

Summary
Ebolaviruses, and the other viral causes of haemorrhagic fevers (VHF) have always posed special problems for diagnostic laboratories. These arise from the rarity of human infections, minimal documented experience with test delivery and interpretation, the paucity of established commercial or in-house assays, the lack of clinical material for test development and validation, the high level containment required for handling live virus, the ongoing evolution of the viruses, and the high personal and public health requirements for accurate diagnosis. This article addresses the current situation and the ongoing challenges associated with delivering timely, high quality and safe testing within Australia for people exposed as part of the current major outbreak of Ebolavirus disease (EVD) in Western Africa. The members of the Public Health Laboratory Network have developed deliverable and reliable nucleic acid detection tests, and also have the laboratory capacity to handle the live viruses if necessary. However delivering and maintaining these services necessitates high levels of experience in developing and applying tests for exotic and emerging infections, strong national and international links and collaborations, ongoing monitoring and reassessment of test design and performance, innovative approaches to generation of positive control material, and a regular quality assurance program.
Keywords: Diagnosis, Ebola, Filovirus, PCR, quality
BACKGROUND
Ebolaviruses, and the other viral causes of haemorrhagic fevers (VHF) with high mortality and the potential for person to person transmission, have always posed special problems for diagnostic laboratories. The current large Ebolavirus disease (EVD) outbreak in West Africa, with cases occurring in returning healthcare workers in a number of developed countries, has raised the level of concern and preparedness in Australia.
The VHF agents are all exotic to Australia and are classed at the highest biosafety level (BSL-4) and require the highest level of physical containment (PC-4). It was recognised in the 1990s that providing the necessary diagnostic support for both suspected and confirmed cases in a timely manner through the small number of PC-4 laboratories nationally was difficult or impossible. In 2002, the Public Health Laboratory Network of Australia pioneered and developed comprehensive guidelines that included a staged approach to testing for patients with suspected VHFs. These have been progressively upgraded1 and allow laboratories to safely conduct testing, other than cultivation of the virus, within lower level and more widely available PC-3 or PC-2 laboratories. PC-4 level support for VHF-specific tests and for virus culture is available at a national level from the National High Security Quarantine Laboratory (NHSQL) at the Victorian Infectious Diseases Reference Laboratory (VIDRL) in Melbourne (Fig. 1).
Fig. 1.

Staff performing Ebolavirus culture under Physical Containment Level 4 conditions at the National High Security Quarantine Laboratory at VIDRL. (Image supplied by Julian Druce.)
The other challenge faced is the intermittent and short-lived nature of previous outbreaks of EVD, which has meant that there has been very limited test development internationally, and even less information on test performance and interpretation. Also, in Australia test development has to be carried out without access to patient material and with tight regulatory restrictions on the importation and distribution of the Ebolaviruses within the country.
This article addresses the current situation and the ongoing challenges associated with delivering timely, high quality and safe testing within Australia for people exposed as part of the current major outbreak of EVD in Western Africa.
Ebolaviruses form a genus within the Filoviridae family (Fig. 2). It is enveloped, with a 19 kb single-stranded negative-sense RNA genome,2 which codes for one non-structural protein (L gene: RNA-dependent RNA polymerase) and six structural proteins: NP gene (major nucleocapsid protein), VP35 (phosphoprotein), VP40 (membrane-associated matrix protein), GP (transmembrane glycoprotein/secreted glycoprotein), VP30 (ribonucleoprotein-associated) and VP24 (membrane associated protein). There are four Ebolavirus species within Africa: Ebolavirus Zaire (EBOV) first found in what is now the Democratic Republic of Congo; Ebolavirus Sudan (SUDV); Ebolavirus Bundibugyo (BDBV) first found in Uganda; and a single case of Ebolavirus Taï Forest (TAFV) infection in the Côte d’Ivoire. Outside Africa, an attenuated species, Ebolavirus Reston (RESTV), was detected in primates from the Philippines, with seroconversion in the primate handlers. The current outbreak of EBOV is due to a strain designated as Makona,3 which appears to have evolved from Central African strains as the virus spread to West Africa in non-human reservoirs over the last decade or more.4
Fig. 2.
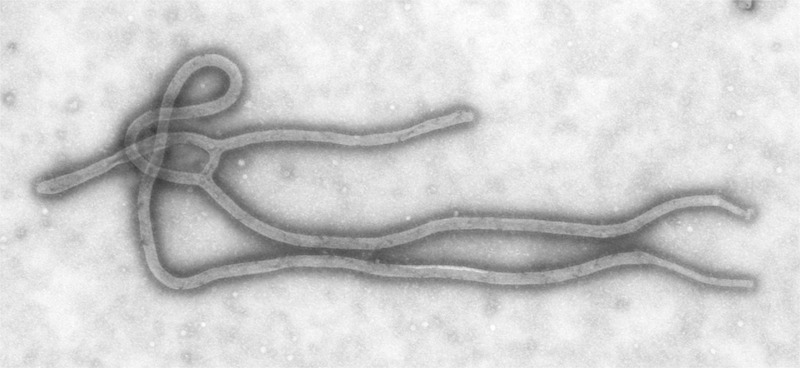
Transmission electron micrograph of an Ebolavirus virion. (Image accessed from the Centers for Disease Control Public Health Image Library, contributed by Cynthia Goldsmith from the CDC.)
DIAGNOSTIC TESTS FOR DETECTION OF EBOV
Virus culture
The filoviruses grow well in Vero and Vero E6 cell lines, although the cytopathic effect may be difficult to detect without passaging.2 Cell culture is less sensitive than polymerase chain reaction (PCR) and can be performed only in a PC-4 laboratory, so it has very limited use for primary diagnosis. However, it is valuable for providing virus for sequencing (including whole genome sequencing) in order to determine the molecular epidemiology, to monitor changes that may affect the sensitivity and specificity of the nucleic acid detection tests, to guide further test development and to provide positive control material for test evaluation. The last includes the development of improved antigen detection tests and serological assays. Occasionally it may be used for sorting out unexpected positive PCR results, for example in cases occurring outside areas of known activity or where the illness is atypical; and for unusual situations where a false negative PCR is suspected, e.g., patients with a strong clinical and exposure history where the negative result may reflect a genetic variant or a faulty assay.
Viral nucleic acid detection
PCR-based assays have now become both the standard and preferred method for the detection of EBOV virus.5 Properly performed, they are the most sensitive tests, are highly specific, and can be performed safely in a standard laboratory environment.
SUDV RNA has been found in a large range of body fluids and tissues in patients with symptomatic diseases, including blood, sweat, saliva, urine, semen and breast milk.6 Blood is regarded as the single most reliable sample for detection and exclusion of EBOV infection.5 A study under field conditions from the SUDV outbreak in Uganda in 2001 showed that RNA was present in the blood on the day of onset of illness, rose over the first 5–6 days of clinical illness and peaked around 3.5 × 106 copies/mL in non-fatal cases and 3.5 × 108 copies/mL in fatal cases. However, it did not reach reliable detectable levels (105 copies/mL) until 72 h after onset of illness, so that a negative PCR in the first 72 h of illness does not exclude infection.7 There are anecdotal reports of similar early false negative PCR results in the current EBOV outbreak.8
Oral fluid is recommended5 if a blood sample cannot be obtained. While it was shown to be as sensitive as serum for diagnosis in eight patients in the 2004 Republic of Congo outbreak, data are limited.9 EBOV has been detected in ocular fluid by PCR and culture for at least 14 weeks,10 semen by PCR for up to 101 days,11 and it also has been reported in breast milk in the absence of detection in blood.12
Transient low levels of EBOV have been detected by PCR (but not virus isolation) in asymptomatic patients in previous outbreaks,13 but they have not yet been described in the current outbreak and are not believed to represent an infectious risk.
A number of targets and assays have been used for EBOV detection: those directed against NP, GP and VP40 gene sequences are species-specific and possibly lineage-specific while those directed at L gene sequences can detect all filoviruses.14 So performance may vary according to the species of the ebolavirus, and not all published assays,14 including some of those approved by the Food and Drug Administration (FDA) in the United States, have been assessed against the current outbreak strain.15 For example, the two Centers for Disease Control (CDC) reverse transcription, real-time PCR assays directed at the NP2 gene and the VP40 gene were evaluated using the 1976 Mayinga strain, the 1995 Kikwit strain and the 2002 Gabon strain of EBOV. They performed well, but the sensitivity for the current Makona strain had to be assumed based on primer and probe sequence homology with the evaluated strains.
Ongoing genetic evolution of EBOV within the current West African outbreak may, of course, also affect assay sensitivity and specificity,4 although there is no evidence of this happening as yet.
Interestingly, a separate small outbreak of EBOV in the Democratic Republic of the Congo in 2014 was due to a different variant of the virus (called Lomela strain), reinforcing the need to be aware of the potential for genetically different strains to emerge.3
Provided that an appropriate process is used, filoviruses are inactivated during specimen extraction and lysis,5,16,17 so that once extraction is performed and the external surfaces are decontaminated, further testing can be carried out at PC-2 level.1,5 The testing laboratory needs to verify that their extraction process is adequate to inactivate virus17 and that appropriately high standards of laboratory practice are applied.1
Due to the lack of availability of clinical samples for test development, the validation of the PCR assays in use internationally is based almost entirely on the use of live virus, gamma-irradiated inactivated virus, or target sequences incorporated into a plasmid. These are diluted in water or used to make mocked clinical samples by diluting in virus-negative specimens, to provide a technical validation of the assay. The limit of detection (LOD) may be determined in a number of ways, but the most meaningful is the level that can be reliably detected 95% of the time, which provides 95% confidence that a negative result on the test means that the level is actually lower than that. LOD may be reported as plaque-forming units (pfu)/mL (if live or inactivated virus is used) or copies/mL (if RNA target is used). Unfortunately there is a variable relationship between pfu/mL and copies/mL18 and assay sensitivity may be affected by the type of specimen used. For example, using gamma-irradiated virus reduces the sensitivity of the assays.
During the Ebolavirus outbreak, the Royal College of Pathologists of Australasia (RCPA) Quality Assurance Program (QAP) Biosecurity program rapidly developed simulated specimens based upon RNA transcripts generated using T7 transcription in vitro and plasmids containing the nucleoprotein (NP), glycoprotein (GP) and L-gene fragments as nucleic acid test targets. The targets selected were known sequences from EBOV strain Mayinga (GenBank accession no. AF086833) and surveys were undertaken in April 2014 and November 2014, with eight participant laboratories. The laboratories used various combinations of these targets for the diagnostic PCRs.
A number of commercial kits for detecting EBOV became available during 2014 and 2015 and a number have been approved for emergency use by the US FDA.15 These assays use a range of platforms and vary in sensitivity and, as for in-house assays, participation in an appropriate external QAP is essential if they are used. None are yet licensed by the Therapeutic Goods Administration for use in Australia.
Hence, for the PCR assays in use:
they have not been validated to the standards usually expected, and we have no realistic likelihood of being able to do so, as access to clinical material remains very difficult;
there are a number of factors that influence the interpretation of the technical validation of these assays;
there is ongoing genetic drift in the circulating viruses that may modify target sequences and affect assay performance.
Therefore, laboratories undertaking PCR testing for Ebolaviruses should be experienced in the development and validation of assays for rare pathogens, and in the use of plasmid targets, UV inactivated materials and other mocked samples; as well as having links to national and international sources of patient material, and to other laboratories performing testing. The method chosen for diagnostic use should have been shown to have performed satisfactorily in a suitable external QAP that is conducted by an organisation with established expertise in generating samples for evaluation, such as the RCPA QAP Biosecurity program.
Ideally, the diagnostic PCR should be directed at more than one reliable target and should use primer and probes known to match all currently circulating strains. That requires ongoing monitoring of sequences of circulating virus, with modification of assays as required, and revalidation of assay performance.
Due to the high personal and public health impact of the diagnosis of EVD in Australia, all samples found to be positive or equivocal on PCR are treated as presumptive positives, but samples must be urgently referred to the National High Security Quarantine Laboratory at the Victorian Infectious Diseases Reference Laboratory in Melbourne, or to an appropriate overseas reference laboratory, for additional testing before this is confirmed. Furthermore, for the first case diagnosed in Australia, which is yet to happen, the result has to be confirmed in a designated overseas reference laboratory.19
The interpretation of PCR results must take into account the nature and timing of the exposure and the clinical history, and the performance of the particular assay used. A negative PCR on blood collected less than 72 h after onset of illness is not currently regarded as reliable for exclusion of EBOV,19,20 although the WHO has accepted a shorter period of 48 h.5 This may change in the future as test sensitivity improves and more technical and field use data become available.
For clearance of patients and determination of non-infectivity, testing for viable virus using a culture method may be helpful. However, cell culture is slow, relatively insensitive and has very restricted availability and it may miss low level viable virus. For those reasons PCR testing has been used as a basis for clearance of patients. Unfortunately, there is a lack of data about the correlation of low RNA levels with infectivity.
For a body fluid that has been shown to be positive for EBOV by PCR, it requires two PCR-negative samples collected at least one day apart for that sample to be declared non-infectious.5
Viral antigen detection
In-house antigen detection tests, mainly enzyme immunoassays, have been developed14 but their poor sensitivity in field use compared with PCR,6,21 their limited availability, and lack of validation data means that they have had very little use.
Antigen appears about 3 days post-onset and increases until death or, in non-fatal cases, persists for 7–16 days.14
Very recently, a rapid immunochromatographic dipstick immunoassay point-of-care test (ReEBOV Antigen Rapid Test Kit; Corgenix, USA) that detects the VP40 antigen has been listed by the World Health Organization.22 It has a reported sensitivity of 91.8% and a specificity of 84.6% compared with PCR, and is intended for presumptive testing of symptomatic individuals, particularly in outbreaks in resource-poor settings without ready access to PCR-based tests.
ANTIBODY DETECTION TESTS
Antibody detection tests have a limited role in the early diagnosis of suspected cases as serological responses are too delayed or entirely absent, particularly in the sicker (and more infectious) patients. IgM usually appears within 1 week of onset, peaks at 2–3 weeks, then persists for 60–80 days.21 IgG persists for years and has been detected more than 10 years after onset,23 but it is not known whether this indicates immunity to further infection. Neutralisation tests, if available, must be performed in a PC-4 laboratory and are generally reserved for research purposes.
CONCLUSIONS
Laboratory diagnosis of infections due to Ebolaviruses has posed a number of challenges to laboratories within Australia and internationally due to the containment requirements for these viruses, the lack of patient material for test development and validation, the evolving nature of the virus, and the limited clinical data to assist test interpretation.
Despite these difficulties, the availability of reliable and timely testing capacity within Australia is essential, and has required innovative approaches to test development and evaluation. Under those circumstances, it is important that testing is conducted within highly experienced laboratories using a range of methods of test validations and accompanied by a rigorous quality assurance program, with regular review and revalidation.
Acknowledgements
Thanks to the staff of the RCPA QAP for Biosecurity at the South East Area Laboratory Service, Randwick, NSW; the Microbiological Security section at PathWest; and the NHSQL at VIDRL for assistance with manuscript preparation and provision of images.
Conflicts of interest and sources of funding: The authors state that there are no conflicts of interest to disclose.
References
- 1.Public Health Laboratory Network. Laboratory procedures and precautions for samples collected from patients with suspected viral haemorrhagic fevers. Canberra: Australian Government Department of Health, 2014. Cited 24 Apr 2015. http://www.health.gov.au/internet/main/publishing.nsf/Content/cda-pubs-other-vhf.htm. [Google Scholar]
- 2.Sanchez A, Geisbert TW, Feldmann H. Knipe DM, Griffin DE, Lamb RA. Filoviridae: Marburg and Ebola viruses. Fields Virology 5th edPhiladelphia: Lippincott Williams & Wilkins; 2007. 1409–1448. [Google Scholar]
- 3.Kuhn JS, Kristian G, Andersen KG, et al. Nomenclature- and database-compatible names for the two Ebola virus variants that emerged in Guinea and the Democratic Republic of the Congo in 2014. Viruses 2014; 6:4760–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gire G, Goba A, Andersen K, et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 2014; 345:1369–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization. Laboratory Guidance for the Diagnosis of Ebola Virus Disease: Interim Recommendations. 19 September 2014; cited 24 Apr 2015. http://www.who.int/csr/resources/publications/ebola/laboratory-guidance/en/. [Google Scholar]
- 6.Onyango CO, Opoka ML, Ksiazek TG, et al. Laboratory diagnosis of Ebola hemorrhagic fever during an outbreak in Yambio, Sudan, 2004. J Infect Dis 2007; 196 Suppl 2:S193–S198. [DOI] [PubMed] [Google Scholar]
- 7.Towner JS, Rollin PE, Bausch DG, et al. Rapid diagnosis of Ebola hemorrhagic fever by reverse transcription-PCR in an outbreak setting and assessment of patient viral load as a predictor of outcome. J Virol 2004; 78:4330–4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chertow DS, Kleine C, Edwards JK, et al. Ebola virus disease in West Africa - clinical manifestations and management. N Engl J Med 2014; 371:2054–2057. [DOI] [PubMed] [Google Scholar]
- 9.Formenty P, Leroy EM, Epelboin A, et al. Detection of Ebola virus in oral fluid specimens during outbreaks of Ebola virus hemorrhagic fever in the Republic of Congo. Clin Infect Dis 2006; 42:1521–1526. [DOI] [PubMed] [Google Scholar]
- 10.Varkey JB, Shantha JG, Crozier I. Persistence of Ebola virus in ocular fluid during convalescence. N Engl J Med 2015; 372:2423–2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez LL, De Roo A, Guimard Y, et al. Persistence and genetic stability of Ebola virus during the outbreak in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis 1999; 179 Suppl 1:S170–S176. [DOI] [PubMed] [Google Scholar]
- 12.Moreau M, Spencer C, Gozalbes JG, et al. Lactating mothers infected with Ebola virus: EBOV RT-PCR of blood only may be insufficient. Euro Surveill 2015; 20:21017. [DOI] [PubMed] [Google Scholar]
- 13.Bellan SE, Pulliam JR, Dushoff J, et al. Ebola control: effect of asymptomatic infection and acquired immunity. Lancet 2014; 384:1499–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reusken C, Niedrig M, Pas S, et al. Identification of essential outstanding questions for an adequate European laboratory response to Ebolavirus Zaire West Africa 2014. J Clin Virol 2015; 62:124–134. [DOI] [PubMed] [Google Scholar]
- 15.US Food and Drug Administration. Emergency Use Authorisation. Cited 27 Apr 2015. http://www.fda.gov/EmergencyPreparedness/Counterterrorism/MedicalCountermeasures/MCMLegalRegulatoryandPolicyFramework/ucm182568.htm. [Google Scholar]
- 16.Blow JA, Dohm DJ, Negley DL, et al. Virus inactivation by nucleic acid extraction reagents. J Virol Methods 2004; 119:195–198. [DOI] [PubMed] [Google Scholar]
- 17.Towner JS, Sealy TK, Ksiazek TG, et al. High-throughput molecular detection of hemorrhagic fever virus threats with applications for outbreak settings. J Infect Dis 2007; 196 Suppl 2:S205–S212. [DOI] [PubMed] [Google Scholar]
- 18.Trombley AR, Wachter L, Garrison J, et al. Comprehensive panel of real-time TaqMan™ polymerase chain reaction assays for detection and absolute quantification of filoviruses, arenaviruses, and new world hantaviruses. Am J Trop Med Hyg 2010; 82:954–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Communicable Disease Network of Australia. Ebola virus disease (EVD): CDNA National Guidelines for Public Health Units. Cited 21 Apr 2015. http://www.health.gov.au/internet/main/publishing.nsf/Content/ohp-ebola-Information-for-Health-Professionals. [Google Scholar]
- 20.Centers for Disease Control. Guidance for collection, transport and submission of samples for Ebola virus testing. Cited 27 Apr 2015. http://www.cdc.gov/vhf/ebola/healthcare-us/laboratories/specimens.html. [Google Scholar]
- 21.Ksiazek TG, Rollin PE, Williams AJ, et al. Clinical virology of Ebola hemorrhagic fever (EHF): virus, virus antigen, and IgG and IgM antibody findings among EHF patients in Kikwit, Democratic Republic of the Congo, 1995. J Infect Dis 1999; 179 Suppl 1:S177–S187. [DOI] [PubMed] [Google Scholar]
- 22.World Health Organization. Public Report for ReEBOV™ Antigen Rapid Test Kit (EA 0011-011-00) 2015. Cited 27 Apr 2014. http://www.who.int/medicines/ebola-treatment/1st_antigen_RT_Ebola/en/. [Google Scholar]
- 23.Ksiazek TG, West CP, Rollin PE, et al. ELISA for the detection of antibodies to Ebola viruses. J Infect Dis 1999; 179 Suppl 1:S192–S198. [DOI] [PubMed] [Google Scholar]