

Abstract
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. It is characterized by neuronal loss and degeneration of the upper motor neurons (UMNs) and lower motor neurons (LMNs), and is usually fatal due to respiratory failure within 3–5 years of onset. Although approximately 5–10 % of patients with ALS have an inherited form of the disease, the distinction between hereditary and apparently sporadic ALS (SALS) seems to be artificial. Thus, genetic factors play a role in all types of ALS, to a greater or lesser extent. During the decade of upheaval, the evolution of molecular genetics technology has rapidly advanced our genetic knowledge about the causes of ALS, and the relationship between the genetic subtypes and clinical phenotype. In this review, we will focus on the possible genotype-phenotype correlation in hereditary ALS. Uncovering the identity of the genetic factors in ALS will not only improve the accuracy of ALS diagnosis, but may also provide new approaches for preventing and treating the disease.
Electronic supplementary material
The online version of this article (doi:10.1186/s40035-015-0036-y) contains supplementary material, which is available to authorized users.
Keywords: Amyotrophic lateral sclerosis (ALS), Genotype, Phenotype, Sporadic ALS (SALS), Familial ALS (FALS)
Introduction
Amyotrophic lateral sclerosis (ALS) is the most common adult-onset motor neuron disease. It is characterized by progressive neuronal loss and degeneration of the upper motor neurons (UMNs) and lower motor neurons (LMNs). The demise of motor neurons causes the central nervous system (CNS) to lose the ability to control voluntary muscle movement, eventually resulting in death due to respiratory failure in the later stages of the disease.
The cause of ALS remains an enigma. However, approximately 5–10 % of patients with ALS have an inherited form of the disease. During the decade of upheaval, the evolution of molecular genetics technology has rapidly advanced our knowledge about the genetic causes of ALS. Familial ALS (FALS) has been attributed to mutations in at least 24 different genes. Some mutations in FALS-related genes have been identified in patients with sporadic ALS (SALS). Because the initial symptoms of ALS vary across patients, a diagnosis of ALS can be established by excluding various diseases mimicking ALS. Smooth and reliable diagnosis is the first step in the good clinical management of patients with ALS. Therefore, genetic testing might be a helpful tool for diagnosing FALS as well as SALS with mutations in FALS-related genes.
It is important, but difficult, to predict which genes are most likely to be implicated in some patients with ALS. A diagnostic algorithm could improve the accuracy of a genetic explanation. Therefore, we review the possible genotype-phenotype relationship in ALS cases with mutations in the FALS-related genes. Uncovering the identity of the genetic factors in ALS will not only improve the accuracy of ALS diagnosis, but may also provide new approaches for preventing and treating the disorder.
Classification of hereditary ALS
Hereditary ALS can be transmitted as a dominant, recessive, or X-linked trait, but the most common type is an adult-onset disorder with autosomal dominant transmission. Autosomal recessive inheritance is rarer and frequently seen in patients with juvenile onset ALS, primary lateral sclerosis (PLS), or spastic paraplegia-like symptoms. X-linked dominantly inherited ALS is a rarely-observed condition, seen in families where male patients tend to show more severe phenotypes. We demonstrate the characteristic phenotypes in each type of FALS, and summarize them in Table 1.
Table 1.
The genotype and phenotype associated with familial ALS-related genes
| Type | Gene | Mode of inheritance | Country | Age at onset (range) | Mean age at onset (years) | Initial symptoms | UMN | Cognitive impairment | Other features |
|---|---|---|---|---|---|---|---|---|---|
| ALS1 | SOD1 | AD, AR, de novo | Japan, Italy, Spain, Korea, UK, USA, Turkey, Sweden, Iran, Porland, Bulgaria, China, France, Germany, Denmark, Pakistan, Canada, and so on | 6–94 | 48 | LL > UL > bulbar | Positive (LMN dominant) | Very rare | Progressive muscular atrophy, progressive bulbar palsy, facial onset sensory motor neuronopathy (FOSMN) syndrome, vocal cord paralysis, cerebellar ataxia, sensory disturbance (vibration), autonomic dysfunction (incontinence, neurogenic bladder), lower back pain |
| ALS2 | Alsin | AR | Tunisia, Saudi Arabia, Kuwait, Italy, Algeria, Hungary, Germany, The Netherlands, Pakistan, Bangladesh, Turkey, Japan, Portugal, France, Cyprus, China | 1–11 | 2 | LL, UL | Positive | None | Juvenile ALS, juvenile primary lateral sclerosis, infantile-onset ascending hereditary spastic paraplegia, generalized dystonia, cerebellar ataxia |
| ALS3 | unknown | AD | |||||||
| ALS4 | SETX | AD | USA, Austria, Belgium, Italy, Afghanistan, China | 1–73 | 19 | LL > UL | Positive | None | Cerebellar ataxia, oculomotor apraxia (type 2), motor neuropathy, thin cervical spinal cord |
| ALS5 | SPG11 | AR | Italy, Turkey, Japan, Canada, Brazil | 7–23 | 16 | Bulbar, LL, UL | Positive | Rare (mental retardation) | Juvenile ALS, hereditary spastic paraparesis, autonomic dysfunction (incontinence) |
| ALS6 | FUS | AD, AR, de novo | Belgium, Italy, Korea, UK, Japan, Turkey, Canada, France, USA, Germany | 13–80 | 45 | UL, bulbar > LL | Positive (LMN dominant) | Rare (mental retardation) | Progressive muscular atrophy, Parkinsonism, essential tremor, schizofrenia, learning disabilities |
| ALS7 | unknown | AD | |||||||
| ALS8 | VAPB | AD | Brazil, UK, France (Japan), The Netherlands | 18–73 | 44 | UL, LL | Negative | None | Progressive muscular atrophy, progressive bulbar palsy, motor neuropathy, postural tremor, autonomic dysfunction (chronic intestinal constipation, sexual dysfunction) |
| ALS9 | ANG | AD | The Neitherland, Ireland, Scotland, UK, USA, Sweden, Italy, France, Germany, China, | 21–86 | 55 | UL, LL, bulbar | Positive | FTD | Parkinsonism, progressive bulbar palsy |
| ALS10 | TDP-43 | AD, AR | Italy, France, UK, China, Germany, Turkey, USA, Belgium, Japan, Porland, Afghaistan, Canada | 20–77 | 54 | UL, LL, bulbar | Positive | FTD (rare) | Parkinsonism, chorea, progressive supranuclear palsy |
| ALS11 | FIG4 | AD | USA | 29–77 | 55 | Bulbar > UL, LL | Positive | None | Hereditary spastic paraparesis, primary lateral sclerosis, personality change |
| ALS12 | OPTN | AD, AR | Japan, Italy, Turkey, The Netherlands, Denmark | 24–83 | 51 | Bulbar, UL, LL | Positive | FTD, AGD | Primary open angle glaucoma, parkinsonism, finger deformity, personality change, depression |
| ALS13 | ATXN2 | AD | USA, Belgium, the Netherlands, Canada, France, China, Germany, Switzerland, Italy, Turkey, Cuba | 21–87 | 60 | UL, LL | Positive | None | Cerebellar ataxia, corticobasal syndrome, Parkinsonism |
| ALS14 | VCP | AD | Italy, USA, The Netherlands, Japan | 36–68 | 48 | LL > UL > bulbar | Positive | FTD | Paget’s Disease, inclusion body myopathy |
| ALS15 | UBQLN2 | SD | USA, Australia, Canada, Italy, Turkey, Belgium, Germany, Bulgaria | M: 14-72, F: 16-77 | 44 | UL, LL, bulbar | Positive | FTD | Primary lateral sclerosis, progressive bulbar palsy, relentlessly progressive choreoathetoid movements, spastic paralysis |
| ALS16 | SIGMAR1 | AD | Saudi Arabia | 1-68 | 1 | LL > UL | Positive | FTD (rare) | Juvenile ALS |
| ALS17 | CHMP2B | AD | Denmark, the Netherlands | 26-73 | 69 | Bulbar, UL, LL, respiratory | Positive (LMN dominant) | FTD | Progressive muscular atrophy, parkinsonism |
| ALS18 | PFN1 | AD | Sephardic Jewish, Italy, USA, China, Belgium | 33-63 | 53 | Limb | N/A | N/A | |
| ALS19 | ERBB4 | AD | Japan, Canada | 45-70 | 61 | UL, bulbar, respiration | Positive | None | |
| ALS20 | HNRNPA1 | AD | N/A | N/A | N/A | N/A | N/A | FTD | Paget’s Disease, inclusion body myopathy |
| ALS21 | MATR3 | AD | USA,UK, Italy, Taiwan | 36-64 | 52 | LL > UL, bulbar | Positive | FTD | Distal myopathy (inclusion body myopathy) |
| ALS-FTD1 | C9ORF72 | AD | Finland, Sardinia, Ireland, UK, Italy, USA,Canada, Germany, the Netherlands, Turkey, Israel, Australia, Japan | 27–80 | 57 | UL, LL, bulbar | Positive | FTD | Parkinsonism, cerebellar ataxia, psychosis, |
| ALS-FTD2 | CHCHD10 | AD | France, USA, Germany, Spain, Italy, Finland | 35-73 | 56 | Bulbar, UL, LL | Positive (LMN dominant) | FTD | Cerebellar ataxia, mitochondrial myopathy, deafness, neurogenic bladder, facial paresis, Parkinsonism |
| TBK1 | AD, de novo | Sweden, Denmark, Germany, France, Portugal | 35-80 | 60 | Bulbar, UL, LL, respiratory | Positive | FTD (50 %) |
AD, autosomal dominant; AR, autosomal recessive; UL, upper limb; LL, lower limb; LMN, lower motor neuron; FTD, frontotemporal dementia
ALS1: Cu/Zn superoxide dismutase 1, soluble (SOD1)
In 1991, Siddique et al. [1] showed the linkage of FALS to chromosome 21q by positional cloning and demonstrated genetic locus heterogeneity in FALS. Rosen et al. [2] then reported a genetic linkage between FALS and a gene encoding cytosolic Cu/Zn superoxide dismutase (SOD1)—a homodimeric metalloenzyme that catalyzes the reaction of toxic superoxide anion O2− to O2 and H2O2. Since SOD1 missense mutations were established as the first causative genes for ALS, the number of known mutations has increased to more than 185 so far (Additional file 1: Table S1). Most cases were inherited in an autosomal dominant manner, but the D90A mutation transmitted the disease in both an autosomal dominant and autosomal recessive manner. Globally, the most frequent SOD1 gene mutation is D90A. However, in the USA, the most frequent mutation was A4V, and in the UK and Japan, the most common mutations were I113T, and H46R, respectively. However, to our knowledge, no SOD1 mutation was reported from Ireland. Regarding clinical features of ALS with the SOD1 mutation, lower limb-onset and predominant LMN involvement are relatively common (Table 1). The D90A-homozygous mutation is associated with slowly progressive paresis in the legs that gradually spreads up to the arms, thoracic and bulbar musculature, with atypical non-motor features such as ataxia, neuralgic, aching pain, heat sensations, and bladder disturbance. Interestingly, it has been reported that patients with SOD1-related FALS greatly differed with respect to the age of onset of weakness, while the duration of the disease appears to be characteristic for each type of mutation. Some SOD1 mutants (D90A-homozygous, E100K, E100G, A89V, L84F, L84V, D76V, H46R, G37R, and G10V) tend to show a uniform phenotype, while other mutants (A4V, C6G, G41S, N86S, D90A-heterozygous, I112M, I113T, L144F, and V148I) have greatly variable phenotypes. The A4V, H43R, L84V, G85R, N86S, and G93A mutations have been associated with rapid disease progression and survival times shorter than 3 years, whereas the cases with G93C, D90A, or H46R mutations exhibit longer life expectancies, up to more than 10 years after disease onset [3–5]. These findings suggest that each type of SOD1 mutation may be associated with a different degree of toxicity. We examined two unrelated FALS families with H46R mutations (Fig. 1). The patients showed a uniform phenotype: the initial symptom was unilateral weakness of the flexor muscles in the distal lower limbs (Fig. 1) [6]. This might be attributed to mitochondrial respiratory chain dysfunction due to mutant SOD1 expression in the muscles as previously reported [7].
Fig. 1.
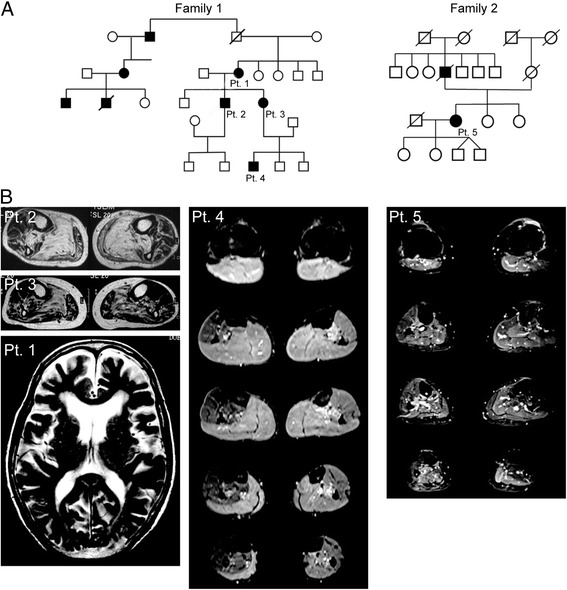
Two FALS families with the SOD1 H46R mutations. a Family trees of Family 1 and 2. All of the patients showed the same uniform feature that initial symptoms were restricted to the flexor muscle group in the unilateral distal leg. b Short-T1 inversion recovery MR images revealed high intensity lesions in the gastrocnemial and soleus muscles of the patient 2, 3, 4, and 5. Brain MRI of patient 1 showed frontal lobe-dominant atrophy
ALS2: Alsin
To date, more than 50 patients with mutations in the Alsin gene have been reported with early onset of the disease (~1 year). These patients generally belong to Middle Eastern, European, and Mediterranean countries, Japan, and China (Additional file 1: Table S1). All patients with ALS2 had homozygous or compound heterozygous mutations in the Alsin gene. Mutations in the Alsin gene cause three distinct disorders: infantile ascending hereditary spastic paraplegia (IAHSP), juvenile primary lateral sclerosis (JPLS), and autosomal recessive juvenile amyotrophic lateral sclerosis (JALS) (Table 1) [8, 9]. A recent study reported patients with ALS2 with nonsense and frameshift mutations in the Alsin gene who presented with generalized dystonia and cerebellar signs [10]. Although the phenotype-genotype correlation remains undetermined so far, most of the mutations predict truncated proteins, which could be unstable in structure and lose their function.
ALS4: Senataxin (SETX)
Senataxin (SETX) was initially identified as a causative gene for severe early-onset ataxia with oculomotor apraxia (AOA2), which is the second most common recessive ataxia after Freidreich's ataxia [11]. Later, heterozygous mutations were found in patients with the autosomal dominant form of juvenile-onset ALS [12]. ALS4 is characterized by slowly progressive distal muscle weakness and atrophy with pyramidal signs, sparing of bulbar and respiratory muscles, and frontal dysfunction (Table 1) [13]. So far, the T3I, L389S, T1118I, C1554G, K2018E, K2029E, R2136H, and I2547T mutations in the SETX gene have been identified in both patients with FALS and those with SALS with widely differing symptoms (Additional file 1: Table S1). In a recent report, a patient with late onset ALS4, bulbar involvement, and predominantly proximal distribution of amyotrophy presented with choreic movements and elevated alpha-fetoprotein levels [14]. In contrast, one study demonstrated that previously published ALS4-related missense mutations are most likely to be non-pathogenic and just polymorphisms [15]. Therefore, we should carefully interpret the significance of SETX missense mutations in the absence of functional assays.
ALS5: Spastic paraplegia 11, autosomal recessive (SPG11)
Mutations in the Spatacsin (SPG11) gene represent the most common form of autosomal recessive hereditary spastic paraplegia with thin corpus callosum (HSP-TCC) [16]. Recently, SPG11 mutations have been identified in patients with the autosomal recessive form of juvenile ALS, indicating a wide clinical spectrum for SPG11 mutations [17]. The SPG11 mutations can be associated with an intrafamilial phenotypic heterogeneity, including atypical ALS and classic HSP-TCC [18]. To our knowledge, at least 28 patients with ALS5 have been described with juvenile onset of the disease, ranging from 7 to 23 years (Table 1 and Additional file 1: Table S1). All patients with ALS5 were associated with slow progression of symptoms with apparent UMN involvement (Table 1). It has been reported that the absence of thin corpus callosum, white matter alterations, cognitive deficits or mental problems clearly differentiates ALS5 from HSP-TCC [17]. At this point, it is unclear why the SPG11 mutations lead to clinical phenotypes resembling ALS or HSP-TCC.
ALS6: Fused in sarcoma/translocated in liposarcoma (FUS/TLS)
Two independent studies have reported that mutations in the fused in sarcoma/translocated in liposarcoma (FUS/TLS) gene were responsible for ~3 % of FALS and <1 % of SALS cases [19, 20]. FUS/TLS mutations, as well as TAR DNA-binding protein (TDP-43) mutations, have been increasingly reported from Asian countries [21, 22]. Some FUS/TLS gene mutations have been observed in patients with the juvenile form of ALS beginning at younger than 25 years [23–25, 22]. Case studies with the R521C mutation in the FUS/TLS gene emphasized the phenotypes of weakness of the neck and proximal muscles, which may be a clinical hallmark of ALS [26]. Most of the reported cases with the FUS/TLS mutation had no cognitive change. However, some of the patients with juvenile ALS with truncating FUS/TLS mutations have had mental retardation [27, 22].
Most ALS-related FUS/TLS mutations are located at the highly conserved regions of exon 15 that include the non-canonical nuclear localization signal (PY-NLS). Recent studies have shown that the mutations that nullify the PY-NLS lead to redistribution of FUS/TLS to the cytoplasm, where it is recruited into stress granules [28–30]. Notably, the degree of cytosolic mislocalization has been shown to be inversely correlated with the age of disease onset [29]. It has been reported that the truncating mutation R495X was associated with an aggressive disease course, whereas the K510R mutation showed a mild phenotype with disease duration ranging from 6 to 8 years [31].
ALS8: Vesicle-associated membrane protein-associated protein B (VAPB)
A mutation in the vesicle-associated membrane protein-associated protein B (VAPB) gene was initially reported in Brazilian families with motor neuron disease with a wide range of phenotypes: late-onset spinal muscular atrophy, atypical ALS, or typical ALS [32]. In addition, several patients showed autonomic abnormalities, including chronic intestinal constipation, and sexual dysfunction [33]. So far, the T46I, P56S, and V234I mutations in the VAPB gene have been described in patients from Brazil, Japan, the United Kingdom, and the Netherlands (Additional file 1: Table S1). Further investigation will be required to understand the phenotype-genotype correlation.
ALS9: Angiogenin (ANG)
A cohort study in Ireland has identified several mutations in the angiogenin (ANG) gene in patients with ALS of Irish and Scottish background, both in familial and sporadic cases [34]. Subsequent clinical studies confirmed the association of these mutations with ALS, and identified new mutations in people with backgrounds from Brazil, China, France, Germany, Italy, Netherlands, Sweden, and the USA (Supplementary Table 1). Frontotemporal dementia (FTD) was also reported in a large FALS pedigree with the K17I ANG mutation [35]. Moreover, a relationship between mutations in the ANG gene and Parkinson’s disease has been revealed [36].
ALS10: TAR DNA-binding protein (TDP-43)
Several groups have identified mutations in a highly conserved region of TDP-43 in SALS and FALS cases [37–40]. Most mutations are located in exon 6, which encodes the conservative glycine-rich domain. The phenotype and genotype analysis study in patients with ALS having TDP-43 gene mutations revealed that they had earlier onset (53.4 years; range 28-78), predominantly upper limb onset (60.7 %), and longer disease duration (63.0 months; range 32.0-77.2), compared with those having SALS [41]. In Caucasians, 51.3 % of the patients had the upper limb onset, whereas 58.8 % of Asian patients had bulbar onset [41].
ALS11: FIG4 homolog, SAC1 lipid phosphatase domain containing (S. cerevisiae) (FIG4)
Mutations in the FIG4 gene are responsible for the recessive form of Charcot-Marie-Tooth disease (CMT4J), with early onset and involvement of both sensory and motor neurons [42]. Subsequently, the same group identified ALS as a rare manifestation of the gene [43]. The phenotype observed in patients with FIG4 mutations is still controversial. Some patients carried a diagnosis of definite or probable ALS, and other patients were diagnosed with PLS, associated with predominant UMN involvement. Personality changes were also reported in patients with ALS11.
ALS12: Optineurin (OPTN)
Maruyama et al. [44] identified mutations in the optineurin (OPTN) gene in 3.8 % of Japanese with FALS and 0.29 % of Japanese with SALS. Mutations in the OPTN gene were also detected in some patients with both FALS and SALS in cohorts of Italian, Danish, French, Turkish, and German patients (Additional file 1: Table S1). As mentioned later, the role of OPTN in the pathogenesis of ALS has been further examined in a recent publication on the TANK-binding kinase (TBK1) gene [45, 46]. The clinical phenotypes of OPTN-related ALS showed relatively slow progression and long duration before respiratory dysfunction, but the onset age of the eight individuals with mutations of OPTN ranged from 30 to 60 years [44]. Brain atrophy with personality change or depression was also observed in patients with ALS12.
ALS13: ataxin 2 (ATXN2)
Long polyglutamine tracts, including more than 34 CAG repeats in the ataxin 2 (ATXN2) gene, have been identified as a cause of spinocerebellar ataxia type 2 (SCA2) [47]. Recent studies revealed that intermediate-length polyglutamine repeats (between 24 and 33) within the ATXN2 gene can be a risk factor for patients with ALS in different ethnic groups [48–50]. However, whether the clinical features of patients with ALS can be affected by ATXN2 intermediate-length repeats is still controversial [49–51].
ALS14: Valosin-containing protein (VCP)
Using exome sequencing, Johnson et al. [52] identified a R191Q mutation in the valosin-containing protein (VCP) gene in an Italian family with autosomal dominantly inherited ALS. Screening of the VCP gene in a cohort of ALS cases identified several mutations including a pathologically proven case of ALS. Mutations in the VCP gene have previously been identified in families with inclusion body myopathy, Paget disease, and frontotemporal dementia (IBMPFD) [53]. The phenotype of patients with VCP mutations shows intrafamilial variations from IBMPFD to FALS [54]. This suggests that motor neuron disease is part of the clinical spectrum of multiple proteinopathy of VCP-associated disease.
ALS15: ubiquilin 2 (UBQLN2)
Recent studies have revealed that ubiquilin 2 (UBQLN2), which regulates the degradation of ubiquitinated proteins, plays a pathogenic role in the X-linked form of ALS with or without FTD [55]. In an original case, the disease was transmitted in a dominant fashion with reduced penetrance without male-to-male transmission of the disease. Age at onset was significantly different between male and female patients, with male patients having earlier age of onset [55]. Mutations in UBQLN2 are not a frequent cause of ALS in the Dutch, French-Canadian, French, Irish, Taiwanese, and Korean population (Additional file 1: Table S1).
ALS16: σNon-opioid receptor (SIGMAR1)
Homozygosity mapping followed by direct sequencing has revealed a mutation in the σNon-opioid receptor (SIGMAR1) gene in patients in a consanguineous family with the autosomal recessive form of juvenile ALS in Saudi Arabia [56]. Furthermore, variants in the 3′-untranslated region (UTR) of the SIGMAR1 gene were reported in patients with frontotemporal lobar degeneration (FTLD) or motor neuron disease with FTLD [57]. However, the same family with the 3′-UTR mutation of the SIGMAR1 gene also had an expansion of a noncoding GGGGCC hexanucleotide repeat in the chromosome 9 open reading frame 72 (C9ORF72) [58]. This indicates that coding and noncoding variants located in the 3'-UTR of the SIGMAR1 gene are not the cause of FTLD-MND.
ALS17: chromatin modifying protein 2B (CHMP2B)
Mutations in the charged multivesicular body protein 2B (CHMP2B) gene have been initially identified in patients with FTD [59]. Although the phenotype is predominantly FTD, ALS has been reported as a rare manifestation of the gene [60, 61]. Neuropathology of the patient with the mutation showed LMN predominant disease with ubiquitylated inclusions in motor neurons [60]. Thus, classical ALS and PMA without corticospinal findings are phenotypes associated with mutations in the CHMP2B gene.
ALS18: profilin 1 (PFN1)
Exome sequencing followed by direct sequencing has shown mutations in the profilin 1 (PFN1) gene, which is a central regulator of actin dynamics in some FALS cases [62]. However, cohort analyses of patients with FALS and those with SALS from France and Quebec, Italy, Germany, the Nordic countries, and the United States suggested that the PFN1 mutation is a rare cause of ALS (Additional file 1: Table S1). In the original report, all patients with ALS18 showed limb symptoms at a relatively younger onset [62].
ALS19: v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4 (ERBB4)
A whole-genome sequencing and parametric linkage analysis identified the mutation in the v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4 (ERBB4) gene in patients of a Japanese family with late-onset, autosomal-dominant ALS [63]. An extensive mutational analysis revealed the same mutation in a Canadian individual with familial ALS and a de novo mutation in a Japanese case [63]. As of this moment, the genotype-phenotype correlation has not been determined.
ALS20: heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1)
Exome sequencing revealed mutations in the heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) gene in patients presenting with ALS and/or multisystem proteinopathy (MSP). These mutations are associated with a rare and complex phenotype associating FTLD, Paget disease of bone, and inclusion body myopathy [64]. Because the clinical information is not fully available, the phenotype of patients with mutant hnRNPA1 is still unclear.
ALS21: matrin-3 (MATR3)
A recent study using exome sequencing revealed mutations in the matrin-3 (MATR3) gene in FALS and FTD cases [65]. Initially, the S85C mutation in the MATR3 gene was reported as the cause of autosomal dominant distal myopathy with vocal cord paralysis (VCPDM) in large multi-generational families [66]. The phenotype observed in some patients carrying MATR3 mutations is still controversial. However, the clinical phenotype might be markedly similar to that observed in patients with mutations in VCP, hnRNPA1, and HNRNPA2B1 as MSP. We examined 2 sisters with VCPDM and S85C mutations in the MATR3 gene (Fig. 2) [67]. Both patients showed no UMN symptoms clinically; however, they showed chronic denervation and renervation on electromyography and muscle biopsy, split hand syndrome, and decremental motor responses to repetitive nerve stimulation, suggesting the involvement of LMNs [67].
Fig. 2.
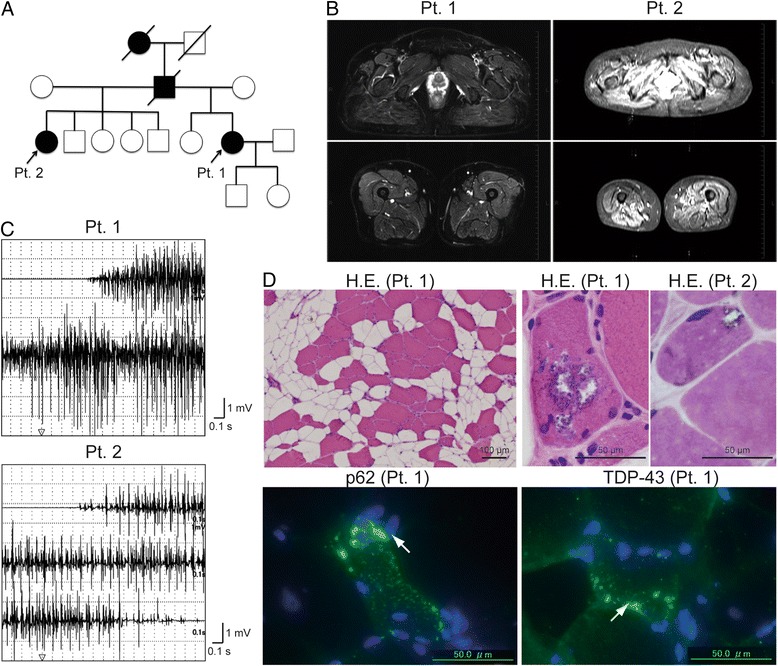
A family with the MATR3 S85C mutation. a Family tree of cases with the MATR3 S85C mutation. The detailed clinical information was previously described [67]. b Short-T1 inversion recovery MR images revealed fatty and degenerative changes in the gluteus, quadriceps, and hamstring muscles of patient 1 and the paraspinal and gluteus muscles of patient 2. c Needle electromyography demonstrated chronic denervation in the vastus lateralis muscles of patients 1 and 2. d Muscle biopsy from patients 1 and 2 showed severe fatty and myopathic changes with rimmed vacuoles. Immunohistochemical analysis demonstrated p62- or TDP-43-positive sarcoplasmic granular staining in degenerating myofibers of patient 1. The observation of chronic denervation and renervation on electromyography and muscle biopsy, split hand syndrome, and decremental motor responses to repetitive nerve stimulation (data not shown) suggest the involvement of lower motor neurons in patients 1 and 2
ALS-FTD1: chromosome 9 open reading frame 72 (C9ORF72)
Two independent studies have discovered an expansion of a noncoding GGGGCC hexanucleotide repeat in the C9ORF72 gene that is associated with disease in a large FTD/ALS kindred linked to chromosome 9p [68, 69]. Analysis of extended clinical series found the C9ORF72 repeat expansion to be the most common genetic abnormality in both familial FTD (11.7 %) and familial ALS (23.5 %) [68]. Another study reported that the C9ORF72 intronic expansion was present in 11 % of the cohort, 43 % of FALS cases, and 7 % of SALS cases [69]. Therefore, C9ORF72 has been thought to be the most common cause of ALS in Caucasians, but rarer in other populations [70]. It is still controversial whether the patients with C9ORF72 expansion have shorter disease duration and relatively rapidly progression. C9ORF72 expansion can also cause parkinsonism and dementia. There is no association between the repeat length of the normal alleles, of the repeat in C9ORF72, and disease phenotype or age at onset in C9ORF72 mutation carriers or non carriers [71].
ALS-FTD2: Coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10)
Whole-exome sequencing identified a missense S59L mutation in the coiled-coil-helix-coiled-coil-helix domain containing 10 (CHCHD10) gene in a large family with a late-onset phenotype including motor neuron disease, cognitive decline resembling FTD, cerebellar ataxia and myopathy [72]. Multiple mitochondrial DNA deletions have been found in the skeletal muscles of patients with ALS-FTD2, suggesting mitochondrial DNA instability. Thus, the phenotype can vary according to affected organs.
TANK-binding kinase 1 (TBK1)
Recently, several studies using exome sequencing of moderate numbers of patients with ALS identified the TBK1 gene as an ALS gene, which is known to bind to and phosphorylate ALS-related proteins such as OPTN and p62 (SQSTM1/sequestosome) [45, 46]. Patients having ALS with the mutations frequently (~50 %) showed cognitive impairment [46]. Another study performing whole-genome sequencing in patients with FTLD-TDP found variants in the TBK1 gene, indicating a key role for the OPTN/TBK1 pathway in ALS and FTD [73].
Importance of genetic testing for ALS diagnosis
We describe the possible correlation between the genotype and phenotype, and aim to provide a clue to the diagnosis of ALS. ALS cases can be divided into 3 groups: juvenile onset less than 10 years or less 25 years, and adult onset type. Cases with juvenile onset were categorized into 2 groups because we could differentiate the genes that cause juvenile ALS alone from the genes that cause both juvenile and adult-onset ALS. ALS cases with juvenile onset less than 10 years include cases with mutations in the SPG11, Alsin, SETX, and SIGMAR1 genes (Fig. 3). When the symptoms are UMN-dominant, SPG and Alsin can be causative genes for ALS. In contrast, SETX might be responsible in cases with LMN-dominant symptoms such as PMA type. In ALS cases with onset from 10 to 24 years, SPG11, FUS, VAPB, SOD1, SETX, ATXN2, ANG, and UBQLN2 should be considered as a cause of ALS (Fig. 3). SPG or UBQLN2 might be a causative gene in UMN-dominant cases whereas FUS, VAPB, SOD1, and SETX should be examined in LMN-dominant cases.
Fig. 3.
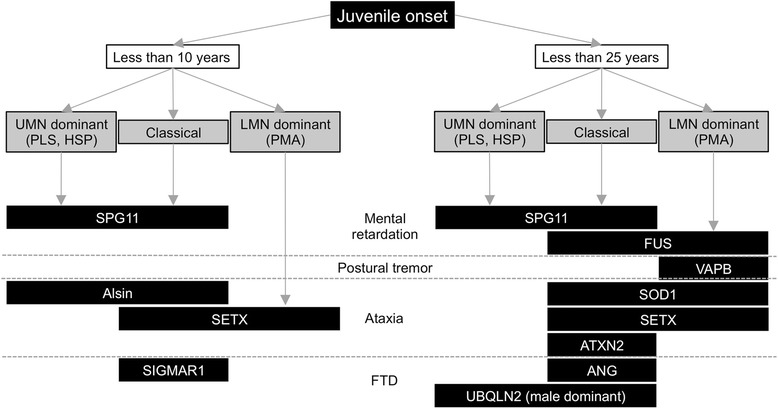
Flowchart for diagnosis of hereditary juvenile-onset ALS
In adult-onset ALS cases, many candidate genes should be excluded (Fig. 4). In patients who suffer from mental retardation, SPG11 may be responsible in UMN-dominant cases and FUS may be responsible in LMN-dominant cases. Coexistence with cerebellar ataxia may suggest the involvement of mutations of SOD1, ATXN2, Alsin, and SETX. Complications of motor neuropathy might occur in cases with mutations in the FIG4, SETX, VAPB, and SOD1 (homozygous D90A) genes. FTD can be present in cases with mutations in the UBQLN2, SIGMAR1, TDP-43, ANG, OPTN, CHMP2B, and C9ORF72 genes. Moreover, parkinsonism can be involved in cases with TDP-43, ANG, OPTN, and CHMP2B mutations. In some cases, muscle biopsy provides useful information for ALS diagnosis. Mitochondrial myopathy is reported in cases with CHCHD10 and SOD1 mutations (Fig. 1). FTD in combination with inclusion body myopathy and Paget’s disease of bone in the patients or families strongly suggests mutations in the VCP, hnRNPA1, or MATR3 genes (Fig. 2).
Fig. 4.

Flowchart for diagnosis of hereditary adult-onset ALS
Although these algorithms might provide some indications of what type of genetic abnormality might be present in a large enough family with somewhat consistent features, most families have a small number of affected individuals with wide variability. Thus, these algorithms may be ineffective. However, ethnic background plays a huge role in determining which genes are most likely. The proportion of ALS caused by a particular gene in a particular population can be a stronger predictor: C9ORF72 intronic expansion is very common in Caucasians, but rare in other populations. Therefore, the algorithms should be optimized based on ethnic backgrounds, and establishment of panels that examine all genes simultaneously would be ideal.
Another limitation is that phenotypes were described in limited number of patients in some genes except SOD1, FUS/TLS, TDP-43, and C9ORF72. This makes it is difficult to draw conclusive genotype-phenotype correlations. Moreover, some of the reported mutations were not necessarily pathogenic, just polymorphisms [15]. Thus, it is difficult to know which reported variants indeed cause the disease; it even more difficult in cases having oligogenic inheritance because their phenotype is derived from the combination of two genes.
Notably, half of the families with FALS do not have a mutation in the identified genes and therefore the genetic test is not necessarily informative for all cases of FALS. At this point, the determination that an individual has FALS is based on a family history rather than a genetic test. If one's family history is unknown or a parent passed away at a young age, testing is appropriate. Those patients with SALS without a family history can also be offered genetic testing. However, it is extremely important that this be done in the context of genetic counseling or after discussion with a neurologist about the implication of finding a mutation, as a mutation would mean that the ALS is hereditary. Although prenatal genetic testing technology exists, the patients and family members should discuss the procedure with their neurologist and genetic counselor for further information on this complex and personal matter [74].
Conclusions
There is no specific test or procedure to establish the diagnosis of ALS. A diagnosis of ALS can be established by ruling out other diseases that mimic ALS thorough comprehensive diagnostic examinations. Earlier diagnosis allows prompt initiation with a specific drug, such as riluzole, and accurate palliative care planning. The recent advances in the genetics of ALS have not only contributed to our understanding of the pathogenesis of ALS, but have also provided a tool for diagnostic procedures in some cases of ALS.
Despite all the progress achieved, the large majority of ALS genes remain unknown. The number of genes known to be involved in ALS is expected to continuously increase with the evolution of molecular genetics technology. Further discovery of the genetic factors in ALS will contribute considerably to the diagnosis, care, prevention, and treatment of ALS.
Acknowledgement
This research was supported by Grants-in-Aid for Science Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 24591269).
Abbreviations
- ALS
Amyotrophic lateral sclerosis
- SALS
Sporadic ALS
- FALS
Familial ALS
- CNS
Central nervous system
- SOD1
Cu/Zn superoxide dismutase
- IAHSP
Infantile ascending hereditary spastic paraplegia
- JPLS
Juvenile primary lateral sclerosis
- JALS
Juvenile amyotrophic lateral sclerosis
- SETX
Senataxin
- SPG11
Spatacsin
- HSP-TCC
Hereditary spastic paraplegia with thin corpus callosum
- FUS/TLS
Fused in sarcoma/translocated in liposarcoma
- PY-NLS
Non-canonical nuclear localization signal
- VAPB
Vesicle-associated membrane protein-associated protein B
- ANG
Angiogenin
- FTD
Frontotemporal dementia
- TDP-43
TAR DNA-binding protein
- CMT4J
Charcot-Marie-Tooth disease
- OPTN
Optineurin
- ATXN2
Ataxin 2
- VCP
Valosin-containing protein
- IBMPFD
Inclusion body myopathy, Paget disease, and frontotemporal dementia
- UBQLN2
Ubiquilin 2
- SIGMAR1
σNon-opioid receptor
- FTLD
Frontotemporal lobar degeneration
- CHMP2B
Charged multivesicular body protein 2B
- PFN1
Profilin 1
- ERBB4
v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 4
- hnRNPA1
Heterogeneous nuclear ribonucleoprotein A1
- MSP
Multisystem proteinopathy
- MATR3
Matrin-3
- VCPDM
Distal myopathy with vocal cord paralysis
- C9ORF72
Chromosome 9 open reading frame 72
- CHCHD10
Coiled-coil-helix-coiled-coil-helix domain containing 10
- TBK1
TANK-binding kinase 1
- LMN
Lower motor neuron
- UMN
Upper motor neuron
Additional file
The genotypes and phenotypes in previously-reported ALS cases with familial ALS-related mutations. UMN, upper motor neuron; LMN, lower motor neuron; UL, upper limb; LL, lower limb; FTD, frontotemporal dementia; N/A, not applicable; IAHSP, infantile ascending hereditary spastic paraplegia; JPLS, juvenile primary lateral sclerosis; JALS, juvenile amyotrophic lateral sclerosis; PMA, progressive muscular atrophy. (XLSX 96 kb)
Footnotes
Competing interests
Both authors declare that they have no competing interests.
Authors’ contributions
SY and YA drafted and revised the manuscript. Both authors read and approved the final manuscript.
References
- 1.Siddique T, Figlewicz DA, Pericak-Vance MA, Haines JL, Rouleau G, Jeffers AJ, et al. Linkage of a gene causing familial amyotrophic lateral sclerosis to chromosome 21 and evidence of genetic-locus heterogeneity. N Engl J Med. 1991;324:1381–4. doi: 10.1056/NEJM199105163242001. [DOI] [PubMed] [Google Scholar]
- 2.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 3.Cudkowicz ME, McKenna-Yasek D, Sapp PE, Chin W, Geller B, Hayden DL, et al. Epidemiology of mutations in superoxide dismutase in amyotrophic lateral sclerosis. Ann Neurol. 1997;41:210–21. doi: 10.1002/ana.410410212. [DOI] [PubMed] [Google Scholar]
- 4.Regal L, Vanopdenbosch L, Tilkin P, Van den Bosch L, Thijs V, Sciot R et al. The G93C mutation in superoxide dismutase 1: clinicopathologic phenotype and prognosis. Arch Neurol. 2006;63: 262-7. doi:10.1001/archneur.63.2.262 [DOI] [PubMed]
- 5.Aoki M, Warita H, Itoyama Y. Amyotrophic lateral sclerosis with the SOD1 mutations. Rinsho Shinkeigaku. 2008;48:966–9. doi: 10.5692/clinicalneurol.48.966. [DOI] [PubMed] [Google Scholar]
- 6.Yamashita S, Kimura E, Yamamoto F, Migita A, Kanda E, Mita S et al. Flexor-dominant myopathic phenotype in patients with His46Arg substitution in the Cu/Zn superoxide dismutase gene. J Neurol Sci. 2009;281: 6-10. doi:10.1016/j.jns.2009.03.010 [DOI] [PubMed]
- 7.Crugnola V, Lamperti C, Lucchini V, Ronchi D, Peverelli L, Prelle A et al. Mitochondrial respiratory chain dysfunction in muscle from patients with amyotrophic lateral sclerosis. Arch Neurol. 2010;67: 849-54. doi:10.1001/archneurol.2010.128 [DOI] [PubMed]
- 8.Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29:166–73. doi: 10.1038/ng1001-166. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y, Hentati A, Deng HX, Dabbagh O, Sasaki T, Hirano M, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–5. doi: 10.1038/ng1001-160. [DOI] [PubMed] [Google Scholar]
- 10.Siddiqi S, Foo JN, Vu A, Azim S, Silver DL, Mansoor A, et al. A novel splice-site mutation in ALS2 establishes the diagnosis of juvenile amyotrophic lateral sclerosis in a family with early onset anarthria and generalized dystonias. PLoS One. 2014;9 doi: 10.1371/journal.pone.0113258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moreira MC, Klur S, Watanabe M, Nemeth AH, Le Ber I, Moniz JC, et al. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat Genet. 2004;36:225–7. doi: 10.1038/ng1303. [DOI] [PubMed] [Google Scholar]
- 12.Chen YZ, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4) Am J Hum Genet. 2004;74:1128–35. doi: 10.1086/421054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Jonghe P, Auer-Grumbach M, Irobi J, Wagner K, Plecko B, Kennerson M, et al. Autosomal dominant juvenile amyotrophic lateral sclerosis and distal hereditary motor neuronopathy with pyramidal tract signs: synonyms for the same disorder? Brain. 2002;125:1320–5. doi: 10.1093/brain/awf127. [DOI] [PubMed] [Google Scholar]
- 14.Saracchi E, Castelli M, Bassi MT, Brighina E, Cereda D, Marzorati L, et al. A novel heterozygous SETX mutation in a patient presenting with chorea and motor neuron disease. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:138–40. doi: 10.3109/21678421.2013.865751. [DOI] [PubMed] [Google Scholar]
- 15.Arning L, Epplen JT, Rahikkala E, Hendrich C, Ludolph AC, Sperfeld AD. The SETX missense variation spectrum as evaluated in patients with ALS4-like motor neuron diseases. Neurogenetics. 2013;14:53–61. doi: 10.1007/s10048-012-0347-4. [DOI] [PubMed] [Google Scholar]
- 16.Stevanin G, Santorelli FM, Azzedine H, Coutinho P, Chomilier J, Denora PS et al. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat Genet. 2007;39: 366-72. doi:10.1038/ng1980 [DOI] [PubMed]
- 17.Orlacchio A, Babalini C, Borreca A, Patrono C, Massa R, Basaran S et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. 2010;133: 591-8. doi:10.1093/brain/awp325 [DOI] [PMC free article] [PubMed]
- 18.Daoud H, Zhou S, Noreau A, Sabbagh M, Belzil V, Dionne-Laporte A et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging. 2012;33: 839 e5-9. doi:10.1016/j.neurobiolaging.2011.11.012 [DOI] [PubMed]
- 19.Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323: 1205-8. doi:10.1126/science.1166066 [DOI] [PubMed]
- 20.Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323: 1208-11. doi:10.1126/science.1165942 [DOI] [PMC free article] [PubMed]
- 21.Suzuki N, Aoki M, Warita H, Kato M, Mizuno H, Shimakura N, et al. FALS with FUS mutation in Japan, with early onset, rapid progress and basophilic inclusion. J Hum Genet. 2010;55:252–4. doi: 10.1038/jhg.2010.16. [DOI] [PubMed] [Google Scholar]
- 22.Yamashita S, Mori A, Sakaguchi H, Suga T, Ishihara D, Ueda A, et al. Sporadic juvenile amyotrophic lateral sclerosis caused by mutant FUS/TLS: possible association of mental retardation with this mutation. J Neurol. 2012;259:1039–44. doi: 10.1007/s00415-011-6292-6. [DOI] [PubMed] [Google Scholar]
- 23.Munoz DG. FUS mutations in sporadic juvenile ALS: another step toward understanding ALS pathogenesis. Neurology. 2010;75: 584-5. doi:10.1212/WNL.0b013e3181ed9ee4 [DOI] [PubMed]
- 24.Baumer D, Hilton D, Paine SM, Turner MR, Lowe J, Talbot K et al. Juvenile ALS with basophilic inclusions is a FUS proteinopathy with FUS mutations. Neurology. 2010;75: 611-8. doi:10.1212/WNL.0b013e3181ed9cde [DOI] [PMC free article] [PubMed]
- 25.Conte A, Lattante S, Zollino M, Marangi G, Luigetti M, Del Grande A et al. P525L FUS mutation is consistently associated with a severe form of juvenile amyotrophic lateral sclerosis. Neuromuscul Disord. 2012;22: 73-5. doi:10.1016/j.nmd.2011.08.003 [DOI] [PubMed]
- 26.Blair IP, Williams KL, Warraich ST, Durnall JC, Thoeng AD, Manavis J, et al. FUS mutations in amyotrophic lateral sclerosis: clinical, pathological, neurophysiological and genetic analysis. J Neurol Neurosurg Psychiatry. 2010;81:639–45. doi: 10.1136/jnnp.2009.194399. [DOI] [PubMed] [Google Scholar]
- 27.Yan J, Deng HX, Siddique N, Fecto F, Chen W, Yang Y et al. Frameshift and novel mutations in FUS in familial amyotrophic lateral sclerosis and ALS/dementia. Neurology. 2010;75: 807-14. doi:10.1212/WNL.0b013e3181f07e0c [DOI] [PMC free article] [PubMed]
- 28.Dormann D, Madl T, Valori CF, Bentmann E, Tahirovic S, Abou-Ajram C et al. Arginine methylation next to the PY-NLS modulates Transportin binding and nuclear import of FUS. EMBO J. 2012;31: 4258-75. doi:10.1038/emboj.2012.261 [DOI] [PMC free article] [PubMed]
- 29.Dormann D, Rodde R, Edbauer D, Bentmann E, Fischer I, Hruscha A et al. ALS-associated fused in sarcoma (FUS) mutations disrupt Transportin-mediated nuclear import. EMBO J. 2010;29: 2841-57. doi:10.1038/emboj.2010.143 [DOI] [PMC free article] [PubMed]
- 30.Kino Y, Washizu C, Aquilanti E, Okuno M, Kurosawa M, Yamada M et al. Intracellular localization and splicing regulation of FUS/TLS are variably affected by amyotrophic lateral sclerosis-linked mutations. Nucleic Acids Res. 2011;39: 2781-98. doi:10.1093/nar/gkq1162 [DOI] [PMC free article] [PubMed]
- 31.Waibel S, Neumann M, Rabe M, Meyer T, Ludolph AC. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology. 2010;75: 815-7. doi:10.1212/WNL.0b013e3181f07e26 [DOI] [PubMed]
- 32.Nishimura AL, Mitne-Neto M, Silva HC, Richieri-Costa A, Middleton S, Cascio D, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–31. doi: 10.1086/425287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marques VD, Barreira AA, Davis MB, Abou-Sleiman PM, Silva WA, Jr, Zago MA, et al. Expanding the phenotypes of the Pro56Ser VAPB mutation: proximal SMA with dysautonomia. Muscle Nerve. 2006;34:731–9. doi: 10.1002/mus.20657. [DOI] [PubMed] [Google Scholar]
- 34.Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C et al. ANG mutations segregate with familial and 'sporadic' amyotrophic lateral sclerosis. Nat Genet. 2006;38: 411-3. doi:10.1038/ng1742 [DOI] [PubMed]
- 35.van Es MA, Diekstra FP, Veldink JH, Baas F, Bourque PR, Schelhaas HJ et al. A case of ALS-FTD in a large FALS pedigree with a K17I ANG mutation. Neurology. 2009;72: 287-8. doi:10.1212/01.wnl.0000339487.84908.00 [DOI] [PubMed]
- 36.van Es MA, Schelhaas HJ, van Vught PW, Ticozzi N, Andersen PM, Groen EJ, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol. 2011;70:964–73. doi: 10.1002/ana.22611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319: 1668-72. doi:10.1126/science.1154584 [DOI] [PMC free article] [PubMed]
- 38.Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40: 572-4. doi:10.1038/ng.132 [DOI] [PubMed]
- 39.Van Deerlin VM, Leverenz JB, Bekris LM, Bird TD, Yuan W, Elman LB et al. TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 2008;7: 409-16. doi:10.1016/S1474-4422(08)70071-1 [DOI] [PMC free article] [PubMed]
- 40.Yokoseki A, Shiga A, Tan CF, Tagawa A, Kaneko H, Koyama A, et al. TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann Neurol. 2008;63:538–42. doi: 10.1002/ana.21392. [DOI] [PubMed] [Google Scholar]
- 41.Corcia P, Valdmanis P, Millecamps S, Lionnet C, Blasco H, Mouzat K et al. Phenotype and genotype analysis in amyotrophic lateral sclerosis with TARDBP gene mutations. Neurology. 2012;78: 1519-26. doi:10.1212/WNL.0b013e3182553c88 [DOI] [PubMed]
- 42.Chow CY, Zhang Y, Dowling JJ, Jin N, Adamska M, Shiga K et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature. 2007;448: 68-72. doi:10.1038/nature05876 [DOI] [PMC free article] [PubMed]
- 43.Chow CY, Landers JE, Bergren SK, Sapp PC, Grant AE, Jones JM et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84: 85-8. doi:10.1016/j.ajhg.2008.12.010 [DOI] [PMC free article] [PubMed]
- 44.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465: 223-6. doi:10.1038/nature08971 [DOI] [PubMed]
- 45.Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–41. doi: 10.1126/science.aaa3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Muller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18:631–6. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 47.Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat Genet. 1996;14:269–76. doi: 10.1038/ng1196-269. [DOI] [PubMed] [Google Scholar]
- 48.Elden AC, Kim HJ, Hart MP, Chen-Plotkin AS, Johnson BS, Fang X et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466: 1069-75. doi:10.1038/nature09320 [DOI] [PMC free article] [PubMed]
- 49.Chen Y, Huang R, Yang Y, Chen K, Song W, Pan P et al. Ataxin-2 intermediate-length polyglutamine: a possible risk factor for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32: 1925 e1-5. doi:10.1016/j.neurobiolaging.2011.05.015 [DOI] [PubMed]
- 50.Liu X, Lu M, Tang L, Zhang N, Chui D, Fan D. ATXN2 CAG repeat expansions increase the risk for Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2013;34: 2236 e5-8. doi:10.1016/j.neurobiolaging.2013.04.009 [DOI] [PubMed]
- 51.Van Damme P, Veldink JH, van Blitterswijk M, Corveleyn A, van Vught PW, Thijs V et al. Expanded ATXN2 CAG repeat size in ALS identifies genetic overlap between ALS and SCA2. Neurology. 2011;76: 2066-72. doi:10.1212/WNL.0b013e31821f445b [DOI] [PubMed]
- 52.Johnson JO, Mandrioli J, Benatar M, Abramzon Y, Van Deerlin VM, Trojanowski JQ et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68: 857-64. doi:10.1016/j.neuron.2010.11.036 [DOI] [PMC free article] [PubMed]
- 53.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 54.Gonzalez-Perez P, Cirulli ET, Drory VE, Dabby R, Nisipeanu P, Carasso RL et al. Novel mutation in VCP gene causes atypical amyotrophic lateral sclerosis. Neurology. 2012;79: 2201-8. doi:10.1212/WNL.0b013e318275963b [DOI] [PMC free article] [PubMed]
- 55.Deng HX, Chen W, Hong ST, Boycott KM, Gorrie GH, Siddique N et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477: 211-5. doi:10.1038/nature10353 [DOI] [PMC free article] [PubMed]
- 56.Al-Saif A, Al-Mohanna F, Bohlega S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol. 2011;70:913–9. doi: 10.1002/ana.22534. [DOI] [PubMed] [Google Scholar]
- 57.Fecto F, Siddique T. SIGMAR1 mutations, genetic heterogeneity at the chromosome 9p locus, and the expanding etiological diversity of amyotrophic lateral sclerosis. Ann Neurol. 2011;70:867–70. doi: 10.1002/ana.22648. [DOI] [PubMed] [Google Scholar]
- 58.Belzil VV, Daoud H, Camu W, Strong MJ, Dion PA, Rouleau GA. Genetic analysis of SIGMAR1 as a cause of familial ALS with dementia. Eur J Hum Genet. 2013;21: 237-9. doi:10.1038/ejhg.2012.135 [DOI] [PMC free article] [PubMed]
- 59.Skibinski G, Parkinson NJ, Brown JM, Chakrabarti L, Lloyd SL, Hummerich H, et al. Mutations in the endosomal ESCRTIII-complex subunit CHMP2B in frontotemporal dementia. Nat Genet. 2005;37:806–8. doi: 10.1038/ng1609. [DOI] [PubMed] [Google Scholar]
- 60.Parkinson N, Ince PG, Smith MO, Highley R, Skibinski G, Andersen PM et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67: 1074-7. doi:10.1212/01.wnl.0000231510.89311.8b [DOI] [PubMed]
- 61.Cox LE, Ferraiuolo L, Goodall EF, Heath PR, Higginbottom A, Mortiboys H, et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS) PLoS One. 2010;5 doi: 10.1371/journal.pone.0009872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu CH, Fallini C, Ticozzi N, Keagle PJ, Sapp PC, Piotrowska K et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature. 2012;488: 499-503. doi:10.1038/nature11280 [DOI] [PMC free article] [PubMed]
- 63.Takahashi Y, Fukuda Y, Yoshimura J, Toyoda A, Kurppa K, Moritoyo H, et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet. 2013;93:900–5. doi: 10.1016/j.ajhg.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim HJ, Kim NC, Wang YD, Scarborough EA, Moore J, Diaz Z, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–73. doi: 10.1038/nature11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Johnson JO, Pioro EP, Boehringer A, Chia R, Feit H, Renton AE, et al. Mutations in the Matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–6. doi: 10.1038/nn.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Senderek J, Garvey SM, Krieger M, Guergueltcheva V, Urtizberea A, Roos A, et al. Autosomal-dominant distal myopathy associated with a recurrent missense mutation in the gene encoding the nuclear matrix protein, matrin 3. Am J Hum Genet. 2009;84:511–8. doi: 10.1016/j.ajhg.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yamashita S, Mori A, Nishida Y, Kurisaki R, Tawara N, Nishikami T, et al. Clinicopathological features of the first Asian family having vocal cord and pharyngeal weakness with distal myopathy due to a MATR3 mutation. Neuropathol Appl Neurobiol. 2015;41:391–8. doi: 10.1111/nan.12179. [DOI] [PubMed] [Google Scholar]
- 68.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72: 245-56. doi:10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed]
- 69.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72: 257-68. doi:10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed]
- 70.Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11: 323-30. doi:10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed]
- 71.Rutherford NJ, Heckman MG, Dejesus-Hernandez M, Baker MC, Soto-Ortolaza AI, Rayaprolu S et al. Length of normal alleles of C9ORF72 GGGGCC repeat do not influence disease phenotype. Neurobiol Aging. 2012;33: 2950 e5-7. doi:10.1016/j.neurobiolaging.2012.07.005 [DOI] [PMC free article] [PubMed]
- 72.Bannwarth S, Ait-El-Mkadem S, Chaussenot A, Genin EC, Lacas-Gervais S, Fragaki K, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137:2329–45. doi: 10.1093/brain/awu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015 doi: 10.1007/s00401-015-1436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fanos JH, Gelinas DF, Miller RG. "You have shown me my end": attitudes toward presymptomatic testing for familial amyotrophic lateral sclerosis. Am J Med Genet A. 2004;129A:248–53. doi: 10.1002/ajmg.a.30178. [DOI] [PubMed] [Google Scholar]