

Abstract
Traumatic brain injury (TBI) has long been recognized to be a risk factor for dementia. This association has, however, only recently gained widespread attention through the increased awareness of ‘chronic traumatic encephalopathy’ (CTE) in athletes exposed to repetitive head injury. Originally termed ‘dementia pugilistica’ and linked to a career in boxing, descriptions of the neuropathological features of CTE include brain atrophy, cavum septum pellucidum, and amyloid-β, tau and TDP-43 pathologies, many of which might contribute to clinical syndromes of cognitive impairment. Similar chronic pathologies are also commonly found years after just a single moderate to severe TBI. However, little consensus currently exists on specific features of these post-TBI syndromes that might permit their confident clinical and/or pathological diagnosis. Moreover, the mechanisms contributing to neurodegeneration following TBI largely remain unknown. Here, we review the current literature and controversies in the study of chronic neuropathological changes after TBI.
Introduction
Compelling epidemiological evidence indicates that a single moderate to severe traumatic brain injury (TBI) is associated with increased risk of development of progressive disorders of cognitive impairment leading to dementia.1–11 Each year in the USA alone, over 1.7 million people sustain a TBI, of which approximately one-quarter are moderate or severe.12 As such, TBI represents a leading cause of disability, particularly in the young,12 and approximately 5.3 million US citizens are currently living with long-term TBI-associated disabilities.13 Despite these substantial numbers, comparatively little is known about the chronic pathologies of TBI and how they might contribute to the later onset of neurodegenerative disease.
Over 85 years ago, the eminent pathologist Harrison S. Martland made careful observations on the ‘punch-drunk’ syndrome, describing chronic motor and neuropsychiatric symptoms in former boxers.14 Through the decades that followed, further case reports and series emerged, indicating that repetitive TBI from boxing might induce a chronic and potentially progressive neuropsychiatric disorder with a neuropathological basis,15–18 termed ‘dementia pugilistica’ by Millspaugh.15 However, little interest was expressed in understanding this disease further until observations emerged of similar neuropathological findings in case series and reports of non-boxing individuals exposed to repetitive mild TBI, including former participants in contact sports other than boxing (American football, ice hockey and wrestling19–27) and military personnel,26–28 and in historical reports of non-sports-related repetitive head injury.29–31
With the appreciation that the pathology was not restricted to boxing, or ‘pugilism’, the term ‘chronic traumatic encephalopathy’ (CTE) was introduced to reflect increasing descriptions of the pathological features in a wider range of exposure situations. This term is now widely accepted in preference to dementia pugilistica, and will be used in the remainder of this article. Not surprisingly, the recent intense media attention on CTE in contact sports participants and war veterans has spawned considerable public concern. However, it is rarely noted that the actual number of purported CTE cases described in the literature is remarkably limited. Moreover, no operational criteria are currently available to confirm either a clinical or a pathological diagnosis of CTE. Indeed, the features that constitute CTE as a distinct disease entity have yet to be defined. Nonetheless, since this term has become so widely used, it is important to review current understanding of the pathology of ‘CTE’, as well as limitations in existing studies and potential avenues for advancement of the field.
TBI as a risk factor for dementia
Repetitive mild TBI
Though long acknowledged anecdotally, Martland’s description in 192814 of the punch-drunk syndrome in boxers provided the first formal account of the chronic neuropsychiatric sequelae of repetitive head injury, with multiple other reports following in the ensuing years.15,16,32,33 In 1969, Roberts assessed 224 randomly selected professional boxers and demonstrated that 17% displayed a “relatively stereotyped” clinical picture,34 which included emotional lability, personality change, memory impairment and dementia, as well as pyramidal and extrapyramidal dysfunction and cerebellar impairment. Subsequent work offered a potential dose–risk association, with increased exposure to TBI from boxing linked to increased risk of later impairment, measured either as radiologically identified structural changes35 or clinical evidence of neurocognitive impairment.36 In keeping with this model, limited evidence suggests that amateur boxers have a lower risk of developing dementia pugilistica than their professional counterparts37 —an observation supported by neuropathological data, although the number of cases described is small.18
Recent studies indicate that the link between repetitive head injury and long-term neurological impairment is not solely the preserve of boxing, with earlier onset of Alzheimer disease (AD)38 and higher “neurodegenerative mortality” reported in former National (American) Football League players.39 To date, there have been 94 reported autopsy cases in non-boxers exposed to repetitive head injury—66 athletes (58 American football players, five ice hockey players and three wrestlers19–27) and 23 military personnel with or without a history of contact sport26–28—in which supportive neuropathological changes were present. Where recorded, the clinical symptoms included neuropsychiatric and behavioural problems, including emotional lability, aggression, poor judgment, depression, suicidal ideation and, in some instances, suicide. Impairments of memory and cognitive function were also reported in several cases.
Notably, although precise numbers are not available, descriptions of motor symptoms in non-boxer athletes are infrequent. This contrasts with studies on boxers, in whom motor, parkinsonian and cerebellar symptoms are commonly reported. However, whether this observation reflects a less severe manifestation, an earlier time point or, indeed, a different clinical phenotype, is not clear. Moreover, the symptomatology described thus far in non-boxer athletes may also reflect the nature of case selection, which in many instances has been achieved via outreach following media attention around notable high-profile early deaths. As such, the incidence of symptoms such as suicidality, emotional lability, aggression and disinhibition may be skewed. In addition, in many cases, details of clinical features were ascertained retrospectively, via interviews with relatives and contacts, which may further confound the results.
Single TBI
Data from several studies suggest that a history of just one moderate to severe TBI is an important risk factor for the later onset of dementia,1–10 although a number of other epidemiological studies have failed to confirm such an association,40–48 perhaps reflecting the retrospective nature of many reports, with potential for recall bias. More-recent prospective studies,9 however, have led to a general acceptance that TBI is a risk factor for dementia.11 Indeed, evidence has emerged not only of a link between TBI and dementia, but also in support of a ‘dose–response’ relationship, with risk of dementia being increased in severe compared with moderate TBI survivors,9 and where there is a history of loss of consciousness compared with no loss of consciousness.7 Furthermore, data suggest that a history of TBI might accelerate the onset of disease.8,49–51
That TBI represents a major risk factor for the later development of neurodegenerative disease is now accepted. However, a consequence of the largely retrospective and serendipitous, observational studies in repetitive and single TBI to date is that a considerable number of questions remain unanswered, not least of which is the prevalence of TBI-related dementia in the population. Furthermore, while TBI-associated dementia is often reported as Alzheimer’s in type in the literature, it is unclear at this stage whether patients with a history of head injury instead develop a clinical phenotype of dementia distinct from ‘typical’ AD. In addition, potential ancillary factors that could contribute to the risk of chronic neurodegeneration after either single or repetitive TBI, such as the mechanism and pathological consequences of acute injury, potential genetic predisposition, and sex or age differences, are largely unknown. Indeed, the possibility remains that certain individuals may be ‘resistant’ to developing dementia following TBI; certainly, not all boxers go on to develop dementia, despite repetitive injury.
The chronic neuropathologies of TBI
While various clinical descriptions of boxers were reported following Martland’s initial account in 1928, it was not until 1954 that Brandenburg and Hallervorden provided the first detailed case report of the neuropathology of CTE.52 Over the next two decades, only a further eight cases were reported by multiple groups,17,32,53–56 and although many of these reports described shared neuropathological features, speculation remained as to whether the findings could reliably be attributed to boxing.
Such concerns were assuaged with the publication of a landmark case series by the eminent London pathologist John A. N. Corsellis, who performed a detailed examination on the brains of 15 former boxers, with retrospectively collected clinical and boxing histories obtained from informants.18 From these observations, and knowledge of existing reports, Corsellis and colleagues outlined the neuropathological findings that comprise the main features of CTE. Although these findings have been described in multiple subsequent reports, this original report remains the seminal neuropathological description of the sequelae of repetitive TBI. Importantly, on the basis of these data and further studies in both repetitive and single injury, we now recognize that the human neuropathology of TBI survival has many facets (meriting the descriptor ‘polypathology’), which are presumed to underlie the clinically described neurodegenerative disease. In this section, we will review the currently recognized chronic pathologies of single and repetitive TBI in humans. A summary of the published observations on the pathology of CTE arising as a result of presumed repetitive injury is provided in Table 1.
Table 1.
Summary of published experience on the neuropathology of chronic traumatic encephalopathy
| Cases and neuropathological findings | Boxing | American football* | Wrestling‡ | Ice hockey | Military (with history of contact sports§) | Military (with history of other TBI||) | Other |
|---|---|---|---|---|---|---|---|
| History of repetitive TBI: studies and cases | |||||||
| Number of studies (studies with controls) | 29 (12)17,21,22,25,27,32, 52–63,69–71,82,85,89,106–108,128 | 8 (1)19–22,24–27 | 4 (2)23,26,27 | 2 (1)22,27 | 3 (2)26–28 | 2 (1)27,28 | 5 (1)27,29–31,70 |
| Number of cases | 72 | 58 | 3 | 5 | 20 | 3 | 5 (1 domestic abuse, 3 self-injury, 1 ‘dwarf throwing’) |
| Incidence of macroscopic changes | |||||||
| Atrophy (global or regional) | 39/47 | 27/50 | 0/3 | 3/5 | 15/18 | 2/3 | 3/4 |
| Ventricular dilation | 32/33 | 35/44 | 0/1 | 3/5 | 15/18 | 2/3 | 2/2 |
| Cavum septum pellucidum | 43/50 | 17/38 | 0/3 | 0/3 | 5/13 | 1/3 | 3/4 |
| Incidence of microscopic changes | |||||||
| Cerebellar pathology | 25/36 | 20/43 | 1/3 | 2/5 | 10/18 | 1/3 | 1/1 |
| White matter change | 20/22 | 9/10 | 1/2 | NR | 1/1 | NR | 1/1 |
| Neuronal loss | 25/28 | 3/3 | 2/2 | NR | 1/1 | NR | 1/1 |
| Substantia nigra pathology | 28/36 | 47/51 | 2/3 | 5/5 | 17/18 | 2/3 | 2/2 |
| Locus coeruleus pathology | 24/28 | 43/46 | 2/3 | 5/5 | 16/18 | 2/3 | 1/1 |
| Tau | 61/68 | 58/58 | 3/3 | 5/5 | 20/20 | 3/3 | 5/5 |
| Amyloid-β plaque | 39/52 | 22/58 | 0/3 | 2/5 | 13/20 | 2/3 | 1/5 |
| Lewy bodies | 1/31 | 12/58 | 0/3 | 2/5 | 6/20 | 0/3 | 0/1 |
| TAR DNA-binding protein 43 | 13/13 | 43/50 | 0/1 | 5/5 | 18/20 | 2/3 | 0/1 |
Number of cases with each pathology is included, based on a clear description of positive or negative findings. There was no presumed absence of findings in reports that only described positive features.
One case overlaps with boxers due to history of both American football and amateur boxing.
One case overlaps with wrestling due to history of American football and wrestling.
All cases overlap with cases included in relevant contact sports columns. One case also had a history of motor vehicle accident.
Other TBI refers to single severe TBI or history of motor vehicle accident.
Abbreviations: NR, not reported; TBI, traumatic brain injury.
Gross pathological features
Brain atrophy
Brain atrophy is a commonly described feature in CTE; indeed, marked cerebral atrophy, particularly of frontal and temporal regions, is often described.17,18,21,32,52–63 In addition, atrophy of the cerebellum, both generalized and focal, has been reported.31,56 However, in cases of non-boxing athletes with CTE, regional atrophy has not been described at the same frequency as in boxers, with overt global atrophy being rarer still.19,20,23–25,27,28 With such small numbers and notable case-selection bias, however, the significance of this observation is not yet clear.
Notably, generalized brain atrophy is a well-characterized feature of survival from single TBI (Figure 1). At autopsy, however, it is not possible to determine whether this atrophy occurred acutely following trauma or provides evidence of progressive pathology. Recent imaging studies have begun to address this question, with the results suggesting that marked atrophy can be observed by 6 months post-injury and may progress for many years.64–68
Figure 1.

Cerebral atrophy following survival from a single moderate to severe TBI. a | Brain of a 24-year-old patient who died within hours of being assaulted. The only macroscopic evidence of TBI is a superficial contusion in the right inferior temporal lobe (arrow), although grade 1 diffuse traumatic axonal injury was observed on histology. b | Brain of a 40-year-old patient who survived 4 years from injury and has a healed contusion in a similar location to that in part a (arrow). In comparison to the previous patient, however, there is notable gyral atrophy, ventricular enlargement, and thinning of the corpus callosum. Abbreviation: TBI, traumatic brain injury.
Cavum septum and ventricular enlargement
One of the most frequent macroscopic observations in autopsy studies in CTE, particularly those involving boxers, is cavum septum pellucidum (CSP). This feature, which was present in 64 of 99 (65%) cases in which it was examined for (Table 1), is often accompanied by a fenestrated septum, communicating hydrocephalus and, in some instances, complete absence of the septum or its detachment from the fornix or corpus callosum.17,19–21,24,25,29,31,32,52–61,63,69–71 In addition to these autopsy studies, various radiological investigations have confirmed the presence of CSP in vivo in boxers with neuropsychiatric symptoms.32,35,55,72 However, as CSP can occur as a ‘normal’ observation in the general population, with estimates ranging from 1–28%,18,73–76 the diagnostic utility of these findings has been questioned.75,77 In Corsellis’ autopsy-based study in boxers, CSP was reported as both qualitatively distinct from and substantially wider than that in non-boxer controls.18 By contrast, CSP has not been reported as a feature of survival following a single TBI.
The mechanism by which CSP arises remains unclear. One proposal is that it develops as a consequence of cerebral atrophy and ventricular dilation, a frequent observation in boxers with CTE.17,18,52–54,56,57,60,61,63 Alternatively, it may arise as a consequence of repeated transient increases in intracranial pressure, producing both the ventricular dilation and septal changes.32 Of further note, periventricular midline structures are highly vulnerable to mechanical strain during trauma.78–81 Thus, the observed abnormalities in the septum might arise as a result of immediate mechanical injury and, perhaps, persistent degeneration.
Histological features
Tauopathy or tau pathology?
The presence of neurofibrillary tangles (NFTs) is one of the most consistent pathologies reported in CTE, and was even documented in the earliest described case (Figure 2).52 Indeed, where sought, NFTs have been observed in at least one brain region in 133 of the 140 autopsy reports of CTE to date, although many of these cases were actually defined as CTE on the basis of this tau pathology. As a consequence, CTE is now commonly referred to as a ‘tauopathy’, implying that the primary pathology of CTE is tau-based. However, the pathology of survival from TBI is increasingly recognized to be multifaceted. As such, ‘tau pathology’ might be a more encompassing and less exclusive descriptor of NFTs after TBI. Of note, in two boxers with CTE, tangles were indistinguishable from those of AD with regard to both isoform ratio and phosphorylation state.59 Remarkably, these remain the only two cases in the literature on which biochemical analyses were performed.
Figure 2.

Tau and amyloid-β pathology after TBI. a,b | Extensive neurofibrillary tangles observed in a boxer.18 The 77-year-old man had participated in over 700 boxing contests during his life and had neuropsychiatric symptoms. Tangles were identified using silver staining (a) and Congo red staining (b). c,d | Neurofibrillary tangles in the parahippocampal gyrus of a 47-year-old man who had sustained a single severe TBI 1 year previously.88 No history of repetitive TBI was recorded. Tangles were identified using immunohistochemistry (c) and thioflavine S staining (d). Scale bars 100 μm. e,f | Sections of the temporal cortex from a 63-year-old man with dementia pugilistica57 who had participated in over 300 boxing contests and was originally noted as plaque-negative.18 Extensive amyloid-β plaques were demonstrated using immunohistochemistry with formic acid pretreatment (e), but not with Congo red (f). Abbreviation: TBI, traumatic brain injury. Permission for parts a and b obtained from Cambridge University Press © Corsellis, J. A. et al. Psychol. Med. 3, 270–303 (1973). Permission for parts c and d obtained from John Wiley and Sons © Johnson, V. E. et al. Brain Pathol. 22, 142–149 (2012). Parts e and f reproduced from Roberts, G. W. et al. The occult aftermath of boxing. J. Neurol. Neurosurg. Psychiatry 53, 373–378 © 1990 with permission from BMJ Publishing Group Ltd.
In Corsellis’ description of the pathology of boxers, NFTs were reported to be most abundant in the medial temporal grey matter.18 By contrast, subsequent observations on NFT distribution in material from both boxers and non-boxers describe a more patchy distribution of pathology, including some cases with relative sparing of the medial temporal lobe, and clusters of NFTs grouped in the neocortex.21,22,27,82 Furthermore, Geddes et al. described a perivascular accentuation of tangles in several cases70,82—a feature that was later observed by other groups.21,22,25–27 Of note, these tau-immunoreactive NFTs and neuropil threads seem to show a preferential distribution in the cerebral cortex, involving the superficial neocortical layers and the depths of sulci. This sulcal distribution has been suggested to be pathognomonic of CTE,83 although examination of CTE cases in parallel with large numbers of non-trauma controls would be required to support this hypothesis. As points of inflection, the sulcal depths may be biomechanically vulnerable to the dynamic forces experienced during trauma, thereby precipitating this pattern of pathology. However, despite numerous studies characterizing pathologies of acute TBI, none has described pathological features in a pattern and distribution reminiscent of the tau pathology in CTE, the biomechanical consequences of TBI instead producing more typically midline to parasagittal pathology (for a review, see Johnson et al.80). Thus, the significance of the distribution of tau pathology in CTE remains unclear.
Recently, a hierarchical distribution of tau pathology in CTE has been proposed, with progression from focal cortical and perivascular clusters of NFTs, often in the frontal lobe (CTE stage I–II pathology), through to high densities of NFTs in widespread cortical areas, medial temporal lobe, deep grey nuclei and brainstem (CTE stage III–IV pathology).27 Given the comparatively small numbers of cases examined and potential case-selection bias in these autopsy-based studies, with limited data available for clinicopathological correlation, meaningful insight into hierarchical distribution and, in particular, the potential progression of tau pathology in CTE, must remain speculative. These descriptions of NFTs in CTE contrast dramatically with the series describing tau staging in AD, where more-robust clinical information is typically available.84
While NFTs have long been described in CTE, until recently such pathology following a single TBI was less well-described, with just two individual autopsy case reports describing AD-like neuropathology, including NFTs, in individuals who sustained a single, severe TBI followed by onset of dementia.85,86 Notably, studies of autopsy material from patients who died acutely (up to 4 weeks) following a single TBI failed to display NFT pathology beyond that of uninjured controls.87 By contrast, more-recent observations on non-selected material from 39 survivors of 1 year or more from a single moderate to severe TBI demonstrated NFTs at greater density and in wider distribution in up to 30% of cases when compared with age-matched controls.88 In this limited number of cases, a hierarchical distribution of NFTs similar to that described in AD was observed. Interestingly, as in CTE, in some cases NFTs were found to be concentrated at the depths of sulci.
Amyloid-β pathologies
The presence of amyloid-β (Aβ) pathology in CTE has emerged as a less consistent feature than tau pathologies. Occasional early studies in boxers described senile plaques,17,52 whereas others reported a complete absence of plaques, with some suggesting that this observation may provide the potential to distinguish CTE from AD.17,53,54 In the original Corsellis et al. study, for example, plaques were observed in only five of 15 cases.18 However, following the advent of Aβ immunohistochemistry, revisitation of Corsellis’ original material by Roberts et al. revealed a further seven cases with plaques—often extensive—in immunohistochemically stained sections, which had previously been identified as plaque-negative using routine silver staining (Figure 2).57
Of the published autopsy series on CTE to date, a total of 66 (53%) of the 124 cases examined for amyloid pathology describe either diffuse or senile plaques (including the revisited Corsellis cases), with 24 (38%) of the 65 non-boxer athletes in more-recent studies reported as showing plaques of any type.19–27 Acknowledged technical issues and conflicting data, however, make it difficult to determine the exact numbers of cases with plaques. Furthermore, given that amyloid plaques may be observed in material from ‘normal’ controls, in particular those greater than 65 years of age, the ability to associate plaques in autopsy-acquired material to a history of trauma demands parallel studies in suitably sized, age-matched control groups. This is a particular issue in existing studies on CTE, which either omit controls entirely or include controls in such small numbers or with sufficient demographic differences to the CTE cases under study as to make them of limited relevance to comparative analysis. Regardless of these limitations, not all reported cases of CTE seem to be plaque- positive.21,22,27,29,31,58,60,61,63,69–71,82,89 Notably, plaques in CTE are typically described as diffuse and do not display the histochemical or morphological features of the neuritic plaques that are characteristic of AD.
With regard to single TBI, amyloid pathologies are perhaps the most extensively researched in the context of potential links with dementia.90 One of the earliest observations in this respect described Aβ plaques in up to 30% of patients who died in the acute phase following a single moderate to severe TBI, both in autopsy series, where plaque pathology exceeded that seen in age-matched controls,91–94 and in surgically acquired material.93 Typically, these plaques are diffuse in nature, can appear within hours of injury, and are observed across all age groups, including young adults. Until recently, the temporal dynamics of these plaques appearing in the acute phase were unknown. However, in observations on autopsy-acquired material from 39 survivors of 1 year or more from TBI, plaques were observed in considerably greater density and wider distribution in the TBI survivors when compared with age-matched, uninjured controls.88 Of particular note, whereas acute plaques following TBI are typically diffuse in nature, similar to those seen in early AD, those observed in long-term survivors from single TBI were more frequently fibrillar; that is, similar to those of established AD.88
As regards the origin of these plaque pathologies following TBI, axonal injury is proposed to have a role, at least in the acute phase. Specifically, diffuse axonal injury (DAI) is an early and frequent event documented in all severities of TBI.78–80 These damaged axons are readily identified in tissue sections as morphologically abnormal profiles, ranging from undulating axons to the classically described swollen axonal bulbs or varicosities.80 Typically, axonal pathology is identified in a stereotypical distribution in the parasagittal white matter and white matter tracts, in and around deep grey nuclei, and in the dorsolateral brainstem.95 These damaged axons can be revealed in tissue sections via immunocytochemical staining for amyloid precursor protein (APP), which accumulates at points of axonal transport interruption within hours of injury.96,97 Interestingly, the enzymes necessary for Aβ cleavage from APP, including presenilin-1 (PS-1) and β-site APP cleaving enzyme (BACE1), were also observed accumulating in damaged axons along with Aβ itself, first in a large animal model of DAI98 and subsequently in humans.99,100 Consequently, axonal pathology provides a potential means for Aβ genesis following trauma.90 Axonal pathology has traditionally been regarded as an event limited to the early phase after injury, resolving within weeks of the insult,101 but more-recent studies have identified ongoing axonal degeneration persisting years and even decades following TBI in some individuals. 81,100 Whether this process contributes to ongoing amyloid plaque deposition following TBI, however, is unclear.
TAR DNA-binding protein
TAR DNA-binding protein (TDP-43) has been identified as the main disease-associated protein in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration,102 although it is also recognized as a secondary feature in a number of other neurodegenerative diseases, including Huntington disease, AD and Parkinson disease.103–105 Recent data indicate that a TDP-43 proteinopathy might also be part of the neuropathological profile following repetitive mild or concussive TBI. Specifically, autopsy studies reported TDP-43 inclusions in multiple brain regions in 61 of 71 cases examined, including in boxers106,107 and retired American football or ice hockey players.22,27 Again, these are highly selected cases and, therefore, do not provide population-based information regarding a TDP-43 proteinopathy in athletes. Included in these studies were eight patients with clinically diagnosed ALS (two retired professional American football players and one former boxer) in whom the authors reported pathological features of CTE coinciding with TDP-43 pathology, leading to a proposed alternative diagnosis, chronic traumatic encephalo-myelopathy (CTEM).22,27 Though regarded as a controversial proposition based on these few observations, this remains an intriguing suggestion that undoubtedly merits further study.
Whereas studies in single or repetitive TBI reveal similarities with respect to amyloid and tau pathology between these two situations, the experience in TDP-43 perhaps suggests a difference. Specifically, in observations on cases surviving a single moderate to severe TBI, cytoplasmic aggregations of abnormally phosphorylated TDP-43 were not observed. However, increased cytoplasmic immunoreactivity to phosphorylation- independent—or physiological—TDP-43 was observed in both acute (n = 23) and long-term (n = 39) survivors of single TBI, perhaps suggesting a physiological role for TDP-43 in response to injury.107
Neuronal loss and the substantia nigra
Neuronal loss has been described in the majority of the brains of boxers and non-boxers with CTE.18–20,23,24,28,31, 52–54,56,59,61–63,71,108 In addition to neuronal dropout in the cerebral cortex, several reports describe specific regions or structures with notable cell loss, including the hippocampus,18,53 locus coeruleus18,19,54,61,63,108 and cerebellum.18,19,23,24,31,52,53,56,71 Varying distributions of cell loss have been described, from widespread and diffuse, to more ‘patchy’ or selective changes.
Acute and chronic neuron dropout has also been examined in single TBI. With the constellation of pathologies recognized in the acute phase following a single moderate to severe TBI, including hypoxic–ischaemic injury, traumatic axonal injury and inflammation, the discovery of marked neuronal loss in several studies that examined acute-survival material is not surprising.109,110 However, growing evidence suggests that neuronal loss continues beyond the immediate acute phase, at least in severely injured patients. In a series of detailed studies in material from patients in the persistent vegetative state included in the Glasgow Traumatic Brain Injury Archive, Maxwell and colleagues demonstrated a continued decrease in neuronal densities in the hippocampus and thalamic nuclei up to 1 year following injury.110,111 In addition, active degeneration of neurons via programmed cell death has been observed, even at 1 year following severe injury.112
One structure of particular interest is the substantia nigra, which, in patients with CTE, has frequently been described as ‘pale’ on gross pathological examination, and as showing depigmentation, NFTs and neuronal dropout on histology, although classic Lewy bodies have rarely been reported (Table 1).18–20,52,54,63 These observations are noteworthy given the incidence of parkinsonian symptoms in the CTE population,113 and may evoke comparisons with tauopathies that can present with parkinsonism, such as progressive supranuclear palsy (PSP). In contrast to CTE, little is known about the vulnerability of the substantia nigra chronically following single TBI.
White matter degeneration
Information on white matter changes in CTE is limited. A small number of reports provide evidence of gliosis, foci of degeneration and/or rarefaction of white matter.17,18,28,60 Some have also described regions of patchy loss of myelin staining,17,18,21,53 while others report an absence of demyelination.54 Notably, recent reports described intra-axonal accumulation of transport-interrupted proteins as axonal pathology in veterans with associated tau pathology.26,27 While interesting, similar axonal pathologies are known to be observed in ‘normal’ controls, presumably as a consequence of various processes, including agonal hypoxic–ischaemic injury.81,114 As such, attribution of pathological relevance to degenerating axons requires careful consideration, matched control observations and adequately sized series.
As noted above, white matter injury and associated axonal pathology is described following all severities of TBI and has been implicated as a mechanism of rapid Aβ genesis.90 In 1956, Sabina Strich first noted that white matter degeneration might not be limited to the acute phase post-injury but, rather, may persist chronically.115 Specifically, by using the Marchi stain to highlight myelin breakdown, evidence in support of active degeneration of white matter tracts was identified in cases of TBI with survival up to 15 months. Using refined techniques, axonal pathology has since been observed even years after a single moderate to severe TBI.81,100 Specifically, white matter loss in the corpus cal-losum parallels axonal degradation, as indicated by APP-immunoreactive profiles.81 Interestingly, the morphology of degenerating axons differs from that observed acutely, and primarily comprises somewhat granular axonal bulbs. In addition, there is notable pallor in myelin staining, with evidence of cytoplasmic myelin granules in regional microglia, indicating active phagocytosis of myelin fragments (Figure 3).100
Figure 3.

Neuroinflammation and white matter degeneration after TBI. a | Extensive reactive microglia (immunostained with antibody CR3/43) in the hippocampus and adjacent sulci of a 65-year-old male with dementia pugilsitica. Scale bar 1 mm. b | Extensive CR3/43-reactive cells with an amoeboid morphology, indicative of macrophages, observed in the atrophic corpus callosum of a 37-year-old male 4 years following a single severe TBI. Scale bar 1 mm for main image (left) and 100 μm for high-magnification image (right). c | Adjacent section to b, stained with Luxol fast blue, indicating chronic white matter change and loss of myelin. Scale bar 1 mm for main image (left) and 100 μm for high-magnification image (right). Abbreviation: TBI, traumatic brain injury.
Neuroinflammation beyond repair
The role of neuroinflammation in the pathophysiology of neurodegenerative disorders, including AD, is currently attracting considerable attention.116–118 Immediately following injury, TBI induces an array of inflammatory responses as a consequence of blood–brain barrier compromise and a complex acute-phase inflammatory cell response, including activation of resident microglia (reviewed elsewhere119). While this acute inflammatory response is to be anticipated, one might also expect this inflammation to resolve following the acute phase, with the brain returned to the quiescent state awaiting further challenge. However, evidence is accumulating that neuroinflammation persists in a proportion of survivors from single moderate to severe TBI, and in some cases continues for many years.81,120 In studies on autopsy-derived material from patients surviving varying intervals following a single TBI, a marked neuroinflammatory response persists in white matter regions such as the corpus callosum, manifesting as amoeboid, activated microglia at markedly higher density than in comparable control, non-injured tissue and associated with thinning of the corpus callosum (Figures 1 and 3).81 These observations on autopsy-acquired tissue are supported by in vivo imaging studies, using the PET ligand PK-11195, in survivors of TBI. In these studies, persistent increased binding was observed in patients with survivals ranging from 11 months to 17 years following injury.121 Though limited in both characterization and number of reports, inflammation has been observed in CTE;17,18,60,122 however, formal assessment is lacking to date.
Whether the observed inflammation following TBI occurs as a response to coincident pathologies, such as ongoing axonal degradation, amyloid pathology and/or NFTs, or, alternatively, is driving these pathologies, remains unknown. Indeed, chronic dysregulation of inflammation has been suggested to underlie the neuro-degenerative pathologies of both single and repetitive TBI.82,123 Further work to elucidate this pathology will be critical to our understanding of long-term outcomes of TBI and, perhaps, may serve as a target for intervention with a long window of therapeutic opportunity.
Cerebellar pathology
Changes in the cerebellum are noteworthy, given the prevalence of cerebellar symptoms in patients with CTE. As noted, loss of cerebellar neurons, including granule cells and Purkinje cells, has been described.18,20,23,24,52,71 In addition, scarring and gliosis of the cerebellum, particularly affecting the tonsilar region, have been observed;17,18,23,24,31,69 these features are suggested to originate from impingement against the foramen magnum, as occurs in herniation.18 Atrophy and demyelination of the folial white matter have also been noted in CTE.18,31,56
Challenges to research
In 1969, a workshop of international experts from multiple disciplines associated with TBI took place in Washington to discuss the “Late Effects of Head Injury”.124 During this workshop, it was recognized that in order to gain adequate insight into the pathophysiology of survival from TBI, concerted efforts directed towards better understanding of the human pathology of TBI, both in the acute setting and with survival, were required. One outcome of this workshop was a call for the establishment of brain banks specifically dedicated to studies in TBI. Over four decades later, the Glasgow Traumatic Brain Injury Archive remains the only comprehensive archive of human brain tissue accrued with the express intent to facilitate studies in TBI over a range of mechanisms, ages and survivals.
Encouragingly, with recent widespread attention on neurodegeneration following TBI, further TBI dedicated banks have emerged, such as the growing archive of material directed towards studies of CTE housed in the Center for the Study of Chronic Traumatic Encephalopathy at Boston University, MA, USA.125 Nonetheless, much of our current understanding of the pathology of survival from TBI, in particular the association with neurodegenerative disease, derives from remarkably few studies in suitably sized TBI cohorts matched to uninjured, control material, or from small, uncontrolled cohorts or descriptive accounts of individual cases—a stark contrast to studies evaluating the neuropathology of other neurodegenerative disorders, such as AD.84,126,127
Undoubtedly, therefore, a pressing need exists for initiatives to expand the current established resources and expertise and to establish further archives of suitably characterized human tissue to advance the field. In particular, material linked to prospectively accrued clinical information will be invaluable. Finally, these precious resources must be openly accessible to suitably designed, international collaborative research to maximize their utility.
Conclusions
Considerable epidemiological evidence supports TBI as a risk factor for the development of dementia. Furthermore, the available data indicate a possible dose and incidence association between TBI and outcome, with the risk increasing following single severe injury versus single moderate injury, and following high-exposure, repetitive mild or concussive injury. According to current autopsy studies, survival from repetitive mild or single moderate to severe TBI is associated with a range of pathologies, best considered as ‘polypathology’, and including tau and Aβ abnormalities, neuroinflammation, white matter degeneration, and neuronal loss. However, the comparatively small numbers of observations in the literature preclude formal characterization of both the clinical syndromes and the pathology of TBI-associated neurodegeneration. As such, robust diagnostic criteria permitting confident differentiation from other, better-characterized neurodegenerative syndromes remain elusive. Indeed, owing to the lack of large-scale controlled studies, our understanding of the pathology of CTE has advanced little since the landmark study by Corsellis et al. in 1973.18 Furthermore, at least some pathological features are common to survival from single and repetitive TBI, raising the possibility that they represent manifestations along a spectrum of common pathology, perhaps with a phenotype influenced by severity and frequency of exposure (see hypothesis outlined in Figure 4).
Figure 4.
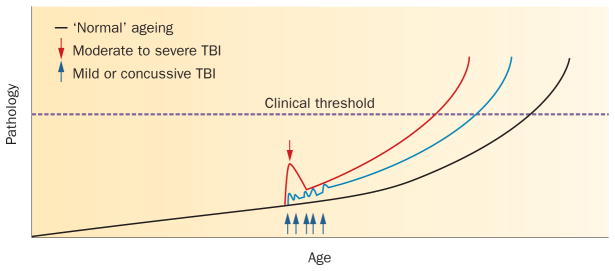
Interaction between TBI and ‘normal’ ageing: a hypothesis. An increasing range of pathologies is recognized in association with survival from TBI, any of which may contribute to the associated clinical syndromes of neurocognitive impairment. In the absence of a history of TBI, many of these same pathologies, such as amyloid-β plaque or neuroinflammatory pathology, may accumulate as a consequence of ’normal’ ageing (black line), with the accumulated pathologies eventually crossing a threshold where clinical symptoms are apparent. Following a single moderate to severe TBI, evidence to date supports an initial spike in pathology immediately after the event, which subsequently resolves. However, it is intriguing to speculate that, in a proportion of patients, there is incomplete resolution of this acute-phase response, with subsequent accelerated accumulation of pathology, leading to the threshold for clinical symptoms being crossed at an earlier age (red line). Similarly, each successive mild TBI may lead to acute pathology, again followed by partial resolution, but ultimately triggering accelerated accumulation of pathology, leading to earlier-onset symptomatology (blue line). Abbreviation: TBI, traumatic brain injury.
Following recent observations of CTE in selected, small series of athletes with a history of repetitive TBI from contact sports other than boxing, and also in military personnel, there has been remarkable public interest in this field. However, considerable work needs to be done to clearly define CTE as a disease entity both pathologically and clinically. Moreover, risk factors for developing these pathologies, such as mechanism, frequency and severity of injury, as well as age, sex, and potential genetic predisposition, remain largely unknown. Undoubtedly, establishment of large-scale, dedicated and networked tissue banks and a movement towards prospective studies will be important to advance our understanding of TBI-associated neurodegeneration and, in turn, targeted developments in therapy.
Key points.
Traumatic brain injury (TBI) represents the strongest environmental risk factor for dementia
Current evidence indicates a possible ‘dose’ and frequency-dependent association between TBI and risk of neurodegenerative disease
The human pathology of survival from TBI is best described as a ‘polypathology’, featuring amyloid-β, tau and TDP-43 pathologies, together with white matter degradation, neuronal loss and neuroinflammation
The chronic pathologies following single and repetitive injuries show similarities, although comparative studies are lacking at present
TBI may offer an opportunity for better understanding of the evolution of pathologies in a wider range of neurodegenerative diseases
There is an urgent need to extend existing tissue banks dedicated to TBI and establish further networked archives to provide broad international research access
Review criteria.
Literature on which this Review is based was acquired via PubMed and MEDLINE (all years), in addition to references cited in relevant papers. Search criteria included terms such as “dementia pugilistica”, “chronic traumatic encephalopathy”, “punch drunk”, “repetitive mild head trauma”, “traumatic brain injury”, “dementia”, “atrophy”, “Alzheimer’s disease”, “tauopathy”, “amyloid beta plaques”, TAR DNA-binding protein”, “diffuse axonal injury”, “cavum septum pellucidum”, “boxing”, “professional football”, “sports injury” and combinations thereof. Full-text articles were used. The majority of articles were in English. Relevant articles not in English were translated.
Acknowledgments
This work was supported by NIH grants NS038104 (D. H. Smith and W. Stewart), NS056202 and AG038911 (D. H. Smith). In addition, we would like to thank Dr Nadia Dahmane and Dr Amaya Wolf for assistance with translation of articles in French and German, respectively.
Footnotes
Competing interests
The authors declare no competing interests.
Author contributions
V. E. Johnson and W. Stewart researched data for the article. All three authors made substantial contributions to discussions of the content, writing the article, and review and/or editing of the manuscript before submission.
Contributor Information
Douglas H. Smith, Penn Center for Brain Injury and Repair and Department of Neurosurgery, Perelman School of Medicine, University of Pennsylvania, 105 Hayden Hall, 3320 Smith Walk, Philadelphia, PA 19104, USA
Victoria E. Johnson, Penn Center for Brain Injury and Repair and Department of Neurosurgery, Perelman School of Medicine, University of Pennsylvania, 105 Hayden Hall, 3320 Smith Walk, Philadelphia, PA 19104, USA
William Stewart, Department of Neuropathology, Southern General Hospital, 1345 Govan Road, Glasgow G51 4TF, UK.
References
- 1.Molgaard CA, et al. Epidemiology of head trauma and neurocognitive impairment in a multi-ethnic population. Neuroepidemiology. 1990;9:233–242. doi: 10.1159/000110778. [DOI] [PubMed] [Google Scholar]
- 2.Mortimer JA, French LR, Hutton JT, Schuman LM. Head injury as a risk factor for Alzheimer’s disease. Neurology. 1985;35:264–267. doi: 10.1212/wnl.35.2.264. [DOI] [PubMed] [Google Scholar]
- 3.Mortimer JA, et al. Head trauma as a risk factor for Alzheimer’s disease: a collaborative re-analysis of case–control studies. EURODEM Risk Factors Research Group. Int J Epidemiol. 1991;20 (Suppl 2):S28–S35. doi: 10.1093/ije/20.supplement_2.s28. [DOI] [PubMed] [Google Scholar]
- 4.Graves AB, et al. The association between head trauma and Alzheimer’s disease. Am J Epidemiol. 1990;131:491–501. doi: 10.1093/oxfordjournals.aje.a115523. [DOI] [PubMed] [Google Scholar]
- 5.O’Meara ES, et al. Head injury and risk of Alzheimer’s disease by apolipoprotein E genotype. Am J Epidemiol. 1997;146:373–384. doi: 10.1093/oxfordjournals.aje.a009290. [DOI] [PubMed] [Google Scholar]
- 6.Salib E, Hillier V. Head injury and the risk of Alzheimer’s disease: a case control study. Int J Geriatr Psychiatry. 1997;12:363–368. doi: 10.1002/(sici)1099-1166(199703)12:3<363::aid-gps515>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 7.Guo Z, et al. Head injury and the risk of AD in the MIRAGE study. Neurology. 2000;54:1316–1323. doi: 10.1212/wnl.54.6.1316. [DOI] [PubMed] [Google Scholar]
- 8.Schofield PW, et al. Alzheimer’s disease after remote head injury: an incidence study. J Neurol Neurosurg Psychiatry. 1997;62:119–124. doi: 10.1136/jnnp.62.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Plassman BL, et al. Documented head injury in early adulthood and risk of Alzheimer’s disease and other dementias. Neurology. 2000;55:1158–1166. doi: 10.1212/wnl.55.8.1158. [DOI] [PubMed] [Google Scholar]
- 10.Fleminger S, Oliver DL, Lovestone S, Rabe-Hesketh S, Giora A. Head injury as a risk factor for Alzheimer’s disease: the evidence 10 years on; a partial replication. J Neurol Neurosurg Psychiatry. 2003;74:857–862. doi: 10.1136/jnnp.74.7.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lye TC, Shores EA. Traumatic brain injury as a risk factor for Alzheimer’s disease: a review. Neuropsychol Rev. 2000;10:115–129. doi: 10.1023/a:1009068804787. [DOI] [PubMed] [Google Scholar]
- 12.Faul M, Xu L, Wald MM, Coronado VG. Traumatic brain injury in the United States: emergency department visits, hospitalizations, and deaths, 2002–2006. Centers for Disease Control and Prevention; 2010. [online], http://www.cdc.gov/traumaticbraininjury/tbi_ed.html. [Google Scholar]
- 13.Thurman DJ, et al. Traumatic brain injury in the United States: a report to Congress. Centers for Disease Control and Prevention; 1999. [online]. http://www.cdc.gov/traumaticbraininjury/tbi_report_to_congress.html. [Google Scholar]
- 14.Martland H. Punch drunk. J Am Med Assoc. 1928;91:1103–1107. [Google Scholar]
- 15.Millspaugh J. Dementia pugilistica. U S Nav Med Bull. 1937;35:297–303. [Google Scholar]
- 16.Critchley M. Medical aspects of boxing, particularly from a neurological standpoint. Br Med J. 1957;1:357–362. doi: 10.1136/bmj.1.5015.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Payne EE. Brains of boxers. Neurochirurgia (Stuttg) 1968;11:173–188. doi: 10.1055/s-0028-1095326. [DOI] [PubMed] [Google Scholar]
- 18.Corsellis JA, Bruton CJ, Freeman-Browne D. The aftermath of boxing. Psychol Med. 1973;3:270–303. doi: 10.1017/s0033291700049588. [DOI] [PubMed] [Google Scholar]
- 19.Omalu BI, et al. Chronic traumatic encephalopathy in a national football league player: part II. Neurosurgery. 2006;59:1086–1092. doi: 10.1227/01.NEU.0000245601.69451.27. [DOI] [PubMed] [Google Scholar]
- 20.Omalu BI, et al. Chronic traumatic encephalopathy in a National Football League player. Neurosurgery. 2005;57:128–134. doi: 10.1227/01.neu.0000163407.92769.ed. [DOI] [PubMed] [Google Scholar]
- 21.McKee AC, et al. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKee AC, et al. TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol. 2010;69:918–929. doi: 10.1097/NEN.0b013e3181ee7d85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Omalu BI, Fitzsimmons RP, Hammers J, Bailes J. Chronic traumatic encephalopathy in a professional American wrestler. J Forensic Nurs. 2010;6:130–136. doi: 10.1111/j.1939-3938.2010.01078.x. [DOI] [PubMed] [Google Scholar]
- 24.Omalu BI, Hamilton RL, Kamboh MI, DeKosky ST, Bailes J. Chronic traumatic encephalopathy (CTE) in a National Football League Player: case report and emerging medicolegal practice questions. J Forensic Nurs. 2010;6:40–46. doi: 10.1111/j.1939-3938.2009.01064.x. [DOI] [PubMed] [Google Scholar]
- 25.Omalu B, et al. Emerging histomorphologic phenotypes of chronic traumatic encephalopathy in American athletes. Neurosurgery. 2011;69:173–183. doi: 10.1227/NEU.0b013e318212bc7b. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein LE, et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med. 2012;4:134ra60. doi: 10.1126/scitranslmed.3003716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKee AC, et al. The spectrum of disease in chronic traumatic encephalopathy. Brain. 2013;136:43–64. doi: 10.1093/brain/aws307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Omalu B, et al. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg Focus. 2011;31:E3. doi: 10.3171/2011.9.FOCUS11178. [DOI] [PubMed] [Google Scholar]
- 29.Hof PR, Knabe R, Bovier P, Bouras C. Neuropathological observations in a case of autism presenting with self-injury behavior. Acta Neuropathol. 1991;82:321–326. doi: 10.1007/BF00308819. [DOI] [PubMed] [Google Scholar]
- 30.Roberts GW, Whitwell HL, Acland PR, Bruton CJ. Dementia in a punch-drunk wife. Lancet. 1990;335:918–919. doi: 10.1016/0140-6736(90)90520-f. [DOI] [PubMed] [Google Scholar]
- 31.Williams DJ, Tannenberg AE. Dementia pugilistica in an alcoholic achondroplastic dwarf. Pathology. 1996;28:102–104. doi: 10.1080/00313029600169653. [DOI] [PubMed] [Google Scholar]
- 32.Mawdsley C, Ferguson FR. Neurological disease in boxers. Lancet. 1963;2:799–801. doi: 10.1016/s0140-6736(63)90498-7. [DOI] [PubMed] [Google Scholar]
- 33.Spillane JD. Five boxers. Br Med J. 1962;2:1205–1210. doi: 10.1136/bmj.2.5314.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roberts G. Brain Damage in Boxers: A Study of the Prevalence of Traumatic Encephalopathy Among Ex-Professional Boxers. Pitman; London: 1969. [Google Scholar]
- 35.Jordan BD, et al. CT of 338 active professional boxers. Radiology. 1992;185:509–512. doi: 10.1148/radiology.185.2.1410364. [DOI] [PubMed] [Google Scholar]
- 36.Jordan BD, Matser EJ, Zimmerman RD, Zazula T. Sparring and cognitive function in professional boxers. Phys Sportsmed. 1996;24:87–98. doi: 10.3810/psm.1996.05.1358. [DOI] [PubMed] [Google Scholar]
- 37.Loosemore M, Knowles CH, Whyte GP. Amateur boxing and risk of chronic traumatic brain injury: systematic review of observational studies. BMJ. 2007;335:809. doi: 10.1136/bmj.39342.690220.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guskiewicz KM, et al. Association between recurrent concussion and late-life cognitive impairment in retired professional football players. Neurosurgery. 2005;57:719–726. doi: 10.1093/neurosurgery/57.4.719. [DOI] [PubMed] [Google Scholar]
- 39.Lehman EJ, Hein MJ, Baron SL, Gersic CM. Neurodegenerative causes of death among retired National Football League players. Neurology. 2012;79:1970–1974. doi: 10.1212/WNL.0b013e31826daf50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandra V, Philipose V, Bell PA, Lazaroff A, Schoenberg BS. Case–control study of late onset “probable Alzheimer’s disease”. Neurology. 1987;37:1295–1300. doi: 10.1212/wnl.37.8.1295. [DOI] [PubMed] [Google Scholar]
- 41.Amaducci LA, et al. Risk factors for clinically diagnosed Alzheimer’s disease: a case–control study of an Italian population. Neurology. 1986;36:922–931. doi: 10.1212/wnl.36.7.922. [DOI] [PubMed] [Google Scholar]
- 42.Broe GA, et al. A case–control study of Alzheimer’s disease in Australia. Neurology. 1990;40:1698–1707. doi: 10.1212/wnl.40.11.1698. [DOI] [PubMed] [Google Scholar]
- 43.Ferini-Strambi L, Smirne S, Garancini P, Pinto P, Franceschi M. Clinical and epidemiological aspects of Alzheimer’s disease with presenile onset: a case control study. Neuroepidemiology. 1990;9:39–49. doi: 10.1159/000110750. [DOI] [PubMed] [Google Scholar]
- 44.van Duijn CM, et al. Head trauma and the risk of Alzheimer’s disease. Am J Epidemiol. 1992;135:775–782. doi: 10.1093/oxfordjournals.aje.a116364. [DOI] [PubMed] [Google Scholar]
- 45.Katzman R, et al. Development of dementing illnesses in an 80-year-old volunteer cohort. Ann Neurol. 1989;25:317–324. doi: 10.1002/ana.410250402. [DOI] [PubMed] [Google Scholar]
- 46.Launer LJ, et al. Rates and risk factors for dementia and Alzheimer’s disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups European Studies of Dementia. Neurology. 1999;52:78–84. doi: 10.1212/wnl.52.1.78. [DOI] [PubMed] [Google Scholar]
- 47.Williams DB, Annegers JF, Kokmen E, O’Brien PC, Kurland LT. Brain injury and neurologic sequelae: a cohort study of dementia, parkinsonism, and amyotrophic lateral sclerosis. Neurology. 1991;41:1554–1557. doi: 10.1212/wnl.41.10.1554. [DOI] [PubMed] [Google Scholar]
- 48.Mehta KM, et al. Head trauma and risk of dementia and Alzheimer’s disease: The Rotterdam Study. Neurology. 1999;53:1959–1962. doi: 10.1212/wnl.53.9.1959. [DOI] [PubMed] [Google Scholar]
- 49.Sullivan P, Petitti D, Barbaccia J. Head trauma and age of onset of dementia of the Alzheimer type. JAMA. 1987;257:2289–2290. doi: 10.1001/jama.1987.03390170045014. [DOI] [PubMed] [Google Scholar]
- 50.Gedye A, Beattie BL, Tuokko H, Horton A, Korsarek E. Severe head injury hastens age of onset of Alzheimer’s disease. J Am Geriatr Soc. 1989;37:970–973. doi: 10.1111/j.1532-5415.1989.tb07283.x. [DOI] [PubMed] [Google Scholar]
- 51.Nemetz PN, et al. Traumatic brain injury and time to onset of Alzheimer’s disease: a population-based study. Am J Epidemiol. 1999;149:32–40. doi: 10.1093/oxfordjournals.aje.a009724. [DOI] [PubMed] [Google Scholar]
- 52.Brandenburg W, Hallervorden J. Dementia pugilistica with anatomical findings [German] Virchows Arch. 1954;325:680–709. doi: 10.1007/BF00955101. [DOI] [PubMed] [Google Scholar]
- 53.Neubuerger KT, Sinton DW, Denst J. Cerebral atrophy associated with boxing. AMA Arch Neurol Psychiatry. 1959;81:403–408. doi: 10.1001/archneurpsyc.1959.02340160001001. [DOI] [PubMed] [Google Scholar]
- 54.Constantinidis J, Tissot R. Generalized Alzheimer’s neurofibrillary lesions without senile plaques. (Presentation of one anatomo-clinical case) [French] Schweiz Arch Neurol Neurochir Psychiatr. 1967;100:117–130. [PubMed] [Google Scholar]
- 55.Ferguson FR, Mawdsley C. Chronic Encephalopathy in Boxers: 8th International Congress of Neurology; Vienna. Vienna: Wiener Medizinischen Akademie; 1965. [Google Scholar]
- 56.Grahmann H, Ule G. Diagnosis of chronic cerebral symptoms in boxers (dementia pugilistica & traumatic encephalopathy of boxers) [German] Psychiatr Neurol. 1957;134:261–283. [PubMed] [Google Scholar]
- 57.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–378. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jordan BD, et al. Apolipoprotein E ε4 and fatal cerebral amyloid angiopathy associated with dementia pugilistica. Ann Neurol. 1995;38:698–699. doi: 10.1002/ana.410380429. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101:518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 60.Saing T, et al. Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma. 2012;29:1054–1070. doi: 10.1089/neu.2011.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nowak LA, Smith GG, Reyes PF. Dementia in a retired world boxing champion: case report and literature review. Clin Neuropathol. 2009;28:275–280. [PubMed] [Google Scholar]
- 62.Areza-Fegyveres R, et al. Dementia pugilistica with clinical features of Alzheimer’s disease. Arq Neuropsiquiatr. 2007;65:830–833. doi: 10.1590/s0004-282x2007000500019. [DOI] [PubMed] [Google Scholar]
- 63.Drachman D, Newall K. Case 12–1999—a 67-year-old man with three years of dementia. N Engl J Med. 1999;340:1269–1277. doi: 10.1056/NEJM199904223401609. [DOI] [PubMed] [Google Scholar]
- 64.Farbota KD, et al. Longitudinal volumetric changes following traumatic brain injury: a tensor-based morphometry study. J Int Neuropsychol Soc. 2012;18:1006–1018. doi: 10.1017/S1355617712000835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross DE, et al. Progressive brain atrophy in patients with chronic neuropsychiatric symptoms after mild traumatic brain injury: a preliminary study. Brain Inj. 2012;26:1500–1509. doi: 10.3109/02699052.2012.694570. [DOI] [PubMed] [Google Scholar]
- 66.Ross DE. Review of longitudinal studies of MRI brain volumetry in patients with traumatic brain injury. Brain Inj. 2011;25:1271–1278. doi: 10.3109/02699052.2011.624568. [DOI] [PubMed] [Google Scholar]
- 67.Tomaiuolo F, et al. Gross morphology and morphometric sequelae in the hippocampus, fornix, and corpus callosum of patients with severe non-missile traumatic brain injury without macroscopically detectable lesions: a T1 weighted MRI study. J Neurol Neurosurg Psychiatry. 2004;75:1314–1322. doi: 10.1136/jnnp.2003.017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Warner MA, et al. Assessing spatial relationships between axonal integrity, regional brain volumes, and neuropsychological outcomes after traumatic axonal injury. J Neurotrauma. 2010;27:2121–2130. doi: 10.1089/neu.2010.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Allsop D, Haga S, Bruton C, Ishii T, Roberts GW. Neurofibrillary tangles in some cases of dementia pugilistica share antigens with amyloid β-protein of Alzheimer’s disease. Am J Pathol. 1990;136:255–260. [PMC free article] [PubMed] [Google Scholar]
- 70.Geddes JF, Vowles GH, Nicoll JA, Revesz T. Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol. 1999;98:171–178. doi: 10.1007/s004010051066. [DOI] [PubMed] [Google Scholar]
- 71.Hof PR, et al. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- 72.Casson IR, et al. Brain damage in modern boxers. JAMA. 1984;251:2663–2667. [PubMed] [Google Scholar]
- 73.Bogdanoff B, Natter HM. Incidence of cavum septum pellucidum in adults: a sign of boxer’s encephalopathy. Neurology. 1989;39:991–992. doi: 10.1212/wnl.39.7.991. [DOI] [PubMed] [Google Scholar]
- 74.Bodensteiner JB, Schaefer GB. Dementia pugilistica and cavum septi pellucidi: born to box? Sports Med. 1997;24:361–365. doi: 10.2165/00007256-199724060-00002. [DOI] [PubMed] [Google Scholar]
- 75.Macpherson P, Teasdale E. CT demonstration of a 5th ventricle—a finding to KO boxers? Neuroradiology. 1988;30:506–510. doi: 10.1007/BF00339691. [DOI] [PubMed] [Google Scholar]
- 76.Schwidde JT. Incidence of cavum septi pellucidi and cavum Vergae in 1,032 human brains. AMA Arch Neurol Psychiatry. 1952;67:625–632. doi: 10.1001/archneurpsyc.1952.02320170043006. [DOI] [PubMed] [Google Scholar]
- 77.Haglund Y, Bergstrand G. Does Swedish amateur boxing lead to chronic brain damage? 2 A retrospective study with CT and MRI. Acta Neurol Scand. 1990;82:297–302. doi: 10.1111/j.1600-0404.1990.tb03307.x. [DOI] [PubMed] [Google Scholar]
- 78.Adams JH, Graham DI, Murray LS, Scott G. Diffuse axonal injury due to nonmissile head injury in humans: an analysis of 45 cases. Ann Neurol. 1982;12:557–563. doi: 10.1002/ana.410120610. [DOI] [PubMed] [Google Scholar]
- 79.Adams JH, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 80.Johnson VE, Stewart W, Smith DH. Axonal pathology in traumatic brain injury. Exp Neurol. doi: 10.1016/j.expneurol.2012.01.013. http://dx.doi.org/10.1016/j.expneurol.2012.01.013. [DOI] [PMC free article] [PubMed]
- 81.Johnson V, et al. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. doi: 10.1093/brain/aws322. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Geddes JF, Vowles GH, Robinson SF, Sutcliffe JC. Neurofibrillary tangles, but not Alzheimer-type pathology, in a young boxer. Neuropathol Appl Neurobiol. 1996;22:12–16. [PubMed] [Google Scholar]
- 83.Baugh CM, et al. Chronic traumatic encephalopathy: neurodegeneration following repetitive concussive and subconcussive brain trauma. Brain Imaging Behav. 2012;6:244–254. doi: 10.1007/s11682-012-9164-5. [DOI] [PubMed] [Google Scholar]
- 84.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 85.Corsellis JA, Brierley JB. Observations on the pathology of insidious dementia following head injury. J Ment Sci. 1959;105:714–720. doi: 10.1192/bjp.105.440.714. [DOI] [PubMed] [Google Scholar]
- 86.Rudelli R, Strom JO, Welch PT, Ambler MW. Posttraumatic premature Alzheimer’s disease. Neuropathologic findings and pathogenetic considerations. Arch Neurol. 1982;39:570–575. doi: 10.1001/archneur.1982.00510210040009. [DOI] [PubMed] [Google Scholar]
- 87.Smith C, Graham DI, Murray LS, Nicoll JA. Tau immunohistochemistry in acute brain injury. Neuropathol Appl Neurobiol. 2003;29:496–502. doi: 10.1046/j.1365-2990.2003.00488.x. [DOI] [PubMed] [Google Scholar]
- 88.Johnson VE, Stewart W, Smith DH. Widespread tau and amyloid-beta pathology many years after a single traumatic brain injury in humans. Brain Pathol. 2012;22:142–149. doi: 10.1111/j.1750-3639.2011.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dale GE, Leigh PN, Luthert P, Anderton BH, Roberts GW. Neurofibrillary tangles in dementia pugilistica are ubiquitinated. J Neurol Neurosurg Psychiatry. 1991;54:116–118. doi: 10.1136/jnnp.54.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Johnson VE, Stewart W, Smith DH. Traumatic brain injury and amyloid-β pathology: a link to Alzheimer’s disease? Nat Rev Neurosci. 2010;11:361–370. doi: 10.1038/nrn2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roberts GW, Gentleman SM, Lynch A, Graham DI. βA4 amyloid protein deposition in brain after head trauma. Lancet. 1991;338:1422–1423. doi: 10.1016/0140-6736(91)92724-g. [DOI] [PubMed] [Google Scholar]
- 92.Roberts GW, et al. β amyloid protein deposition in the brain after severe head injury: implications for the pathogenesis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1994;57:419–425. doi: 10.1136/jnnp.57.4.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikonomovic MD, et al. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203. doi: 10.1016/j.expneurol.2004.06.011. [DOI] [PubMed] [Google Scholar]
- 94.Huber A, Gabbert K, Kelemen J, Cervos-Navarro J. Density of amyloid plaques in brains after head trauma. J Neurotrauma. 1993;10 (Suppl 1):S180. [Google Scholar]
- 95.Adams JH, et al. Diffuse axonal injury in head injury: definition, diagnosis and grading. Histopathology. 1989;15:49–59. doi: 10.1111/j.1365-2559.1989.tb03040.x. [DOI] [PubMed] [Google Scholar]
- 96.Gentleman SM, Nash MJ, Sweeting CJ, Graham DI, Roberts GW. β-amyloid precursor protein (βAPP) as a marker for axonal injury after head injury. Neurosci Lett. 1993;160:139–144. doi: 10.1016/0304-3940(93)90398-5. [DOI] [PubMed] [Google Scholar]
- 97.Sherriff FE, Bridges LR, Sivaloganathan S. Early detection of axonal injury after human head trauma using immunocytochemistry for β-amyloid precursor protein. Acta Neuropathol. 1994;87:55–62. doi: 10.1007/BF00386254. [DOI] [PubMed] [Google Scholar]
- 98.Smith DH, et al. Accumulation of amyloid beta and tau and the formation of neurofilament inclusions following diffuse brain injury in the pig. J Neuropathol Exp Neurol. 1999;58:982–992. doi: 10.1097/00005072-199909000-00008. [DOI] [PubMed] [Google Scholar]
- 99.Smith DH, Chen XH, Iwata A, Graham DI. Amyloid β accumulation in axons after traumatic brain injury in humans. J Neurosurg. 2003;98:1072–1077. doi: 10.3171/jns.2003.98.5.1072. [DOI] [PubMed] [Google Scholar]
- 100.Chen XH, Johnson VE, Uryu K, Trojanowski JQ, Smith DH. A lack of amyloid β plaques despite persistent accumulation of amyloid β in axons of long-term survivors of traumatic brain injury. Brain Pathol. 2009;19:214–223. doi: 10.1111/j.1750-3639.2008.00176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gultekin SH, Smith TW. Diffuse axonal injury in craniocerebral trauma. A comparative histologic and immunohistochemical study. Arch Pathol Lab Med. 1994;118:168–171. [PubMed] [Google Scholar]
- 102.Neumann M, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 103.Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Geser F, Martinez-Lage M, Kwong LK, Lee VM, Trojanowski JQ. Amyotrophic lateral sclerosis, frontotemporal dementia and beyond: the TDP-43 diseases. J Neurol. 2009;256:1205–1214. doi: 10.1007/s00415-009-5069-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Neumann M, Kwong LK, Sampathu DM, Trojanowski JQ, Lee VM. TDP-43 proteinopathy in frontotemporal lobar degeneration and amyotrophic lateral sclerosis: protein misfolding diseases without amyloidosis. Arch Neurol. 2007;64:1388–1394. doi: 10.1001/archneur.64.10.1388. [DOI] [PubMed] [Google Scholar]
- 106.King A, et al. Abnormal TDP-43 expression is identified in the neocortex in cases of dementia pugilistica, but is mainly confined to the limbic system when identified in high and moderate stages of Alzheimer’s disease. Neuropathology. 2010;30:408–419. doi: 10.1111/j.1440-1789.2009.01085.x. [DOI] [PubMed] [Google Scholar]
- 107.Johnson VE, Stewart W, Trojanowski JQ, Smith DH. Acute and chronically increased immunoreactivity to phosphorylation-independent but not pathological TDP-43 after a single traumatic brain injury in humans. Acta Neuropathol. 2011;122:715–726. doi: 10.1007/s00401-011-0909-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mann DM, Yates PO, Hawkes J. The pathology of the human locus ceruleus. Clin Neuropathol. 1983;2:1–7. [PubMed] [Google Scholar]
- 109.Shaw K, et al. TUNEL-positive staining in white and grey matter after fatal head injury in man. Clin Neuropathol. 2001;20:106–112. [PubMed] [Google Scholar]
- 110.Maxwell WL, et al. There is differential loss of pyramidal cells from the human hippocampus with survival after blunt head injury. J Neuropathol Exp Neurol. 2003;62:272–279. doi: 10.1093/jnen/62.3.272. [DOI] [PubMed] [Google Scholar]
- 111.Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol. 2006;65:478–488. doi: 10.1097/01.jnen.0000229241.28619.75. [DOI] [PubMed] [Google Scholar]
- 112.Williams S, et al. In situ DNA fragmentation occurs in white matter up to 12 months after head injury in man. Acta Neuropathol. 2001;102:581–590. doi: 10.1007/s004010100410. [DOI] [PubMed] [Google Scholar]
- 113.Royal College of Physicians of London. Committee on Boxing. Report on the Medical Aspects of Boxing. Royal College of Physicians of London; London: 1969. [Google Scholar]
- 114.Reichard RR, Smith C, Graham DI. The significance of β-APP immunoreactivity in forensic practice. Neuropathol Appl Neurobiol. 2005;31:304–313. doi: 10.1111/j.1365-2990.2005.00645.x. [DOI] [PubMed] [Google Scholar]
- 115.Strich SJ. Diffuse degeneration of the cerebral white matter in severe dementia following head injury. J Neurol Neurosurg Psychiatry. 1956;19:163–185. doi: 10.1136/jnnp.19.3.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Perry VH, Nicoll JA, Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol. 2010;6:193–201. doi: 10.1038/nrneurol.2010.17. [DOI] [PubMed] [Google Scholar]
- 117.Brettschneider J, et al. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE. 2012;7:e39216. doi: 10.1371/journal.pone.0039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brettschneider J, et al. Microglial activation and TDP-43 pathology correlate with executive dysfunction in amyotrophic lateral sclerosis. Acta Neuropathol. 2012;123:395–407. doi: 10.1007/s00401-011-0932-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurotherapeutics. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gentleman SM, et al. Long-term intracerebral inflammatory response after traumatic brain injury. Forensic Sci Int. 2004;146:97–104. doi: 10.1016/j.forsciint.2004.06.027. [DOI] [PubMed] [Google Scholar]
- 121.Ramlackhansingh AF, et al. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70:374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- 122.Adams CW, Bruton CJ. The cerebral vasculature in dementia pugilistica. J Neurol Neurosurg Psychiatry. 1989;52:600–604. doi: 10.1136/jnnp.52.5.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Blaylock RL, Maroon J. Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy-A unifying hypothesis. Surg Neurol Int. 2011;2:107. doi: 10.4103/2152-7806.83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Walker A, Caveness W, Critchley M, editors. The Late Effects of Head Injury. Charles C. Thomas; Springfield, IL: 1969. [Google Scholar]
- 125.Center for the Study of Traumatic Encephalopathy—Boston University. 2012 [online] www.bu.edu/cste/
- 126.Braak H, Braak E. Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging. 1997;18:351–357. doi: 10.1016/s0197-4580(97)00056-0. [DOI] [PubMed] [Google Scholar]
- 127.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–969. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 128.Tokuda T, Ikeda S, Yanagisawa N, Ihara Y, Glenner GG. Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid β-protein and tau protein. Acta Neuropathol. 1991;82:280–285. doi: 10.1007/BF00308813. [DOI] [PubMed] [Google Scholar]