

Abstract
X-Linked Charcot-Marie-Tooth disease is an inherited peripheral neuropathy arising in patients with mutations in the gene encoding connexin 32 (Cx32). Cx32 is expressed at the paranodes and Schmidt-Lantermann incisures of myelinating Schwann cells in which it is believed to form a reflexive pathway between the abaxonal and adaxonal cytoplasmic domains. Patients with the Val181Ala (V181A) mutation have a severe peripheral neuropathy. Experiments using a nude mouse xenograft system show that Schwann cells expressing only this mutant form of Cx32 are profoundly impaired in their ability to support the earliest stages of regeneration of myelinated fibers. Coupling between paired Xenopus oocytes expressing V181A is reduced compared with the coupling between oocytes expressing wild-type human Cx32 (32WT), and protein levels assayed by Western blot are substantially lower. Immunocytochemisty shows that Neuro2a cells expressing the V181A mutant have very few gap junction plaques compared with cells expressing 32WT; Cx32 protein levels are lower in these cells than in those expressing 32WT. Because failure of normal regeneration is evident before formation of myelin, loss of function of Cx32 may impact on the function of precursors of the myelinating Schwann cell before the formation of the hypothesized reflexive pathway. The Glu102Gly (E102G) mutation leads to a milder phenotype. Early regeneration is normal in grafts with Schwann cells expressing the E102G mutant. The only abnormality detected in the behavior of its channel is increased sensitivity to acidification-induced closure, a property that may lead to reduced gap junction coupling during periods of metabolic stress. This restricted functional abnormality may explain the relatively mild phenotype seen in the xenograft model and in E102G patients.
Keywords: connexin 32, xenograft myelin, Charcot-Marie-Tooth disease, peripheral neuropathy, myelin regeneration
Introduction
CMTX, the X-linked form of Charcot-Marie-Tooth disease, is an inherited peripheral neuropathy arising in patients with a mutation in the gene encoding connexin (Cx) 32, a gap junction protein expressed in a number of tissues including brain (Dermietzel et al., 1997), liver (Milks et al., 1988), and peripheral nerve (Bergoffen et al., 1993). In peripheral nerve, Cx32 localizes to the paranodal and incisural regions of the myelinating Schwann cell (Scherer et al., 1995), in which it is thought to form reflexive gap junctions that shorten the diffusion pathway between perinuclear and adaxonal cytoplasm (Bergoffen et al., 1993; Oh et al., 1997). Cx29 is also expressed in the myelinating Schwann cell but does not appear to form gap junctions (Sohl et al., 2001; Altevogt et al., 2002; Li et al., 2002). Dye transfer from perinuclear to adaxonal cytoplasm occurs in both the wild-type and Cx32 knock-out (KO) mice (Balice-Gordon et al., 1998); this finding calls into question the dependence of the reflexive pathway on Cx32. The age of onset of CMTX is variable. In many cases, the earliest symptoms are noted in the second or early third decade of life; likewise, the severity in affected males varies widely, with some mutations causing particularly severe disease (Abrams and Bennett, 2000). Peripheral nerve specimens from patients with CMTX are characterized by axonal loss, partially failed regeneration, and myelin abnormalities (Senderek et al., 1998; Senderek et al., 1999; Tabaraud et al., 1999; Hahn et al., 2001). A mouse with targeted ablation of the gene for Cx32 shows similar pathological changes in myelination after 3 months of age (Anzini et al., 1997; Scherer et al., 1998).
Little is known about the fundamental role or roles of Cx32 in myelinating Schwann cells, and, therefore, the relevance of these findings to the pathogenesis of CMTX has remained obscure. Similarly, the reasons for the variability in latency of onset of symptoms in this disorder and in severity of disease caused by various mutations are not well explained.
Here, we report on mutations in Cx32 in which a valine at position 181 (in the second extracellular loop) is replaced by an alanine (V181A) or the glutamate at position 102 (in the cytoplasmic loop) is replaced by a glycine (E102G). Patients with the V181A mutation have a severe X-linked peripheral neuropathy (Abrams et al., 2000). The biophysical behavior of the V181A channel seems similar to that of the channel formed by wild-type human Cx32 (32WT). Nonetheless, (human) Schwann cells expressing only this mutant form of Cx32 are profoundly impaired in their ability to support the regeneration of (mouse) myelinated fibers (MFs). This reduced ability is correlated with reduced Cx32 expression of the mutant in Xenopus oocytes and in transfected mammalian cells relative to 32WT. Another mutation, in which glutamate at position 102 is replace by glycine (E102G), leads to a milder phenotype in several different families studied (Ionasescu et al., 1996). In contrast with the findings for V181A, early regeneration is normal in grafts with Schwann cells expressing this mutant form of Cx32 and protein expression levels are like those of 32WT in exogenous systems. The only detectable abnormality in this channel is increased sensitivity to closure by cytoplasmic acidification.
Materials and Methods
Nerve biopsies and xenografts
Xenografts generated from three CMTX patients (one patient with the V181A mutation and two from a family with E102G mutation) and three healthy controls were used for morphometric analysis. Methods of surgical procedures and tissue harvesting for data collection were published previously (Sahenk and Chen, 1998). Briefly, xenografts were generated by grafting an ∼6 mm length of a human fascicle in between the cut ends of the sciatic nerve in the anesthetized nude mice. The research protocols were approved by the Institutional Review Board for the use of human subjects and the Animal Care and Use Committee of the Ohio State University. Distal graft segments (4 mm distal to the proximal host-graft junction), obtained at 2, 4, 8, and 16 weeks after surgical procedures, were used to generate temporal profiles of myelination-associated regeneration of axons of different size, growing through the xenografts. Six xenografts from V181A, 12 from E102G, and 12 from controls were analyzed.
Morphometric analysis
Quantitative analysis at the light microscopic level was performed on 1-μm-thick cross-sections using a microscope-mounted video camera at ×1600 magnification and Bioquant TCW98 version 3.50 6ML image analysis (R&M Biometrics Inc., Nashville, TN). Five randomly selected frames (frame area, 14,285 μm2; one centrally located and four from the periphery of the nerve) from each specimen were analyzed. Automatic bordering of the inner edge of the myelin in MFs provided direct axonal perimeter measurements (although vacuolization at the inner edge of the myelin may have introduced occasional errors). Axonal diameter for each fiber was calculated as the diameter of a circle of the same perimeter. Fibers with a diameter <0.5 μm, as well as those located in the periphery of the graft, were excluded from the analysis. Automatic counting of MFs in each frame allowed calculation of MF density, which is expressed as the number per millimeter squared of fascicular area. Data from three xenografts for each time point were combined to generate MF diameter histograms. Sural nerve biopsy specimens from one patient with each Cx32 mutation and a control donor were used for morphometry. The ages of the subjects at the time of biopsy were 37 for V181A, 38 for E102G, and 36 for control.
Generation of mutants
Mutants were produced as described previously (Abrams et al., 2001). Briefly, the gene for human Cx32 was amplified by PCR from human chromosomal DNA. Mutants were produced by oligonucleotide-directed PCR, screened by restriction digestion, and sequenced using primers encompassing the entire cloned PCR product and flanking restriction sites.
In vitro transcription of RNA and expression in oocytes
RNA was transcribed using the Message-Machine kit with T7 RNA Polymerase (Ambion, Austin, TX) according to manufacturer's instructions. RNA was column purified and eluted in nuclease-free H20 containing antisense phosphorothioate oligonucleotide complementary to Cx38 at a concentration of 0.3 pmol/nl. Cx38 is the only known endogenous connexin expressed in oocytes that forms cell-cell channels, and the antisense oligonucleotide prevents endogenous coupling between paired oocytes. RNA quality was evaluated by gel electrophoresis, and concentration was quantitated by absorbance at 260 nm and evaluation of relative intensity on ethidium-stained agarose gels. The RNA was diluted, aliquoted for single use, and stored at -70°C.
Oocyte preparation and injection. Xenopus oocytes were harvested and prepared as described previously (Verselis et al., 1994; Abrams et al., 2001), then injected with 0.1 μl of a solution containing 0.22 μg/μl RNA and 0.3 pmol/nl antisense oligonucleotide to Cx38 in nuclease-free H20. After 24 hr at 18°C, oocytes were devitilinized manually and paired. Determinations of resting conductance and conductance-voltage relationships were made between 22 and 28 hr after pairing. RNA was diluted as necessary to reduce junctional conductance and minimize series resistance artifacts (Wilders and Jongsma, 1992).
Oocyte expression and dual two-electrode voltage clamping. Junctional conductances between paired oocytes were determined using dual two-electrode voltage clamp as described previously (Harris et al., 1981; Rubin et al., 1992; Verselis et al., 1994). Both cells of a pair were voltage clamped at an identical potential near their resting potentials (usually -30 mV) to yield a transjunctional voltage gradient (Vj) of zero. Simple “instantaneous” conductance measurements were made by pulsing from Vj = 0 to ±50 and ±100 mV. Values for 32WT and mutant-injected cells (both with coinjected Cx38 antisense) were compared using the Kruskal-Wallis test with Dunn's correction for multiple comparisons. For determination of steady-state conductance-voltage (Gj-Vj) relationships, cells were held between each pulse at Vj = 0 for 60 sec, and cell 1, defined as the cell being pulsed, was pulsed in 10 mV increments from -120 to +120 mV. Any resulting change in current in cell 2, the apposed cell, is attributable to junctional current. Currents are displayed with positive up for both cells; thus, a positive Vj step in cell 1 causes a downward deflection in cell 2. Each voltage pulse was preceded generally by a short (∼500 msec) pulse to -20 mV for normalization. Data were collected as described (Abrams et al., 2001). Instantaneous (t = 0) and steady-state (t = ∞) conductances were determined by fitting each current trace to a sum of exponentials of the form A1e -t/τ(1) +.... Ane -t/τ(n) + C, where C is the steady-state current, A1... n are initial current amplitudes of the component exponentials, and τ(1) to τ(n) are their time constants. Typically, two or three exponentials were required to fit a trace. Dividing the current at t = 0 or t = ∞ by the applied voltage gives the instantaneous or steady-state junctional conductance.
Transfection of Neuro2a cell lines
DNA for transfection was produced by cloning the connexin of interest into the pIRES-EGFP2 vector (Clontech, Palo Alto, CA). Stable transfections were performed using Lipofectin (Invitrogen, Rockville MD) according to the manufacturer's instructions. Cells were dissociated and replated onto number 0 glass coverslips at least 3 hr before recording.
Recording from transfected mammalian cell lines
Dual whole-cell patch clamping was performed as described previously (Oh et al., 1997). The standard pipette solution was (in mm): 140 KCl, 2 EGTA, 1 CaCL2, 3 MgCl2, and 3 HEPES, pH7.2. The bath solution was (in mm): 150 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 0.9 NaH2PO4, 5 glucose, and 5 HEPES, pH 7.2. Permeability to Lucifer yellow was determined by selecting an isolated cell pair and establishing gigaohm seals with each of the cells. One pipette contained recording solution and 1 mm Lucifer yellow, whereas the other contained recording solution alone. The patch beneath the dye-containing pipette was then ruptured, allowing dye to diffuse into the cell. When the two cells were coupled via gap junctions permeable to Lucifer yellow, dye transfer between the two cells was observed. At the end of each dye transfer experiment, junctional conductance was measured to exclude false negative results, and 2 mm heptanol was applied to exclude the possibility of a cytoplasmic bridge between the cells.
Indirect immunofluoresence on fixed cells
Cells were plated overnight on poly-d-lysine-treated coverslips, fixed with 4% paraformaldehyde with HOECHST 33258 (Molecular Probes, Eugene, OR) at 4 μg/ml for 15 min at room temperature, and blocked in ICC buffer (0.1% Triton X-100, 2% BSA, 1% normal goat serum, and PBS). The coverslips were incubated in mouse anti-Cx32 (C76.C7; a gift from Dr. Elliot Hertzberg, Albert Einstein College of Medicine, Bronx, NY) diluted 1:200 in ICC buffer. This antibody is to a C-terminal epitope that would not be affected by the mutations. The coverslips were then washed, incubated with Alexa 546-conjugated secondary antibody (Molecular Probes), washed, mounted with Prolong Antifade (Molecular Probes), and imaged using scanning laser confocal microscopy. Images presented are maximum intensity projections from five contiguous optical sections produced using the IPLab program (Scanalytics, Fairfax, VA). Monlayers of Neuro2a cells stably expressing 32WT or the V181A or E102G mutants and stained with anti-Cx32 were used to quantitate intercellular puncta (the light microscopic correlate of gap junction plaques). For each connexin, five randomly selected high-power (400×) fields were photographed at identical exposure with the Magnafire digital CCD camera (Olympus, Melville, NY). Intercellular puncta and cells were counted manually, and a puncta/cell ratio was determined for each image. Cell counts were made by counting Hoechst-stained nuclei. The individual ratios were then averaged and analyzed for statistical significance using ANOVA.
Western blotting of mammalian cell line and oocyte extracts
Cells were plated in 12-well (22 mm) plates and grown to 80% confluence. The cells were washed on ice with PBS and lysed in NP-40 buffer (1% NP-40, 50 mm Tris, and 0.5% SDS) with freshly added PMSF (25 mm), leupeptin (10 μg/ml), aprotonin (10 μg/ml), and DTT (0.5 mm) for 30 min. Extracts from 5 × 104 cell samples were diluted with sample buffer (2% SDS, 62.5 mm Tris, pH 6.8, 5% glycerol, 5% β-mercaptoethanol, and 0.25% bromophenol blue), incubated at room temperature 10 min, loaded onto a 10% SDS-polyacrylamide gel, electrophoresed, and transferred to Immobilon-P membrane (Millipore, Billerica, MA). The blots were probed with either a mouse monoclonal antibody to the C terminus of Cx32 (1:1500) or rabbit anti-enhanced green fluorescent protein (EGFP) (1:300; Clontech), followed by incubation with peroxidase-conjugated secondary antibodies (KPL Laboratories, Gaithersburg, MD). The Western blots were visualized by enhanced chemiluminescence (Pierce, Rockford, IL) according to the manufacturer's instructions. The following modifications were used for oocyte protein extracts. Oocytes were homogenized in 20 μl/oocyte of 100 mm NaCl, 20 mm Tris-HCl, pH 7.4, 1% Triton X, and protease inhibitor mixture (Sigma, St. Louis, MO). One oocyte equivalent was loaded per lane. Blots were probed with a monoclonal antibody to residues 106-124 of the cytoplasmic loop of Cx32 (1:1000; Sigma). Relative Western blot band intensities were determined from digitally scanned images. Adobe Photoshop (Adobe Systems, San Jose, CA) was used to invert the images, and the histogram function was used to determine the average pixel intensity of a fixed area large enough to encompass each of the bands of interest. Because the area for each of the determinations was fixed, the ratios of the average pixel intensities for each band are equal to the ratios of band intensities. For blots of oocyte extracts, the ratios given are simply those of mutant or control with respect to WT. For blots of Neuro2a extracts, the intensity of each Cx32 immunoreactive band was first normalized to the intensity of the EGFP immunoreactive band from the same line. For each gel, the ratios for the 32WT-expressing lines were averaged. This average value was then used to normalize each of the individual Cx32/EGFP ratios for that gel. All normalized values for 32WT-, E102G-, or V181A-expressing lines were then averaged, and differences were examined for statistical significance using ANOVA.
Results
Descriptions of patients
A 37-year-old man with onset of disease in early childhood and multiple surgical procedures on his feet presented with severe distal extremity weakness, more pronounced in lower extremities, muscle atrophy, and prominent vibration and position sense loss at toes. He first displayed gait difficulties at the age of 1 year. The clinical examination at age 5 showed that he had difficulty walking on his heels and toes; bilateral cavovarus feet with hammer-toes were noted. Weakness and intrinsic atrophy in the hands were seen. Electrodiagnostic studies at that time revealed normal nerve conduction velocities (NCVs). Repeat studies at age 13 showed a marked drop in NCVs (peroneal CV, 19 m/sec; ulnar, 38 m/sec; median, 30 m/sec; normal NCVs: peroneal, >39 msec; median and ulnar, >49 m/sec). Three maternal male cousins and a maternal great uncle were also affected. Direct sequencing of the Cx32 gene revealed a novel missense mutation (T to C change at position 604), causing an amino acid substitution of Val with Ala at position 181. In contrast, the patient with the E102G mutation presented at age 34 with muscle atrophy of the hands and feet and a history of gait difficulties beginning at age 18. Electrodiagnostic studies showed reduced NCVs. (ulnar, 36 m/sec; median, 34 m/sec). Biopsies were obtained from each patient.
Sural nerve biopsies
The prominent features of the biopsy from the patient harboring the V181A mutation are shown in Figure 1a. In contrast to what is seen in control biopsies (Fig. 1c), the fiber density distribution is much narrower and has an increase in smaller diameter fibers. Specifically, the patient with the V181A mutation has a MF density of 4822/mm2, whereas the age-matched control subject biopsy has a density of 8038/mm2. Large diameter MFs (>4 μm) comprise only 3% of the total MFs in the V181A biopsy compared with 60% of the total MFs in the control biopsy. Regeneration-associated onion bulbs (arrowheads), an absence of large fibers with normal caliber myelin, and many thinly MFs figure prominently. Scattered examples of fibers with an enlarged region of the periaxonal space (arrows) are also noted (Fig. 1a, inset). This finding is unlikely to be an artifact because: (1) the tissue is otherwise well preserved; and (2) the findings are similar to those described in other biopsies from patients with CMTX (Hahn et al., 2001). The biopsy from a patient with the E102G mutation (Fig. 1b) also shows a MF density distribution that is abnormally shifted toward smaller diameter fibers. The MF density is 6174/mm2, with large MFs constituting 11% of the total. Regeneration-associated onion bulbs are seen, but fibers with enlarged periaxonal spaces are rare. MF size distributions for the V181A, E102G, and control biopsies are shown in Figure 1c. Thus, analysis of the biopsies of the patients harboring the E102G and V181A mutations confirm the clinical observation that the V181A mutation leads to a more severe neuropathy than does the E102G mutation. Furthermore, the enlarged periaxonal spaces frequently seen in the biopsy of the patient with the V181A mutation are only rarely seen in the biopsy from the patient with the E102G mutation.
Figure 1.

Sural nerve biopsies from male patients harboring mutations in Cx32. a, Sural nerve biopsy from a patient with the V181A mutation. Prominent features include the presence of thinly myelinated smaller-diameter fibers and regeneration-associated onion bulbs (arrowheads). Scattered enlarged periaxonal spaces (arrows) were seen in this biopsy specimen. The inset shows a fiber associated with an enlarged periaxonal space and onion bulb. b, Sural nerve biopsy from a patient with the E102G mutation. Fibers with normal myelin thickness as well as regeneration-associated onion bulbs (arrowhead) were seen in this biopsy. c, MF size distribution in sural nerve biopsies. Quantitative morphological analyses of the nerves were performed as described in Materials and Methods. The fiber loss was more severe in the V181A nerve with an MF density of 4872/mm2 in comparison with the E102G nerve (6174/mm2) and an age-matched control nerve (8038/mm2). In both mutations, there were larger numbers of small-diameter myelinated axons, often in clusters, suggesting active regeneration, and there were fewer axons >4 μm. The large-diameter fibers in the V181A mutation comprised only 3% of the total MFs in the V181A nerve compared with 11% in the E102G nerve and 60% in the control nerve. Scale bars, 10 μm.
Sural nerve xenografts in nude mice
To further investigate the CMTX-inducing mutations, we replaced segments of the sciatic nerves of nude mice with fascicles from the patients' sural nerves. This xenograft allows for the examination of the effects of the specific mutations expressed in donor Schwann cells on regeneration of host axons that are (presumably) normal in their competence to regenerate through mouse Schwann cells. Myelination of nude mouse axons regenerating through xenografts of control human nerve shows striking similarities to the myelination process during the postnatal period in rats (Fraher, 1972, 1973; Hahn et al., 1987). Serial examination of axon size (diameter) distribution histograms generated from the distal graft segments (∼4 mm distal to proximal mouse sciatic nerve-graft junction) revealed that size differentiation of MFs within the grafts is established as early as 2 weeks (Sahenk et al., 2003), although myelination of the smallest MFs (0.5-2 μm) is delayed by ∼2 weeks compared with fibers >4 μm.
Representative areas of sections from the distal graft segments of control, V181A, and E102G xenografts at 2, 4, 8, and 16 weeks are shown in Figures 2 and 3. The results of morphometric analysis of five randomly selected areas from xenografts at each time point are shown in Figure 4. As is evident from inspection of the micrographs and shown in Figure 4e, the overall MF densities for V181A are less at 2 and 4 weeks than those for the control and E102G, but the difference is reduced at 8 weeks. For 2, 4, 8, and 16 weeks, normalized total MF counts are 0, 19, 39, and 36%, respectively, when compared with control xenografts. At the earliest time examined, 2 weeks, there was little or no regeneration in the V181A xenograft in contrast to that of the E102G or control (Fig. 4a). At 4 weeks, all MF densities up to 4 μm were lower than those for the control or E102G grafts, but the densities of MFs >4 μm were all similar. Pathological radial growth (increase in diameter) of large fibers, thought to be caused by disordered axonal transport (Sahenk and Chen, 1998), may explain this finding. The presence of large naked axons (Fig. 3a, arrow) with no myelin in the 2 week graft distal segment suggests that the delayed regeneration is attributable to a defect in onset of myelination. In fact, none of the axons were myelinated, and many appeared to be participating in premyelinating Schwann cell-axon complexes. As with the biopsy specimen from the patient with the V181A mutation, the 8 and 16 week graft distal sections contained MFs with large periaxonal and intramyelinic clear spaces (Fig. 3c,d, arrowheads), some with fine granular material (data not shown). As with the biopsy specimen, these are very similar to artifacts that can be induced during processing. However, their high frequency and similarity to findings in pathological specimens from mice lacking Cx32 suggests that they may result from an underlying defect related to loss of Cx32 function.
Figure 2.
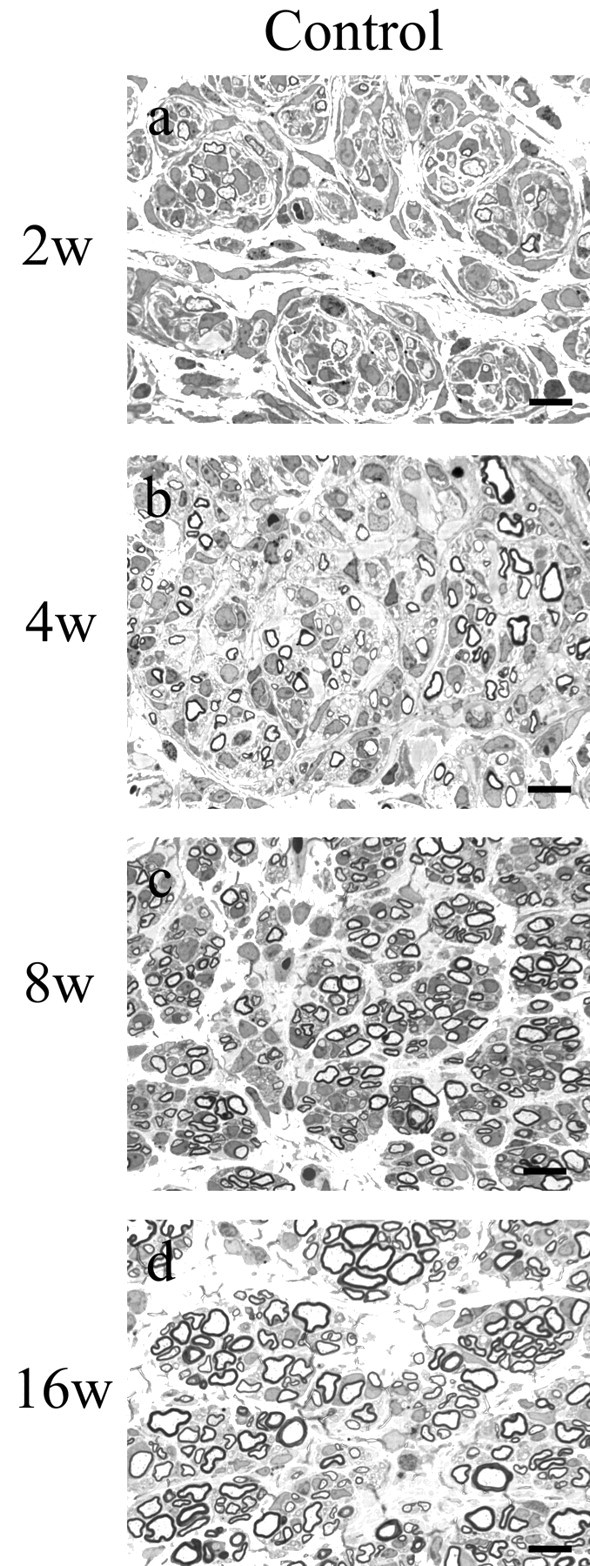
Regeneration in control xenografts at 2 (a), 4 (b), 8 (c), and 16 (d) weeks after surgical procedures. One-micrometer-thick, toluidine blue-stained cross-sections taken ∼1 mm proximal to the distal graft host junction are shown. Sural nerve was obtained from the normal donor, and nerve xenografts were generated as described in Materials and Methods. By 2 weeks, many myelinated axons, predominantly in the 2-4 μm size range, had regenerated into the graft. The density of all fiber sizes increased at 4 and 8 weeks, but a relative increase in the number of very small (<2 μm) MFs was noted. At 16 weeks, there was a substantial increase in the number of large (>4 μm)-diameter fibers (for histograms, see Fig.4a-d). Scale bars, 10 μm.
Figure 3.

Regeneration in V181A and E02G xenografts at 2, 4, 8, and 16 weeks after surgical procedures. One-micrometer-thick, toluidine blue-stained cross-sections taken ∼1 mm proximal to the distal graft host junction are shown. a-d, V181A xenografts at 2 (a), 4 (b), 8 (c), and 16 (d) weeks. Regeneration was significantly delayed compared with the E102G and control xenografts. Occasional fibers showed regions of increased periaxonal space (c, d, arrowheads). e-h, E102G xenografts at 2 (e), 4 (f), 8 (g), and 16 (h) weeks. Axons efficiently regenerated into and through the xenograft. Enlarged regions of periaxonal space noted for the V181A xenografts were not present in the E102G grafts. Although more prominent in the more proximal portion of the graft (Sahenk and Chen, 1998), abnormally enlarged axons were also seen in the distal segments of the E102G grafts. These were most prominent at 8 weeks. Scale bars, 10 μm.
Figure 4.

MF size distribution in xenografts. Quantitative morphological analysis of xenografts at 2, 4, 8, and 16 weeks. a-d, MF size distribution histograms at 4, 8, and 16 weeks after grafting revealed significantly different patterns of regeneration-associated myelination for these two Cx32 mutations. At 2 and 4 weeks, regeneration and myelination of all fiber sizes through the V181A grafts were significantly retarded compared with E102G and controls grafts. At 8 and 16 weeks, V181A densities approached those for E102G but remained below those in control grafts. e, Total MF densities for control and E102G were similar at 2 and 4 weeks. At later times, the densities for control, but not E102G, MFs continued to increase. MF counts for V181A xenografts were reduced at all time points.
The MF densities for the E102G xenografts are somewhat less affected than are those for the V181A xenografts, with overall normalized MF counts of 144, 98, 56, and 44% of control xenografts at 2, 4, 8, and 16 weeks, respectively. Total MF densities are similar to those for control at 2 and 4 weeks (Fig. 4e), suggesting that early regeneration is accomplished in an efficient manner. However, at 8 weeks, densities of MFs >4 μm are actually increased with respect to control, a finding that may be accounted for by pathological radial growth associated with abnormalities in neurofilament packing (Sahenk and Chen, 1998). Absolute fiber counts for MFs <4 μm increase between 8 and 16 weeks, whereas those for fibers >4 μm drop markedly, suggesting that pathological radial growth may eventually lead to loss of the largest MFs and that regeneration is likely continuing from a more proximal site.
Expression of gap junctions formed by V181A, but not E102G, is reduced compared with 32WT
Each gap junction cell-cell channel is composed of two gap junction hemichannels or connexons, one anchored in each of two apposed cell membranes. Gap junction hemichannels, in turn, are composed of six connexin molecules. Homomeric hemichannels contain six identical subunits, whereas heteromeric hemichannels contain differing subunits. A homotypic cell-cell channel contains two identical apposed hemichannels, whereas in a heteromeric channel the two hemichannels differ. Xenopus oocytes were injected with mRNA for V181A, E102G, or 32WT and paired homotypically to evaluate the channel-forming ability of the mutants (Fig. 5a). One day after pairing, the mean (instantaneous) junctional conductance between oocytes injected with RNA for E102G and paired homotypically was similar to that between paired oocytes expressing 32WT. Paired oocytes expressing the V181A mutant showed an average junctional conductance significantly reduced to 6% of those expressing 32WT (p < 0.01), whereas E102G-expressing oocytes showed a nonsignificant reduction to 70%. These differences are also seen at longer intervals after pairing. We used semiquantitative Western blot analysis of total protein extracts from oocytes (Fig. 5b) to examine the expression levels of Cx32. V181A-expressing oocytes had levels of Cx32 immunoreactive protein that were significantly reduced to 40 ± 8.4% of 32WT (p < 0.05), whereas the E102G-expressing oocytes had levels of Cx32 immunoreactive protein 130 ± 29% of 32WT (not significantly different), suggesting that the smaller junctional conductance of the V181A mutant was attributable, at least in part, to reduced steady-state levels of Cx32 V181A protein; the inability of the protein to traffic to the cell membrane and be incorporated into gap junctions or an alteration in the biophysical properties of the channel could have also contributed to the lower conductance. Investigations to evaluate these possibilities are described below.
Figure 5.

Expression of 32WT and the V181A and E102G mutants in Xenopus oocytes. a, Graphic representation of junctional conductance of homotypically paired oocytes injected with antisense oligonucleotide to Cx38 (Anti38) or mRNA for 32WT, or the V181A or E102G mutants determined as described in Materials and Methods; n = 44, 26, 37, and 15, respectively. b, Representative Western blot of total protein extracts from sister oocytes of those injected for assessment of junctional conductance. Levels of expression of V181A were significantly lower than those of 32WT (see Results for details).
The instantaneous and steady-state macroscopic junctional Gj-Vj relationships for channels formed by V181A and E102G are similar to those for the 32WT channel
Junctional conductance between paired oocytes expressing 32WT is maximal when no voltage gradient exists between the two cells (i.e., when the transjunctional voltage, Vj, is 0 mV). As shown in Figure 6a, 32WT induced junctional current decay when the absolute value of Vj is increased above ∼30 mV. These current traces can be used to compute normalized instantaneous and steady-state Gj-Vj relationships such as that shown for 32WT in Figure 6b. Applying the same analysis to oocytes expressing V181A and E102G shows that these mutants have junctional currents (Fig. 6c,e) and instantaneous and steady-state Gj-Vj relationships (Fig. 6d,f) virtually identical to those of 32WT. Some of the findings for E102G have been reported previously by Ressot et al. (1998), and Oh et al. (1997) showed similar results for rat Cx32 containing this mutation. This result suggests that the reduced junctional conductance seen for V181A is not caused by altered gating but rather to a reduction in the number of functional channels in the membrane (Abrams et al., 2001). The lack of an effect of the V181A and E102G mutations on voltage gating is also supported by heterotypic pairings between the V181A and E102G mutants and wild-type Cx26 (26WT). The resulting instantaneous and steady-state Gj-Vj relationships (Fig. 6h,i) are only slightly different from those generated for the heterotypic 32WT/26WT channels (Fig. 6g). This suggests that the V181A, E102G, and 32WT hemichannels function similarly when paired with 26WT.
Figure 6.

Representative current traces and average normalized Gj-Vj relationships for 32WT and the E102G and V181A mutants. a, c, e, Xenopus oocytes were injected with the noted mRNAs and paired as described in Materials and Methods. Junctional currents recorded from cell 1 in response to voltage steps to cell 2 in 20 mV increments from ±20 to ±120 are shown. b, d, f, Average instantaneous (▵) and steady-state (▪) Gj-Vj relationships for 32WT and the two mutants determined from currents like those in a, c, and e and plotted as mean ± SEM. g-i, Average normalized Gj-Vj relationships for 32WT and the two mutants paired heterotypically with 26WT. Xenopus oocytes were injected with the noted mRNAs and paired heterotypically with 26WT. Junctional currents in response to a series of voltage steps were obtained and analyzed. The resulting instantaneous (▵) and steady-state (▪) Gj-Vj relationships were averaged and plotted as mean ± SEM.
The channels formed by V181A and E102G exhibit single-channel conductances and dye permeabilities similar to those of the 32WT channel
To further evaluate the functional implications of the V181A and E102G mutations, we examined the single-channel conductance and dye permeability of the junctions formed by the V181A and E102G mutants in Neuro2a cells. Both the V181A and E102G mutants showed predominant unitary conductance changes of ∼55 pS in 140 mm KCl (Fig. 7a,b), a value very similar to that seen for 32WT (Oh et al., 1997) in 140 mm CsCl. Chimeras formed by fusion of EGFP (Trexler et al., 2000) to the C terminal of 32WT (32WT-EGFP), V181A (V181A-EGFP), and E102G (E102G-EGFP) were used to evaluate the effects of these mutations on permeability to Lucifer yellow (Fig. 8). The EGFP fusion proteins allowed for selection of cell pairs coupled by gap junction plaques. (These plaques were much less common in the V181A-EGFP-expressing cells than in the 32WT-EGFP- or E102G-EGFP-expressing cells.) As for 32WT-EGFP (Trexler et al., 2000), channels formed by the V181A and E102G mutants were permeable to Lucifer yellow. Because Lucifer yellow is a fairly large divalent anion (molecular weight, 443), this finding suggests that these mutant channels are unlikely to exclude small signaling molecules on the basis of size. Additional support for similarity of the permeability of 32WT, E102G, and V181A channels is lack of significant effect of these mutations on the instantaneous Gj-Vj relationships produced by pairing with 26WT. Changes in fixed charges in the pore of a mutant Cx32 hemichannel (which could lead to changes in selectivity) would be predicted to change the instantaneous rectification of the heterotypic cell-cell channel formed by pairing the mutant with 26WT (Oh et al., 1999). Furthermore, changes in pore size without changes in total fixed charge may also affect instantaneous rectification by changing charge density (Bukauskas et al., 2002).
Figure 7.

Single-channel recordings of the cell-cell channel formed by the V181A and E102G mutants. Ramps were applied to cell 1 from -120 to +120 mV, and currents were recorded in cell 2. a, Single-channel currents from cells expressing the V181A mutant. b, Single-channel currents from cells expressing the E102G mutant. c, Graphic representation of the voltage waveform applied to cell 1. The sizes of the major transitions (arrows) for both channels (∼50-55 pS) were similar to what we (data not shown) and others (Oh et al., 1997) have observed for 32WT.
Figure 8.

Dye transfer between Neuro2a cells transfected with the V181A*EGFP or E102G*EGFP mutants. Pairs of cells expressing fluorescent plaques, a morphological correlate of EGFP-tagged gap junctions, were selected. Using the dual patch-clamp technique, high-resistance seals were established with each of the adjacent cells expressing V181A*EGFP (a) and E102G*EGFP (d). One pipette (patched to cell 1, denoted by an asterisk) contained both recording solution and ∼1 mm Lucifer yellow, whereas the other pipette, patched to cell 2, contained only recording solution. The patch under the Lucifer yellow-containing pipette was then ruptured, allowing dye to diffuse into cell 1. b, e, Fluorescent images obtained a few seconds after rupture. c,f, Fluorescent images obtained ∼30 sec later for V181A and 90 sec later for E102G, showing substantial amounts of Lucifer yellow in cell 2 of each pair. At the end of each experiment, the membrane under pipette 2 was ruptured, and junctional conductance was measured (in this case, 50 nS for the V181A-expressing pair and 25 nS for the E102G-expressing pair). In addition, the presence of a cytoplasmic bridge was excluded by demonstrating that the conductance between cell 1 and cell 2 could be reduced to undetectable levels by application of a bath solution containing 1% heptanol (v/v). Similar findings were obtained with 32WT*EGFP. In no case did untransfected Neuro2a cells lacking cytoplasmic bridges show detectable levels of dye transfer.
The channel formed by E102G, but not V181A, exhibits increased sensitivity to acidification when compared with the 32WT channel
Intracellular acidification of cells coupled via gap junctions reduces junctional conductance (Spray et al., 1981). In mammalian cells coupled by gap junctions, such as transfected cells expressing Cx32, acidification with CO2-saturated bath solution leads to a very rapid and complete loss of junctional conductance, with recovery after reperfusion with CO2-free solution (Trexler et al., 1999). In oocytes, the reduction in Cx32-induced junctional conductance is somewhat slower and is generally incomplete (Wang and Peracchia, 1996), possibly because of the large size of the oocytes and inaccessibility of the appositional membrane. The effect of acidification of an oocyte pair expressing 32WT is shown in Figure 9a. In agreement with observations published previously (Wang and Peracchia, 1996; Ressot et al., 1998), there was an initial small increase in junctional conductance, followed by a slow decline in junctional conductance over many minutes. Reversal of acidification led to a nearly immediate, but slow, increase in junctional conductance. In several cases, the final level of junctional conductance was substantially greater than that at the beginning of the experiment, a change ascribable to continued formation of channels. The sensitivity of the V181A-induced conductance to CO2 seems similar to that for 32WT (Fig. 9b) in that the time courses are virtually superimposable until the 17.5 min time point, with differences between the values for WT and V181A smaller than the error bars. The differences that occur after 17.5 min may involve pH-dependent recruitment of a nonjunctional pool of connexins, which may be larger in the 32WT-expressing cells. In contrast, as reported previously (Ressot et al., 1998), the channel formed by the E102G mutant showed an increased sensitivity to acidification compared with 32WT, with a more rapid and complete loss of junctional conductance and a more delayed onset of recovery when CO2 was removed from the bath (Fig. 9c). As for the cells expressing 32WT, the E102G-expressing cells also show variable degrees of overshoot with removal of CO2 from the bath solution.
Figure 9.

Effect of bath acidification with CO2 on junctional conductance of 32WT and the V181A and E102G mutants. Oocytes were injected with the mRNAs for 32WT, V181A, and E102G and paired homotypically. Both cells were clamped at -30 mV, and junctional conductances were monitored by measuring the current responses in cell 2 to a ±10 mV pulse applied to cell 1 (frequency, 0.5 Hz). After establishing a stable baseline conductance for at least 2 min, the bath was acidified by continuous perfusion with bath solution saturated with CO2 (arrows). Twelve minutes later, the perfusate was changed to a standard bath solution at pH 7.6 (arrowheads). Junctional conductances were determined at 30 sec intervals and plotted for times beginning 1 min before application of CO2-saturated solution. As shown, the effects of acidification and realkalinization on the 32WT-induced (a) and V181A-induced (b) junctional conductances were nearly identical, whereas the E102G-induced (c) junctional conductance declines more rapidly and fully; the E102G-induced conductance also shows a longer delay in onset of recovery when CO2 is removed from the bath. The overshoot seen with realkalinization of the WT and E102G was quite variable; overshoot was not as prominent with the V181A mutant, in which overall protein levels are lower. Overshoot is likely an effect of pH on junctional conductance distinct from any direct effect on channel gating.
Cx32 protein levels are lower in transfected cell lines expressing V181A than in lines expressing E102G and 32WT
Detailed biophysical analyses of the V181A mutant indicated that altered gating did not account for the reduced levels of functional expression in oocytes. However, Western blotting of oocyte extracts showed a reduced level of Cx32 immunoreactive protein for this mutant. We performed Western blot analysis of protein extracts from transfected Neuro2a cells to confirm that the reduction seen in oocytes was also seen in a mammalian cell type. The vector used in these transfections (pIRES-EGFP2) contains the EGFP gene downstream of an internal ribosome entry site (IRES), which in turn is just downstream from the cloning site into which the gene of interest (32WT or a mutant form of Cx32) was inserted. Thus, each mRNA transcript for Cx32 had an attached transcript for EGFP just downstream of an IRES. As shown in Figure 10a, Cx32 immunoreactivity was substantially lower in protein extracts from the stably transfected V181A-expressing cells than in extracts from the 32WT-expressing cells, whereas these same cell lines had comparable levels of EGFP. Thus, reduction in protein levels for V181A is unlikely because of a lower number of incorporated copies of the transfected construct or reduced transcriptional efficiency. The lower levels for V181A might have resulted from reduced translational efficiency or mRNA stability; a reduced half-life of the protein, possibly associated with misfolding and destruction, could also have contributed. In contrast, Cx32 immunoreactivity in protein extracts from cells expressing Cx32WT and E102G was similar (Fig. 10b). Semiquantitative analysis of Cx32 immunoreactive protein was performed as described in the Materials and Methods. Levels of Cx32 immunoreactive protein were significantly lower for V181A than for 32WT (0.26 ± 0.081 for V181A and 1.0 ± 0.18 for 32WT; p < 0.01), whereas levels of E102G (0.80 ± 0.21) were not statistically different from 32WT. (Twelve independent clonal lines were examined for 32WT and V181A, and three were examined for E102G.)
Figure 10.
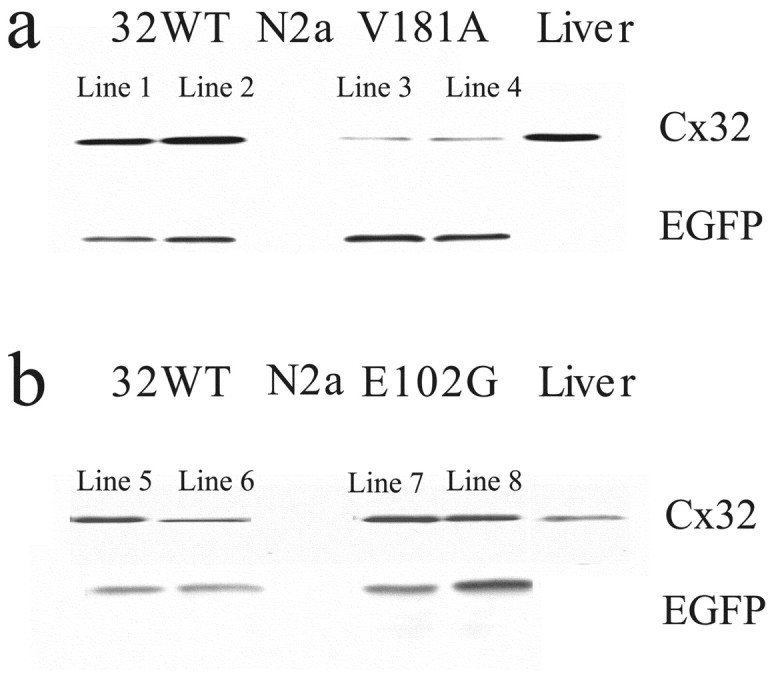
Expression levels of 32WT and E102G in transfected Neuro2a cells were significantly higher than that of V181A expressed in stably transfected Neuro2A cells, as determined by Western blot. Neuro2A cells were stably transfected with 32WT or the V181A or E102G mutants in the pIRES-EGFP vector as described in Materials and Methods. Protein extracts from 5 × 104 cells/well were loaded in each lane, and blots were probed with Cx32 antibody as described in Materials and Methods. a, Comparison of two cell lines expressing Cx32WT (1 and 2) or the V181A mutation (3 and 4). As shown, levels of EGFP expression in the four lines are comparable. However, the level of expression of V181A is much lower than that of 32WT. b, Comparison of two lines expressing Cx32WT (5 and 6) or the E102G mutation (7 and 8). As shown, levels of expression of EGFP in the two lines are comparable as are levels of 32WT and E102G.
The distribution of V181A protein differs from that of 32WT or E102G proteins
Cx32 immunoreactivity was examined in fixed Neuro2a cells transfected with DNA encoding 32WT or the V181A or E102G mutants. The fluorescence levels for V181A (Fig. 11b) were low compared with those for either 32WT (Fig. 11a) or E102G (Fig. 11c). 32WT (Fig. 11a) was distributed in what appeared to be intercellular puncta as well as in the cytoplasm in a mixed diffuse and punctate distribution. The apparent intercellular puncta are likely to be gap junction plaques, whereas the intracellular puncta presumably are vesicles containing connexins being transported to the surface for insertion or from the surface for degradation. V181A was distributed predominantly in the cytoplasm (Fig. 11b) with rare intracellular or intercellular puncta. Like 32WT, the E102G mutant (Fig. 11c) showed large numbers of intercellular plaques and intracellular puncta. We quantitated the numbers of intercellular puncta (plaques) for five randomly selected high-power fields of Neuro2a cells expressing 32WT and the V181A and E102G mutants. 32WT-expressing cells showed 29 ± 4.0 plaques/100 cells, E102G-expressing cells showed 21 ± 3.7 plaques/100 cells, and V181A-expressing cells showed 3.0 ± 2.5 plaques/100 cells. The difference between 32WT and V181A is statistically significant (p < 0.001), whereas the difference between 32WT and E102G is not. We also examined the distribution of EGFP-tagged forms of 32WT and the two mutants. The findings were similar to those for the non-EGFP-tagged forms of 32WT and the mutants (data not shown).
Figure 11.

Immunohistochemical localization of Cx32 in stably transfected Neuro2a cells expressing 32WT or the E102G or V181A mutants. a, 32WT is distributed in intercellular and intracellular puncta as well as, too a lesser extent, diffusely in the cytoplasm in a predominantly perinuclear distribution. The intercellular puncta are likely to be gap junction plaques. b, Like 32WT, the E102G mutant shows large intercellular plaques; interestingly, the number of intracellular puncta seems reduced. c, V181A is distributed diffusely, predominantly in the perinuclear cytoplasm with extremely rare intracellular or intercellular puncta. d, These control cells expressing Cx32WT were processed in a manner identical to those shown in a-c, except that mouse IgG was substituted for the Cx32-specific antibody. Similar results were obtained using Cx32-specific antibody with untransfected Neuro2a cells.
Differences between steady-state distribution of 32WT and V181A may be attributable to degradation of a substantial percentage of the mutant by cellular quality control machinery before it can be trafficked to (and subsequently from) the cell membrane. However, we cannot currently distinguish between this and other possibilities such as inability of the mutant to enter into a protected compartment, in which it would have a greater half-life. Regardless of the mechanism, decreased steady-state levels of the V181A mutant may provide an explanation for the lower levels of functional expression in oocytes, lower level of Cx32 in transfected Neuro2a cells, and much smaller number of gap junction plaques detected by immunocytochemistry.
Discussion
Here, we describe the clinical features of two patients with X-linked inherited neuropathies. The V181A mutation leads to a severe neuropathy as shown by early onset of symptoms and profound loss of large MFs in biopsied nerve. In contrast, the E102G mutation gives rise to a less severe clinical syndrome with later onset and a smaller reduction in number of large MFs. The different biological effects of these two mutations are further emphasized by application of the xenograft model. In the case of the V181A mutation, there is a substantial delay in myelination in the distal graft segment compared with human WT control or to the E102G xenografts. This delay seems to be related to defects in the onset of myelination, because virtually all axons (including some of fairly large caliber) present in the distal portion of the V181A graft at 2 weeks are unmyelinated. Furthermore, those axons that do succeed in regenerating develop axonal cytoskeletal abnormalities and seem to undergo additional cycles of degeneration and regeneration. Scattered adaxonal clear spaces are seen in the biopsy from the patient harboring the V181A mutation but are much more prominent in the V181A xenografts at 8 and 16 weeks. We believe that these spaces, many of which are associated with collapsed, atrophic axons, may represent an early form of axonal degeneration. These findings are present from 4 weeks after grafting but are more prominent at 12 weeks (data not shown). We have seen similar features during regeneration after crush of the sciatic nerve of the Cx32 KO mice (Z. Sahenk and C. K. Abrams, unpublished observations). This pathological feature has also been reported in both human biopsy tissue from patients with CMTX (Hahn et al., 2001) and in the Cx32 KO mouse (Anzini et al., 1997; Scherer et al., 1998). In contrast, in the case of the E102G mutant, early regeneration and myelination seems to proceed normally, with axonal loss occurring only after development of axoskeletal abnormalities as noted above and fully described previously (Sahenk and Chen, 1998). Studies on the functional properties of these two mutants reveal that the biophysical properties of the intercellular channel formed by the V181A mutant, including instantaneous and steady-state Gj-Vj relationships for homotypic junctions, instantaneous and steady-state Gj-Vj relationships for the heterotypic pairing with Cx26, single-channel conductance, permeability to Lucifer yellow, and response to acidification and realkalinization, are similar to those for 32WT. However, the steady-state levels of protein and functional expression are reduced. Thus, all functions normally subsumed by Cx32, including those involved in nerve regeneration, would be predicted to be disrupted, and a severe clinical phenotype would be expected.
Protein and functional expression levels of the E102G mutant are similar to WT; the only functional abnormality is an altered sensitivity to acidification-induced closure and a delay in recovery of conductance with realkalinization. For this mutant, loss of function may occur only at times of metabolic stress, when tissue acidosis is likely. This limited loss of function might lead to a milder disease. Regardless, the finding that the E102G mutant is particularly sensitive to pH-mediated closure may have important implications for patients harboring this or other pH-sensitive mutations because of the possible effects of vigorous exercise or other conditions causing relative ischemia of the peripheral nerve.
One interpretation of the findings in the V181A xenografts is that expression of Cx32 is required for efficient nerve regeneration. This might seem unlikely because of previous reports that after crush Cx32 protein and mRNA levels drop precipitously in the nerve segments distal to the crush (Scherer et al., 1995; Chandross et al., 1996). Nonetheless, the Cx32 message is clearly present in the proximal segment at 4 and 8 d, even though the message encoding P0, an essential myelin protein, is extremely low to undetectable. During this interval, one would expect regeneration to be taking place just distal to the crush but not further out. The Cx32 message is nearly undetectable in the segment 2-4 cm distal to the crush at 12 d, a time likely to precede or coincide with the onset of regeneration in this segment. In addition, Cx32 protein levels seem to be detectable at low levels between 8 and 12 d just distal to the crush, with an increase in Cx32 protein levels between 4 and 8 d, which could coincide with events preceding reinnervation such as Schwann cell proliferation. As shown by Scherer et al. (1995), Cx32 protein levels subsequently increase to near normal levels between 28 and 60 d in the segment 2-4 cm distal to the crush. RNA levels normalize between 12 and 24 d more proximally (0-2 cm distal to the crush) and between 24 and 58 d in the distal 2-4 cm. Findings in Chandross et al. (1996) are similar; differences may be attributable to differences in experimental methodology. Thus, it seems that low levels of Cx32 expression may accompany nerve regeneration. A possible role for Cx32 in the regenerative process has been proposed by Dezawa et al. (1998). They identified “small-scale gap junction-like structures” between regenerating axons and adjacent Schwann cells in rat sciatic nerve by electron microscopy. Cx32 immunoreactivity was identified within these structures by immunoelectron microscopy, and biocytin dye transfer was demonstrated between axons and their adjacent Schwann cells. Tetzlaff (1982) identified gap junctions between proliferating Schwann cells within bands of Büngner between 2 and 8 d after crush injury. This finding provides additional support for a possible role for gap junction proteins in nerve regeneration. An alternative explanation to a direct role of Cx32 in regeneration is that absence or depletion of Cx32 before injury can lead to changes in Schwann cells that make them less competent to respond when required to participate in the regenerative process. Reduced regenerative competence is also seen in xenografts from patients with inherited neuropathies because of duplications or deletions of PMP22 (Sahenk et al., 1998). In either case, the findings reported here have important implications for the pathophysiology of CMTX and the role that abnormalities in the regenerative process may play in the clinical expression of this disease.
The specific residues that are altered in the two mutants examined here are highly conserved. With regard to the valine at position 181, all known connexins contain a valine, leucine, or isoleucine at this position. Valine, leucine, and isoleucine have a hydrophobic side chain substantially larger than alanine. The loss of a bulky side group in the second extracellular loop may affect its ability to appropriately associate with organellar or plasma membranes. Alternatively, substitution of an alanine at this position may create instability in the protein structure that leaves it more susceptible to degradation. The E102G mutant, located in the cytoplasmic loop connecting the second and third extracellular domains, is also highly conserved. The cytoplasmic loop is thought to play an important role in pH-mediated gating (Wang and Peracchia, 1996). Consistent with this hypothesis, the CMTX mutant del111-116 also shows an increased sensitivity to pH-mediated gating (Ressot et al., 1998). Thus, it is not surprising that a mutation in a titratable residue in the cytoplasmic loop would lead to altered dependence of gating on pH.
Overall, our results suggest that Cx32 has multiple roles in the Schwann cell. For example, Cx32 may have a role in the early regeneration of nerve fibers through regions of previous nerve injury. Because no compact myelin is present at this time, such a role would be independent of a reflexive pathway. Cx32 may also participate in a reflexive gap junction pathway, shortening the diffusion distance between adaxonal and perinuclear cytoplasm. However, the importance of the reflexive pathway and the role of Cx32 remain to be elucidated. The hypothesis of multiple roles for Cx32 can explain the divergent effects of different mutant forms of Cx32; two examples are shown here. Multiple roles for Cx32 may also contribute to the variability in clinical manifestations of the disorder between families with different mutations.
Footnotes
This work was supported by National Institutes of Health Grant 1K08 NS02149-01 (C.K.A.) and by Muscular Dystrophy Association Research grants (C.K.A. and Z.S.). We thank all patients and volunteers who participated in the xenograft studies. We also thank Angela Bukauskas and Geng Qui Liu for technical assistance.
Correspondence should be addressed to Dr. Charles K. Abrams, Department of Neuroscience, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10463. E-mail: mmfreidi@ix.netcom.com.
Copyright © 2003 Society for Neuroscience 0270-6474/03/2310548-10$15.00/0
References
- Abrams CK, Bennett MVL ( 2000) Hereditary human diseases caused by connexin mutations. In: Gap junctions—molecular basis of cell communication in health and disease (Peracchia C, ed) pp 423-459. New York: Academic.
- Abrams CK, Freidin MM, Dobrenis K, Bargiello TA, Verselis VK, Bennett MVL, Sahenk Z ( 2000) Pathogenesis of CMTX: analysis of a new connexin 32 mutation leading to an inability to regenerate large caliber axons. Ann Neurol 48: 439. [Google Scholar]
- Abrams CK, Freidin MM, Verselis VK, Bennett MVL, Bargiello TA ( 2001) Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res 900: 9-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altevogt BM, Kleopa KA, Postma FR, Scherer SS, Paul DL ( 2002) Connexin 29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J Neurosci 22: 6458-6470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzini P, Neuberg DH, Schachner M, Nelles E, Willecke K, Zielasek J, Toyka KV, Suter U, Martini R ( 1997) Structural abnormalities and deficient maintenance of peripheral nerve myelin in mice lacking the gap junction protein connexin 32. J Neurosci 17: 4545-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Bone LJ, Scherer SS ( 1998) Functional gap junctions in the schwann cell myelin sheath. J Cell Biol 142: 1095-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergoffen J, Trofatter J, Pericak-Vance MA, Haines JL, Chance PF, Fischbeck KH ( 1993) Linkage localization of X-linked Charcot-Marie-Tooth disease. Am J Hum Genet 52: 312-318. [PMC free article] [PubMed] [Google Scholar]
- Bukauskas FF, Bukauskiene A, Verselis VK ( 2002) Conductance and permeability of the residual state of connexin 43 gap junction channels. J Gen Physiol 119: 171-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandross KJ, Kessler JA, Cohen RI, Simburger E, Spray DC, Bieri P, Dermietzel R ( 1996) Altered connexin expression after peripheral nerve injury. Mol Cell Neurosci 7: 501-518. [DOI] [PubMed] [Google Scholar]
- Dermietzel R, Farooq M, Kessler JA, Althaus H, Hertzberg EL, Spray DC ( 1997) Oligodendrocytes express gap junction proteins connexin 32 and connexin 45. Glia 20: 101-114. [PubMed] [Google Scholar]
- Dezawa M, Mutoh T, Dezawa A, Adachi-Usami E ( 1998) Putative gap junctional communication between axon and regenerating Schwann cells during mammalian peripheral nerve regeneration. Neuroscience 85: 663-667. [DOI] [PubMed] [Google Scholar]
- Fraher JP ( 1972) A quantitative study of anterior root fibres during early myelination. J Anat 112: 99-124. [PMC free article] [PubMed] [Google Scholar]
- Fraher JP ( 1973) A quantitative study of anterior root fibres during early myelination. II. Longitudinal variation in sheath thickness and axon circumference. J Anat 115: 421-444. [PMC free article] [PubMed] [Google Scholar]
- Hahn AF, Chang Y, Webster HD ( 1987) Development of myelinated nerve fibers in the sixth cranial nerve of the rat: a quantitative electron microscope study. J Comp Neurol 260: 491-500. [DOI] [PubMed] [Google Scholar]
- Hahn AF, Ainsworth PJ, Bolton CF, Bilbao JM, Vallat JM ( 2001) Pathological findings in the x-linked form of Charcot-Marie-Tooth disease: a morphometric and ultrastructural analysis. Acta Neuropathol (Berl) 101: 129-139. [DOI] [PubMed] [Google Scholar]
- Harris AL, Spray DC, Bennett MVL ( 1981) Kinetic properties of a voltage-dependent junctional conductance. J Gen Physiol 77: 95-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionasescu V, Ionasescu R, Searby C ( 1996) Correlation between connexin 32 gene mutations and clinical phenotype in X-linked dominant Charcot-Marie-Tooth neuropathy. Am J Med Genet 63: 486-491. [DOI] [PubMed] [Google Scholar]
- Li X, Lynn BD, Olson C, Meier C, Davidson KG, Yasumura T, Rash JE, Nagy JI ( 2002) Connexin 29 expression, immunocytochemistry and freeze-fracture replica immunogold labelling (FRIL) in sciatic nerve. Eur J Neurosci 16: 795-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milks LC, Kumar NM, Houghten R, Unwin N, Gilula NB ( 1988) Topology of the 32-kd liver gap junction protein determined by site-directed antibody localizations. EMBO J 7: 2967-2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Ri Y, Bennett MVL, Trexler EB, Verselis VK, Bargiello TA ( 1997) Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19: 927-938. [DOI] [PubMed] [Google Scholar]
- Oh S, Rubin JB, Bennett MVL, Verselis VK, Bargiello TA ( 1999) Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J Gen Physiol 114: 339-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ressot C, Gomes D, Dautigny A, Pham-Dinh D, Bruzzone R ( 1998) Connexin 32 mutations associated with X-linked Charcot-Marie-Tooth disease show two distinct behaviors: loss of function and altered gating properties. J Neurosci 18: 4063-4075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin JB, Verselis VK, Bennett MVL, Bargiello TA ( 1992) Molecular analysis of voltage dependence of heterotypic gap junctions formed by connexins 26 and 32. Biophys J 62: 183-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahenk Z, Chen L ( 1998) Abnormalities in the axonal cytoskeleton induced by a connexin 32 mutation in nerve xenografts. J Neurosci Res 51: 174-184. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Chen L, Mendell JR ( 1998) Effects of PMP22 depilation and deletions on the axonal cytoskeleton. Ann Neurol 45: 16-24. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Serrano-Munuera C, Chen L, Kakabadze I, Najagara HN ( 2003) Evidence for impaired axonal regeneration in PMP22 duplication: studies in nerve xenografts. J Periph Nerv Syst 8: 116-127. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Deschenes SM, Xu YT, Grinspan JB, Fischbeck KH, Paul DL ( 1995) Connexin 32 is a myelin-related protein in the PNS and CNS. J Neurosci 15: 8281-8294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer SS, Xu YT, Nelles E, Fischbeck K, Willecke K, Bone LJ ( 1998) Connexin 32-null mice develop demyelinating peripheral neuropathy. Glia 24: 8-20. [DOI] [PubMed] [Google Scholar]
- Senderek J, Bergmann C, Quasthoff S, Ramaekers VT, Schroder JM ( 1998) X-linked dominant Charcot-Marie-Tooth disease: nerve biopsies allow morphological evaluation and detection of connexin 32 mutations (Arg15Trp, Arg22Gln). Acta Neuropathol (Berl) 95: 443-449. [DOI] [PubMed] [Google Scholar]
- Senderek J, Hermanns B, Bergmann C, Boroojerdi B, Bajbouj M, Hungs M, Ramaekers VT, Quasthoff S, Karch D, Schroder JM ( 1999) X-linked dominant Charcot-Marie-Tooth neuropathy: clinical, electrophysiological, and morphological phenotype in four families with different connexin 32 mutations(1). J Neurol Sci 167: 90-101. [DOI] [PubMed] [Google Scholar]
- Sohl G, Eiberger J, Jung YT, Kozak CA, Willecke K ( 2001) The mouse gap junction gene connexin 29 is highly expressed in sciatic nerve and regulated during brain development. Biol Chem 382: 973-978. [DOI] [PubMed] [Google Scholar]
- Spray DC, Harris AL, Bennett MVL ( 1981) Gap junctional conductance is a simple and sensitive function of intracellular pH. Science 211: 712-715. [DOI] [PubMed] [Google Scholar]
- Tabaraud F, Lagrange E, Sindou P, Vandenberghe A, Levy N, Vallat JM ( 1999) Demyelinating X-linked Charcot-Marie-Tooth disease: unusual electrophysiological findings. Muscle Nerve 22: 1442-1447. [DOI] [PubMed] [Google Scholar]
- Tetzlaff W ( 1982) Tight junction contact events and temporary gap junctions in the sciatic nerve fibers of the chicken during Wallerian degeneration and subsequent regeneration. J Neurocytol 11: 839-858. [DOI] [PubMed] [Google Scholar]
- Trexler E, Bukaushas FF, Bennett MVL, Bargiello TA, Verselis VK ( 1999) Rapid and direct effects of pH on connexins revealed by the connexin 46 hemichannel preparation. J Gen Physiol 113: 721-742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trexler EB, Bukauskas FF, Kronengold J, Bargiello TA, Verselis VK ( 2000) The first extracellular loop domain is a major determinant of charge selectivity in connexin 46 channels. Biophys J 79: 3036-3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verselis VK, Ginter CS, Bargiello TA ( 1994) Opposite voltage gating polarities of two closely related connexins. Nature 368: 348-351. [DOI] [PubMed] [Google Scholar]
- Wang X, Peracchia C ( 1996) Connexin 32/38 chimeras suggest a role for the second half of inner loop in gap junction gating by low pH. Am J Physiol 271: C1743-C1749. [DOI] [PubMed] [Google Scholar]
- Wilders R, Jongsma HJ ( 1992) Limitations of the dual voltage clamp method in assaying conductance and kinetics of gap junction channels. Biophys J 63: 942-953. [DOI] [PMC free article] [PubMed] [Google Scholar]