

Abstract
Imines, carbon-nitrogen double bonds, are fundamentally important functional groups in organic chemistry. This is largely due to the fact that imines act as electrophiles in C–C bond forming reactions towards carbon nucleophiles, thereby serving one of the most widely used precursors for the formation of amines in both synthetic and biosynthetic settings.1–5 If the carbon atom of the imine could be rendered electron-rich, the imine could react as a nucleophile instead of as an electrophile. Such a reversal in the electronic characteristics of the imine functionality would facilitate the development of new chemical transformations that convert imines into amines via C–C bond forming reactions with carbon electrophiles, thereby creating new opportunities for the efficient synthesis of amines. The development of asymmetric ‘umpolung’ reactions of imines remains an uncharted ground, in spite of the far-reaching impact of such reactions in organic synthesis. Here we report the discovery and development of new chiral phase transfer catalysts that promote the highly efficient asymmetric umpolung reactions of imines and enals. These catalysts mediate the deprotonation of imines and direct the 2-azaallylanions thus formed to react in a highly chemoselective, regioselective, diastereoselective and enantioselective fashion with enals. The reaction tolerates a broad range of imines and enals, and can be carried out in high yield with as little as 0.01 mol % catalyst with a moisture and air-tolerant operational protocol. These umpolung reactions provide a conceptually new and practical approach towards chiral amino compounds.
Umpolung reactions create new activities by reversing the inherent polarity of common organic functionalities such as carbonyls and consequently allow the development of new reactions of distinct bond connections.6 The successful development of numerous C–C forming umpolung reactions with carbonyls as acyl anion equivalents has greatly expanded the repertoire of organic synthesis.7–9 The power of carbonyl umpolung reactions was tapped for asymmetric synthesis through the successful development of efficient chiral catalysts for enantioselective Stetter reactions and other asymmetric reactions.10 In contrast C–C bond forming umpolung reactions of imines are rarely reported.11–14 Aiming at the realization of highly efficient catalytic asymmetric umpolung reactions of imines, we embarked on a search for catalysts to both promote the formation of carbanions from imines and direct the carbanions thus formed to react with carbon electrophiles to generate chiral amines in an asymmetric fashion.
We recently reported that modified cinchona alkaloids such as Q-2 could promote highly enantioselective isomerization of trifluoromethyl imines (Fig. 1).15, 16 This reaction presumably proceeds through first the formation of 2-azaallylanion 3 then a highly enantioselective protonation of 3. This discovery prompted us to postulate that, if the 2-azaallylanions 3 could be made to react with carbon electrophiles in a stereoselective manner, novel C–C bond forming asymmetric reactions transforming imines 1 into enantioenriched amines could be realized (Fig. 1). Although numerous catalytic asymmetric C–C bond forming reactions with enolates derived from glyoxylateimines14 and glycine imines17 have been documented for the synthesis of amino acids, only two catalytic asymmetric C–C bond forming reactions with 2-azaallylanions are reported.18, 19 The Pd-catalyzed cross coupling of 2-azaallylanions with aryl halides and triflates remains the sole example of highly enantioselective C–C bond forming reactions with 2-azaallylanions.18
Figure 1.
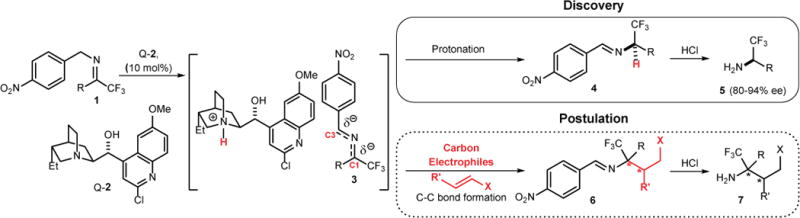
Design of a catalytic C–C bond forming umpolung reaction of imines.
Guided by these considerations, we investigated cinchona alkaloid-derived organocatalysts 2, 11 and 12 for the reaction of imine 1A and crotonaldehyde (8a) (Table 1). None of them was active toward the desired C–C bond forming reaction; only the isomerized imine 4A was detected. These catalysts promoted the deprotonation of trifluoromethyl imine 1A to form the 2-azaallyl anion 3, but were unable to direct the conjugate addition of 3 to crotonaldehyde. Presumably, the protonated cinchona alkaloids formed on deprotonation of 1A rapidly protonates 3 to form 4A. As the 2-azaallylanion 3 were shown to engage in protonation in the presence of a proton donor, we surmise that a novel class of catalysts must be developed to afford the required chemoselectivity in favor of the C–C bond formation over the protonation.
Table 1.
Attempts with chiral base catalysts

| ||||
|---|---|---|---|---|
| Entry | T (°C) | catalyst | conversion (%) | 9/4 |
| 1 | rt | Q-2 | 84 | 0/100 |
| 2 | rt | QD-11 | 32 | 0/100 |
| 3 | rt | QD-12 | 9 | 0/100 |
Conditions: 10 mol % cat., 16 h.
We decided to explore chiral phase transfer catalysts.20 Under phase transfer catalysis conditions stronger bases could be explored for the deprotonation of imine 1 to form 2-azaallylanion 3. Furthermore, in the absence of a protonated cationic species, 3 should be less prone to protonation and therefore more likely to engage in the addition to 8a. A cinchonine-derived phase transfer catalyst 13 was first investigated to promote the reaction of 1A and 8a in toluene and aqueous KOH at room temperature. The desired amine 9Aa was formed, albeit in miniscule amounts (entry 1, Table 2). Importantly, the chemoselectivity for the C–C bond formation could be improved with catalyst 14 bearing PYR, a bulky heteroaryl group, although both the reaction conversion and the chemoselectivity remained poor (entry 2). Subsequently, we found that a reaction at lower temperature afforded significantly improved conversion and chemoselectivity. The absence of 10Aa, which would be formed by conjugate addition from the other end of the 2-azaallylanion, is noteworthy. However, amine 9Aa was formed with moderate diastereoselectivity and poor enantioselectivity.
Table 2.
Screening and optimization of chiral phase transfer catalysts

| ||||||
|---|---|---|---|---|---|---|
| Entry | T (°C) | catalyst | conversion (%) | 9/4; 9/10 | d.r. of 9 | ee (%) |
| 1 | rt | C-13 | 41 | 2/98; – | – | – |
| 2 | rt | C-14 | 18 | 11/89; – | – | – |
| 3 | −20 | C-14 | 58 | 37/63; >95/5 | 82/18 | 39 |
| 4 | −20 | C-15 | 54 | 36/64; >95/5 | 67/33 | 18 |
| 5 | −20 | C-16 | 84 | 34/66; >95/5 | 76/24 | 40 |
| 6* | −20 | C-16 | 41 | 32/68; >95/5 | 74/26 | 39 |
| 7* | −20 | C-17 | 14 | 67/33; >95/5 | 87/13 | 68 |
| 8* | −20 | C-18 | 40 | 74/26; >95/5 | 86/14 | 77 |
| 9* | −20 | C-19 | 39 | 45/55; >95/5 | 96/4 | 55 |
| 10* | −20 | C-20 | 66 | 68/32; >95/5 | 91/9 | 85 |
| 11* | −20 | C-21a | 88 | 94/6; >95/5 | 91/9 | 91 |
| 12* | −20 | C-21b | 99 | 99/1; >95/5 | 93/7 | 96 |
| 13† | −20 | C-21b | 97 | 99/1; >95/5 | 93/7 | 95 |
| 14* | −20 | TBAB | 31 | 4/96; – | – | – |
Conditions: 10 mol % cat., 10 mol % KOH(aq.), 16 h. TBAB, Tetra-n-butylammonium bromide.
1.0 mol % cat., 10 mol % KOH(aq.), 2 h
0.2 mol % of C-21b used, 5 h.
Introducing an additional interaction between a conformational well-defined phase transfer catalyst and the anionic nucleophile has proven to be a useful strategy to enhance catalytic selectivity.21 We hypothesized that a cinchonine-derived phase transfer catalyst bearing a properly located aromatic group with suitable electronic properties might interact with 2-azaallylanion 3A via both ionic and π–π interactions,22–24 thereby mediating the model umpolung reaction in a highly chemo-, regio-, diastereo- and enantio-selective fashion. Analogues C-15 and C-16 bearing electron withdrawing and donating N-benzyl substituents, respectively, were examined. We found that C-16 afforded only improved conversion whereas C-15 was worse than 14 (entries 4–5). Interestingly, we observed that a decrease in the loading of C-16 did not affect negatively on the catalytic selectivities (entries 6 vs. 5). We therefore decreased the catalyst loading from 10 mol% to 1 mol% in our subsequent catalyst screening and optimization studies. We next turned to C-17, an analogue containing a biphenyl group. C-17 afforded dramatically improved chemo-, diastereo- and enantioselectivity, thereby allowing amine 9Aa to be formed as the major product (entry 7 vs. 6).
Assuming the improved catalysis resulted from a π–π interaction between the biaryl moiety of C-17 and 3A, we designed and synthesized catalyst C-18. We reasoned that the presence of the C2-symmetric terphenyl moiety could render C-18 a more efficient catalyst than C-17. This working hypothesis received support from the superior performance of C-18 in catalytic activity as well as chemo- and enantio-selectivity (entry 8 vs. 7). Further tuning of the terphenyl moiety was initially attempted by introducing electron-withdrawing and –donating groups on the 3- and 5-phenyl groups. Catalyst C-20 (entry 10) bearing an electron-rich terphenyl group performed better than C-19 (entry 9) which contained an electron-deficient terphenyl moiety. However, C-20 furnished higher stereoselectivity but lower chemoselectivity than those by C-18 (entry 10 vs. 8).
We next examined catalyst C-21a, which was designed to create an electron-rich terphenyl moiety with an electron-donating substituent in a position not causing obstructive steric interference between the catalyst and 2-azaallyanion 3. Gratifyingly, C-21a not only turned out to be much more active, but also afforded 9Aa with synthetically useful chemo-, regio-, diastereo- and enantioselectivity (entry 11). Catalyst C-21b with a more electron-donating and bulky OTBS group was more active and selective, even in a loading of only 0.2 mol% producing imine 9Aa rapidly with almost complete chemoselectivity and excellent stereoselectivity. We attributed the superiority of C-21b over C-21a to two factors resulting from the substitution of the 4-methoxy with the 4-OTBS group: 1) the terphenyl moiety is more electron rich due to the presence of the more electron donating 4-OTBS group; 2) the terphenyl moiety has less conformational flexibility due to steric hindrance of the rotation of the 3,5-phenyl rings by the bulky 4-OTBS group. Both factors could reinforce the π–π interaction between the 3A and the catalyst C-21b.
Only a trace of 9Aa was formed from 1A and 8a using tetrabutylammonium bromide (TBAB) as the quaternary ammonium salt (entry 14); which confirmed that the structural characteristics of C-21b were responsible for both the catalytic activity and selectivity observed for the umpolung reaction between imine 1A and enal 8a. To ascertain that 2-azaallyl anion 3 originated only from imine 1 rather than also from the isomerized imine 4, we established that no reaction occurred between 4A and 8a under the optimized conditions. It should be noted that amine 9Aa may also form via a [3+2] cycloaddition between 1A and 8a followed by a retro-Mannich reaction. However, we did not detect the formation of the [3+2] adduct when monitoring the reaction by 1H and 19F NMR analyses.
Our investigation of the substrate scope began with the reaction of 1A and 8a with 0.2 mol% of C-21b (entry 1, Table 3). The reaction proceeded to full conversion within 5 h with excellent chemo-, regio-, diastereo- and enantio-selectivities. The optically active amine 9Aa was then converted to the more stable N-benzyl aminoalcohol 22Aa by reducing first the aldehyde with NaBH4 and then the imine with NaBH4 and acetic acid, which could be readily isolated as a single diastereomer in good yield. Reactions of 8a with a series of trifluoromethyl imines (1B–E) bearing simple and functionalized linear alkyl substituents consistently proceeded in high yield and excellent chemoselectivity and stereoselectivity. The reaction tolerated an imine bearing a β-branched alkyl substituent (1F). The reaction accepted larger β-alkyl groups on the enal (entries 7–8). Cinnamaldehyde (8d) reacted with 1A to give a 68:32 mixture of the desirable amine 9Ad and the regioisomer 10Ad. Nonetheless, 9Ad was produced with high chemo-, diastereo- and enantioselectivity in synthetically useful yield (entry 9).
Table 3.
Substrate scope for umpolung reactions of trifluoromethyl imines with enals

| ||||||
|---|---|---|---|---|---|---|
| Scope of imines in reactions with crotonaldehyde (8a, R2 = Me)
| ||||||
| Entry | R1 | time (h); conversion (%) |
9/4; 9/10 | d.r. of 9 | yield (%)* | ee (%)† |
| 1 |
|
5; 99 | >95/5; >95/5 | 93/7 | 81 (22Aa) | 95 |
| 2 |
|
5; 97 | >95/5; >95/5 | 91/9 | 84 (22Ba) | 94 |
| 3 |
|
5; 98 | >95/5; >95/5 | 91/9 | 83 (22Ca) | 96 |
| 4 |
|
5; 99 | >95/5; >95/5 | 91/9 | 75 (22Da) | 96 |
| 5 |
|
7; 94 | >95/5; >95/5 | 91/9 | 72 (22Ea) | 96 |
| 6 |
|
12; 98 | 91/9; >95/5 | 93/7 | 54 (22Fa) | 95 |
| Scope of β-substituted enals in reactions with imine 1A
| ||||||
|---|---|---|---|---|---|---|
| Entry | R2 | time (h); conversion (%) |
9/4; 9/10 | d.r. of 9 | yield (%)* | ee (%)† |
| 7 | CH3CH2; 8b | 5; 99 | 89/11; >95/5 | >95/5 | 64 (22Ab) | 95 |
| 8 | CH3(CH2)5; 8c | 12; 93 | 86/14; >95/5 | >95/5 | 51 (22Ac) | 96 |
| 9 | Ph; 8d | 8; 93 | >95/5; 68/32 | >95/5 | 51 (22Ad) | 91 |
| Scope of imines in reactions with acrolein (8e, R2 = H)
| |||||
|---|---|---|---|---|---|
| Entry | R1 | time (h); conversion (%) |
9/4; 9/10 | yield (%)* | ee (%)† |
| 10ˆ |
|
3; 95 | >95/5; >95/5 | 89 (22Ae) | 92 |
| 11ˆ |
|
3; 99 | >95/5; >95/5 | 82 (22Be) | 91 |
| 12ˆ |
|
3; 97 | >95/5; >95/5 | 84 (22De) | 91 |
| 13ˆ |
|
3; 99 | >95/5; >95/5 | 90 (22Fe) | 92 |
| 14 | Ph; 1G | 3; 99 | 94/6; >95/5 | 71 (23Ge) | 94 |
| 15 | p-MeOC6H4; 1H | 3; 94 | 92/8; >95/5 | 67 (23He) | 94 |
| 16 | p-CF3C6H4; 1I | 3; 99 | 88/12; >95/5 | 78 (23Ie) | 92 |
| 17 |
|
1; 99 | >95/5; >95/5 | 90 (23Je) | 93 |
Conditions: imine 1 (0.2 mmol), aldehyde 8 (0.4 mmol), C-21b (0.2 mol%), KOH (2.2 uL, 50 wt% aq., 10 mol%), PhMe (2.0 mL). Conversion, Regioselectivity (9/10) and d.r. of 9 were determined by 1H NMR analysis of the crude umpolung reaction mixture. Chemoselectivity (9/4) was determined by 19F NMR analysis.
Overall yield for the transformation of imine 1 to either 22 or 23.
ee of 22 or 23 was determined by HPLC analysis.
Reaction was performed at −10 °C.
We next examined the reactions of trifluoromethylated imines 1 with acrolein (8e). We found that at −10 °C the reaction between 1A and 8e proceeded cleanly and in a highly enantioselective fashion to furnish the corresponding amine 9Ae as the only detectable product by NMR analysis of the crude reaction mixture. The reactions of acrolein (8e) with trifluoromethyl imines 1 bearing a variety of alkyl, aryl and alkenyl substituents were equally successful, affording the corresponding trifluoromethylated amines 9 containing a tetrasubstituted stereocenter25, 26 in high optical purity (entries 11–17). Alkyl trifluoromethylated amines (9Ae–9Fe) were converted to N-benzyl aminoalcohols 22 (entries 10–13). Aryl and alkenyl amines 9Ge–9Je were converted to aminoalcohols 23 by reduction of the aldehyde with NaBH4 and hydrolysis of the imine with aqueous HCl (entries 14–17). In all these cases, the aminoalcohols 22 and 23 were obtained in good yields and high optical purity.
A gram-scale reaction of 1A with 8a with 0.01 mol % of C-21b went to completion without deterioration in selectivity (Fig. 2a). This remarkable catalytic efficiency indicates the utility of this new reaction in preparative-scale organic synthesis.27 To demonstrate the synthetic versatility of this reaction, we converted chiral aminoaldehyde 9Aa to aminoalcohol 23Aa and pyrrolidine 24Aa as shown in Figure 2a. Similarly, the phenyl substituted product 9Ge was converted to pyrrolidine 24Ge (Fig. 2b). The absolute configurations of 24Aa and 24Ge were determined by X-ray crystallography.
Figure 2.

Gram scale reaction and synthetic applications.
We are interested in extending the scope to simple imines, which would greatly expand the reach of this asymmetric umpolung reaction in organic synthesis. However, 2-azaallylanions 26 derived from aryl imines 25 are substantially less stable than those derived from the corresponding trifluoromethyl imines 1. Furthermore regioselectivity control for the electrophilic reaction with an unsymmetrically substituted 1,3-diaryl-2-azaallylanions 26 might prove difficult. (Fig. 3a). For example, deprotonation of phenyl imine 25A should form 2-azaallylanion 26A, which is flanked by the phenyl and the 4-nitrophenyl rings (Fig. 3b). Thus, there is an inherent electronic bias for an electrophile to react with 26A by attacking preferentially the more electron-rich C3.28 Nonetheless, the remarkable catalytic efficiency of C-21b made us hopeful that it could provide powerful catalytic activity and selectivity to overcome this undesirable substrate bias while still affording the required stereoselectivity for an efficient asymmetric imine umpolung reaction.
Figure 3.
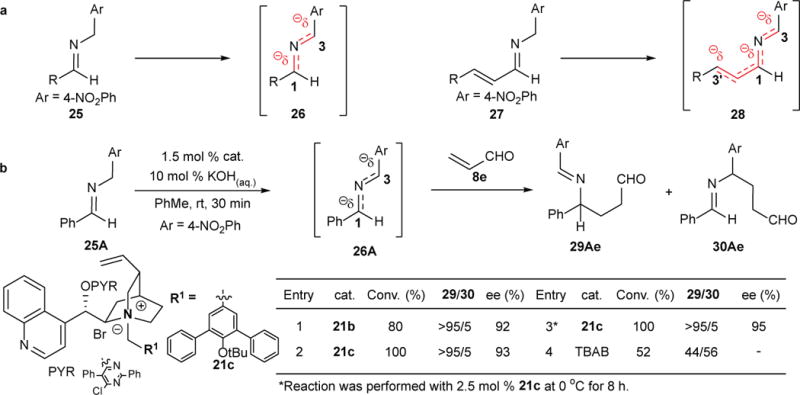
Asymmetric umpolung reactions of aryl and unsaturated aldimines.
Accordingly, we investigated the reaction of phenyl aldimine 25A with acrolein (8e) applying the conditions established with trifluoromethyl imines 1. As expected, 25A was far less reactive than 1A; only a trace amount of the desired product 29Ae was detected. With a substantially increased catalyst loading (entry 1, Fig. 3b), the reaction progressed to high conversion and in excellent enantioselectivity. A new catalyst bearing a 4-OtBu group (C-21c) was found to be more active and afforded better enantioselectivity (entry 2); allowing a clean and complete reaction to occur at 0 °C in excellent enantioselectivity with 2.5 mol % of C-21c (entry 3). Amine 29Ae was converted to the Boc-protected aminoalcohol 31Ae in high optical purity and good yield in three steps (entry 1, Table 4). Subsequently, we established that the umpolung reaction tolerated a broad range of aryl and heteroaryl aldimines of varying steric and electronic properties (entries 2–8, Table 4). Electron-rich aryl imines such as 25H appeared to be less active, but the umpolung reaction with C-21c still went to completion with high chemoselectivity, regioselectivity and enantioselectivity.
Table 4.
Substrate scope for umpolung reactions of aryl aldimines with acrolein (8e)

| |||||
|---|---|---|---|---|---|
| Entry | R | time (h) | 29/30 | yield of 31 (%)* | ee (%)† |
| 1 | Ph; 25A | 8 | >95/5 | 55 | 93 |
| 2 | o-CH3C6H4; 25B | 8 | >95/5 | 51 | 94 |
| 3ˆ | 2-Naphthyl; 25C | 8 | 90/10 | 54 | 94 |
| 4 | 2-Thienyl; 25D | 8 | >95/5 | 53 | 95 |
| 5ˆ | p-BrC6H4; 25E | 5 | >95/5 | 52 | 95 |
| 6 | o-BrC6H4; 25F | 5 | >95/5 | 56 | 95 |
| 7ˆ | p-MeO2CC6H4; 25G | 8 | 83/17 | 53 | 90 |
| 8# | p-MeOC6H4; 25H | 18 | >95/5 | 45 | 95 |
Conditions: Reactions were performed with 25 (0.20 mmol), 8e (0.40 mmol), 21c (2.5 mol%) and KOH (2.2 uL, 50 wt% aq., 10 mol%) in PhMe (2.0 mL) until full conversion. Regioselectivity (29/30) was determined by 1H analysis of the crude umpolung reaction mixture.
Overall yield for the transformation of imine 25 to 31.
ee of 31 was determined by HPLC analysis.
Reaction was performed in PhMe/CH2Cl2 = 2/1 solution (3.0 mL).
5.0 mol% C-21c used.
Due to the synthetic versatility of the olefin and amine functionalities, chiral allylic amines are highly valuable chiral building blocks.29 If we could extend the substrate scope to α,β-unsaturated imines 27, the impact of the imine umpolung reactions would be further enlarged. However, the 2-azaallylanions 28 derived from α,β-unsaturated imines 27 were expected to be even less stable than those derived from arylaldimines.30 Furthermore, the conjugation of an azaallylanion with an olefin renders 28 a more challenging nucleophile from the viewpoint of achieving catalytic control of regioselectivity (Fig. 3a). Gratifyingly, C-21c provided highly selective catalysis to efficiently promote the umpolung reaction of 27A and 8e (entry 1, Table 5). Importantly, the efficiency of C-21c remained undiminished for reactions involving a variety of α,β-unsaturated imines bearing di- and trisubstituted olefins (entries 2–6). As allylic amines could be readily hydrogenated to the corresponding aliphatic amines (Table 5), these results established this imine umpolung reaction as a useful method for the asymmetric synthesis of both chiral allylic and aliphatic amines.
Table 5.
Substrate scope for umpolung reactions of alkenyl aldimines with acrolein (8e)

| |||||
|---|---|---|---|---|---|
| Entry | Alkenyl | time (h) | 32/33 | yield of 34 (%)* | ee (%)† |
| 1 |
|
16 | 86/14 | 51 | 92 |
| 2 |

|
16 | 95/5 | 50 | 92 |
| 3# |

|
24 | 82/18 | 46 | 95 |
| 4 |
|
12 | 77/23 | 44 | 92 |
| 5# |
|
24 | 83/17 | 41 | 90 |
| 6 |

|
6 | 95/5 | 37ˆ | 90 |
Conditions: Reactions were performed with 25 (0.20 mmol), 8e (0.40 mmol), 21c (2.5 mol%) and KOH (2.2 uL, 50 wt% aq., 10 mol%) in PhMe (2.0 mL) until full conversion. Regioselectivity (29/30) was determined by 1H analysis of the crude umpolung reaction mixture.
Overall yield for the transformation of imine 25 to 34.
ee of 34 was determined by HPLC analysis.
5.0 mol% C-21c used.
Overall yield for a four-step transformation of (Z)-3-bromobut-2-enal to 34Fe, see SI for details.
We have identified a new class of tunable chiral phase transfer catalysts and demonstrated their unique ability to promote C–C bond forming reactions with 2-azaallylanions in a highly chemoselective, regioselective, diastereoselective and enantioselective fashion. This discovery unleashes the potential of imines as nucleophiles; thereby allowing the realization of catalytic asymmetric umpolung reactions of imines. These umpolung reactions provide a fundamentally new approach towards chiral amino compounds. With a simple operational protocol and low catalyst loading, this transformation also provides a practical method for organic synthesis.
Supplementary Material
Acknowledgments
We are grateful for financial support from the National Institute of General Medical Science (NIH, GM-61591). We are grateful to Mark Bezpalko and Prof. Bruce Foxman for X-ray crystallographic characterizations of structures. Chao Fei and Bin Hu are acknowledged for the help in substrates preparation.
Footnotes
Supplementary Information is available in the online version of the paper.
Author Contributions Y. Wu, L. Hu and Z. Li performed the experiments and analyzed data. Y. Wu and L. Deng conceived the idea and prepared this manuscript with feedbacks from L. Hu and Z. Li.
Author Information The authors declare no competing financial interests. Readers are welcome to comment on the online version of the paper.
References
- 1.Nugent TC. Chiral Amine Synthesis: Methods, Developments and Applications. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim, Germany: 2010. [Google Scholar]
- 2.Robak MT, Herbage MA, Ellman JA. Synthesis and Applications of tert-Butanesulfinamide. Chem Rev. 2010;110:3600–3740. doi: 10.1021/cr900382t. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi S, Mori Y, Fossey JS, Salter MM. Catalytic Enantioselective Formation of C–C Bonds by Addition to Imines and Hydrazones: A Ten-Year Update. Chem Rev. 2011;111:2626–2704. doi: 10.1021/cr100204f. [DOI] [PubMed] [Google Scholar]
- 4.Silverio DL, et al. Simple organic molecules as catalysts for enantioselective synthesis of amines and alcohols. Nature. 2013;494:216–221. doi: 10.1038/nature11844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 3. John Wiley & Sons Ltd; West Sussex, United Kingdom: 2009. [Google Scholar]
- 6.Seebach D. Methods of Reactivity Umpolung. Angew Chem, Int Ed. 1979;18:239–258. [Google Scholar]
- 7.Seebach D, Corey EJ. Generation and synthetic applications of 2-lithio-1,3-dithianes. J Org Chem. 1975;40:231–237. [Google Scholar]
- 8.Smith AB, Adams CM. Evolution of Dithiane-Based Strategies for the Construction of Architecturally Complex Natural Products. Acc Chem Res. 2004;37:365–377. doi: 10.1021/ar030245r. [DOI] [PubMed] [Google Scholar]
- 9.Brehme R, Enders D, Fernandez R, Lassaletta JM. Aldehyde N,N-Dialkylhydrazones as Neutral Acyl Anion Equivalents: Umpolung of the Imine Reactivity. Eur J Org Chem. 2007:5629–5660. [Google Scholar]
- 10.Vora HU, Rovis T. Asymmetric N-heterocyclic carbene (NHC) catalyzed acyl anion reactions. Aldrichim Acta. 2011;44:3–11. [PMC free article] [PubMed] [Google Scholar]
- 11.Reich BJE, Justice AK, Beckstead BT, Reibenspies JH, Miller SA. Cyanide-Catalyzed Cyclizations via Aldimine Coupling. J Org Chem. 2004;69:1357–1359. doi: 10.1021/jo035245j. [DOI] [PubMed] [Google Scholar]
- 12.Ogle JW, Zhang J, Reibenspies JH, Abboud KA, Miller SA. Synthesis of Electronically Diverse Tetraarylimidazolylidene Carbenes via Catalytic Aldimine Coupling. Org Lett. 2008;10:3677–3680. doi: 10.1021/ol8012765. [DOI] [PubMed] [Google Scholar]
- 13.Liu X, Gao A, Ding L, Xu J, Zhao B. Aminative Umpolung Synthesis of Aryl Vicinal Diamines from Aromatic Aldehydes. Org Lett. 2014;16:2118–2121. doi: 10.1021/ol500522d. [DOI] [PubMed] [Google Scholar]
- 14.Matsumoto M, Harada M, Yamashita Y, Kobayashi S. Catalytic imine-imine cross-coupling reactions. Chem Commun. 2014;50:13041–13044. doi: 10.1039/c4cc06156j. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Deng L. Asymmetric Synthesis of Trifluoromethylated Amines via Catalytic Enantioselective Isomerization of Imines. J Am Chem Soc. 2012;134:14334–14337. doi: 10.1021/ja306771n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Li J, Xiao X, Xie Y, Shi Y. An efficient synthesis of optically active trifluoromethyl aldimines via asymmetric biomimetic transamination. Chem Commun. 2013;49:1404–1406. doi: 10.1039/c2cc37423d. [DOI] [PubMed] [Google Scholar]
- 17.Jakubowska A, Kulig K. Progress in the glycine equivalent based α-amino acids synthesis. Curr Org Synth. 2013;10:547–563. [Google Scholar]
- 18.Zhu Y, Buchwald SL. Ligand-Controlled Asymmetric Arylation of Aliphatic α-Amino Anion Equivalents. J Am Chem Soc. 2014;136:4500–4503. doi: 10.1021/ja501560x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian X, et al. Palladium-Catalyzed Decarboxylative Generation and Asymmetric Allylation of α-Imino Anions. Org Lett. 2014;16:5228–5231. doi: 10.1021/ol502693r. [DOI] [PubMed] [Google Scholar]
- 20.Shirakawa S, Maruoka K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew Chem, Int Ed. 2013;52:4312–4348. doi: 10.1002/anie.201206835. [DOI] [PubMed] [Google Scholar]
- 21.Ooi T, Ohara D, Tamura M, Maruoka K. Design of New Chiral Phase-Transfer Catalysts with Dual Functions for Highly Enantioselective Epoxidation of α,β-Unsaturated Ketones. J Am Chem Soc. 2004;126:6844–6845. doi: 10.1021/ja048600b. [DOI] [PubMed] [Google Scholar]
- 22.Dolling UH, Davis P, Grabowski EJJ. Efficient catalytic asymmetric alkylations. 1. Enantioselective synthesis of (+)-indacrinone via chiral phase-transfer catalysis. J Am Chem Soc. 1984;106:446–447. [Google Scholar]
- 23.Bandini M, Bottoni A, Eichholzer A, Miscione GP, Stenta M. Asymmetric Phase-Transfer-Catalyzed Intramolecular N-Alkylation of Indoles and Pyrroles: A Combined Experimental and Theoretical Investigation. Chem–Eur J. 2010;16:12462–12473. doi: 10.1002/chem.201000560. [DOI] [PubMed] [Google Scholar]
- 24.Knowles RR, Lin S, Jacobsen EN. Enantioselective Thiourea-Catalyzed Cationic Polycyclizations. J Am Chem Soc. 2010;132:5030–5032. doi: 10.1021/ja101256v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu P, Snapper ML, Hoveyda AH. Catalytic Asymmetric Alkylations of Ketoimines. Enantioselective Synthesis of N-Substituted Quaternary Carbon Stereogenic Centers by Zr-Catalyzed Additions of Dialkylzinc Reagents to Aryl-, Alkyl-, and Trifluoroalkyl-Substituted Ketoimines. J Am Chem Soc. 2008;130:5530–5541. doi: 10.1021/ja8001343. [DOI] [PubMed] [Google Scholar]
- 26.Nie J, Guo HC, Cahard D, Ma JA. Asymmetric Construction of Stereogenic Carbon Centers Featuring a Trifluoromethyl Group from Prochiral Trifluoromethylated Substrates. Chem Rev. 2010;111:455–529. doi: 10.1021/cr100166a. [DOI] [PubMed] [Google Scholar]
- 27.Giacalone F, Gruttadauria M, Agrigento P, Noto R. Low-loading asymmetric organocatalysis. Chem Soc Rev. 2012;41:2406–2447. doi: 10.1039/c1cs15206h. [DOI] [PubMed] [Google Scholar]
- 28.Bordwell FG, Algrim D, Vanier NR. Acidities of anilines and toluenes. J Org Chem. 1977;42:1817–1819. [Google Scholar]
- 29.Hartwig JF, Stanley LM. Mechanistically Driven Development of Iridium Catalysts for Asymmetric Allylic Substitution. Acc Chem Res. 2010;43:1461–1475. doi: 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jaun B, Schwarz J, Breslow R. Determination of the basicities of benzyl, allyl, and tert-butylpropargyl anions by anodic oxidation of organolithium compounds. J Am Chem Soc. 1980;102:5741–5748. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.