, Nonoyama S, Eguchi M, et al. WASP is involved in proliferation and differentiation of human haemopoietic progenitors in vitro. Br J Haematol. 1999;107(2):254–262. doi: 10.1046/j.1365-2141.1999.01694.x. [DOI] [PubMed] [Google Scholar]
- 25.Mangalpally KK, Siqueiros-Garcia A, Vaduganathan M, Dong JF, Kleiman NS, Guthikonda S. Platelet activation patterns in platelet size sub-populations: differential responses to aspirin in vitro. J Thromb Thrombolysis. 2010;30(3):251–262. doi: 10.1007/s11239-010-0489-x. [DOI] [PubMed] [Google Scholar]
- 26.Harrison P, Wilbourn B, Debili N, et al. Uptake of plasma fibrinogen into the alpha granules of human megakaryocytes and platelets. J Clin Invest. 1989;84(4):1320–1324. doi: 10.1172/JCI114300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zucker MB, Mosesson MW, Broekman MJ, Kaplan KL. Release of platelet fibronectin (cold-insoluble globulin) from alpha granules induced by thrombin or collagen; lack of requirement for plasma fibronectin in ADP-induced platelet aggregation. Blood. 1979;54(1):8–12. [PubMed] [Google Scholar]
- 28.Aslan JE, McCarty OJ. Rho GTPases in platelet function. J Thromb Haemost. 2013;11(1):35–46. doi: 10.1111/jth.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oury C, Sticker E, Cornelissen H, De Vos R, Vermylen J, Hoylaerts MF. ATP augments von Willebrand factor-dependent shear-induced platelet aggregation through Ca2+-calmodulin and myosin light chain kinase activation. J Biol Chem. 2004;279(25):26266–26273. doi: 10.1074/jbc.M402032200. [DOI] [PubMed] [Google Scholar]
- 30.Rottner K, Hall A, Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol. 1999;9(12):640–648. doi: 10.1016/s0960-9822(99)80286-3. [DOI] [PubMed] [Google Scholar]
- 31.Dwivedi S, Pandey D, Khandoga AL, Brandl R, Siess W. Rac1-mediated signaling plays a central role in secretion-dependent platelet aggregation in human blood stimulated by atherosclerotic plaque. J Transl Med. 2010;8:128. doi: 10.1186/1479-5876-8-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McCarty OJ, Larson MK, Auger JM, et al. Rac1 is essential for platelet lamellipodia formation and aggregate stability under flow. J Biol Chem. 2005;280(47):39474–39484. doi: 10.1074/jbc.M504672200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Notarangelo LD, Ochs HD. Wiskott-Aldrich Syndrome: a model for defective actin reorganization, cell trafficking and synapse formation. Curr Opin Immunol. 2003;15(5):585–591. doi: 10.1016/s0952-7915(03)00112-2. [DOI] [PubMed] [Google Scholar]
- 34.Shcherbina A, Cooley J, Lutskiy MI, Benarafa C, Gilbert GE, Remold-O’Donnell E. WASP plays a novel role in regulating platelet responses dependent on alphaIIbbeta3 integrin outside-in signalling. Br J Haematol. 2010;148(3):416–427. doi: 10.1111/j.1365-2141.2009.07959.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laszik Z, Jansen PJ, Cummings RD, Tedder TF, McEver RP, Moore KL. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood. 1996;88(8):3010–3021. [PubMed] [Google Scholar]
- 36.Rivera-Nieves J, Burcin TL, Olson TS, et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med. 2006;203(4):907–917. doi: 10.1084/jem.20052530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.da Costa Martins P, García-Vallejo JJ, van Thienen JV, et al. P-selectin glycoprotein ligand-1 is expressed on endothelial cells and mediates monocyte adhesion to activated endothelium. Arterioscler Thromb Vasc Biol. 2007;27(5):1023–1029. doi: 10.1161/ATVBAHA.107.140442. [DOI] [PubMed] [Google Scholar]
- 38.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279(5350):509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 39.Hung WC, Chen SH, Paul CD, et al. Distinct signaling mechanisms regulate migration in unconfined versus confined spaces. J Cell Biol. 2013;202(5):807–824. doi: 10.1083/jcb.201302132. [DOI] [PMC free article] [PubMed] [Google Scholar]