


Abstract
IS200 is found throughout Enterobacteriaceae and transposes at a notoriously low frequency. In addition to the transposase protein (TnpA), IS200 encodes an uncharacterized Hfq-binding sRNA that is encoded opposite to the tnpA 5'UTR. In the current work we asked if this sRNA represses tnpA expression. We show here that the IS200 sRNA (named art200 for antisense regulator of transposase IS200) basepairs with tnpA to inhibit translation initiation. Unexpectedly, art200-tnpA pairing is limited to 40 bp, despite 90 nt of perfect complementarity. Additionally, we show that Hfq and RNA secondary structure in the tnpA 5'UTR each repress tnpA expression in an art200-independent manner. Finally, we show that disrupting translational control of tnpA expression leads to increased IS200 transposition in E. coli. The current work provides new mechanistic insight into why IS200 transposition is so strongly suppressed. The possibility of art200 acting in trans to regulate a yet-unidentified target is discussed as well as potential applications of the IS200 system for designing novel riboregulators.
INTRODUCTION
Small non-coding RNAs (sRNAs) play an important role in regulating many physiological processes in bacteria, including but not limited to metabolism, stress response and virulence (reviewed in (1–4)). Most sRNAs regulate gene expression through complementary basepairing with target mRNAs, which usually affects translation and often transcript stability. The best studied class of sRNAs are expressed in trans relative to their target mRNA, and accordingly have only partial sequence complementarity. The chaperone protein Hfq is important for the function of most trans-sRNAs, protecting the sRNA from degradation and facilitating pairing between sRNAs and their target(s). Conversely, cis-encoded sRNAs (also called antisense RNA, asRNA) are expressed from the same loci as mRNAs on the opposite strand of DNA. This results in perfect and usually extended complementarity between asRNAs and their target mRNAs. Hfq is typically thought to be dispensable for asRNA regulation (5,6), although there are a few systems where this is not the case (7,8).
The first sRNAs discovered in bacteria were asRNAs involved in plasmid and transposon copy-number control (9,10). In the case of IS10, translation of the transposase mRNA (RNA-IN) is inhibited by the cis-encoded asRNA, RNA-OUT. Pairing between these RNAs initiates between the 5' end of RNA-IN and the terminal loop domain of RNA-OUT. Propagation of the paired species to ultimately include 35 intermolecular base-pairs blocks 30S ribosome binding to RNA-IN (11,12). Since RNA-OUT can act in trans on all copies of IS10 in a cell, the strength of antisense regulation increases with IS10 copy-number and accordingly plays an important role in limiting transposition (13). Identification of new functional sRNAs has been aided by the development of RNA-Seq coupled with Hfq immunoprecipitation (Hfq-IP)(14). By sequencing sRNAs that interact with Hfq, it is possible to separate putative functional sRNAs from spurious transcription products. One surprising observation from these Hfq-IP experiments is that Hfq interacts with a number of cis-encoded sRNAs. The first study to use Hfq-IP for identifying sRNAs in Salmonella enterica serovar Typhimurium (hereafter Salmonella) found that about 3% of Hfq-bound RNA mapped antisense to protein coding regions (15). More strikingly, asRNAs made up the second largest class (25%) of Hfq-binding sRNAs in Mycobacterium smegmatis although the significance of this is unclear as no Hfq orthologs have been identified in Mycobacterium species so far (16). Escherichia coli may express up to 300 functional asRNAs, although only 67 were detected in an Hfq-IP (17,18). Hfq may therefore play a previously unappreciated role in antisense regulation. Alternatively, the subset of asRNAs that interact with Hfq may be trans-sRNAs that just happen to be expressed antisense to protein coding genes.
We have previously shown that Hfq facilitates antisense pairing between the IS10 transposase mRNA (RNA-IN) and the cis-encoded sRNA, RNA-OUT (7). We were interested in determining if Hfq regulated other transposons by a similar mechanism and accordingly searched Hfq-IP data sets for evidence of Hfq-interacting RNAs that are antisense to transposase mRNAs. IS200 encodes an sRNA (STnc490) that is antisense to 90 nucleotides (nt) of the transposase 5' untranslated region (5'UTR) in Salmonella (15,19). The closely related IS1541 element from Yersinia pestis also expresses STnc490 (20). Promoters for asRNAs can arise stochastically, but the conserved expression of STnc490 suggests this is a functional asRNA (21).
IS200 elements are ubiquitous in Enterobacteriaceae and have been identified throughout Eubacteria and Archaea (22–26). IS200 was first identified as a polar insertion mutant in the hisD gene of Salmonella (hisD984; (27)). Repeated attempts to measure IS200 transposition under various laboratory conditions were unsuccessful, and environmental samples of Salmonella collected 30 years apart showed no evidence of transposition (22,28–29). However, IS200 does transpose during long-term stab culture and there is evidence that the closely related IS1541 element is active during mouse infection by Y. pestis (27,30). Taken together these observations have led to the conclusion that IS200 is a mostly dormant transposable element (22,31). A reasonable presumption would be that most IS200 elements are inactive remnants of the active transposon. However, sequence comparison of ‘genomic’ IS200 elements and rare transposition products revealed that the sequence of ‘active’ and ‘inactive’ elements is almost identical (23). It therefore seems likely that the ‘native’ state of IS200 elements is ‘off’ for transposition although specific conditions might lead to sporadic transposition.
Transposition of many bacterial transposons is limited by the expression of the transposase genes they encode (32–34). IS200, the smallest fully autonomous insertion sequence known, contains a single open reading frame (ORF) that encodes a transposase protein, TnpA (Figure 1A). Transcription of the tnpA gene is limited by an intrinsically weak promoter and a bi-directional rho-independent terminator in the ‘left end’ that protects against impinging transcription (23,35). The left end contains a second inverted repeat that comprises a portion of the 5'UTR of tnpA. This is most likely a cis-regulatory element that represses tnpA translation by sequestering the Shine-Dalgarno (SD) sequence (35). Antisense control of tnpA expression would therefore be an additional level of regulation for transposase protein.
Figure 1.
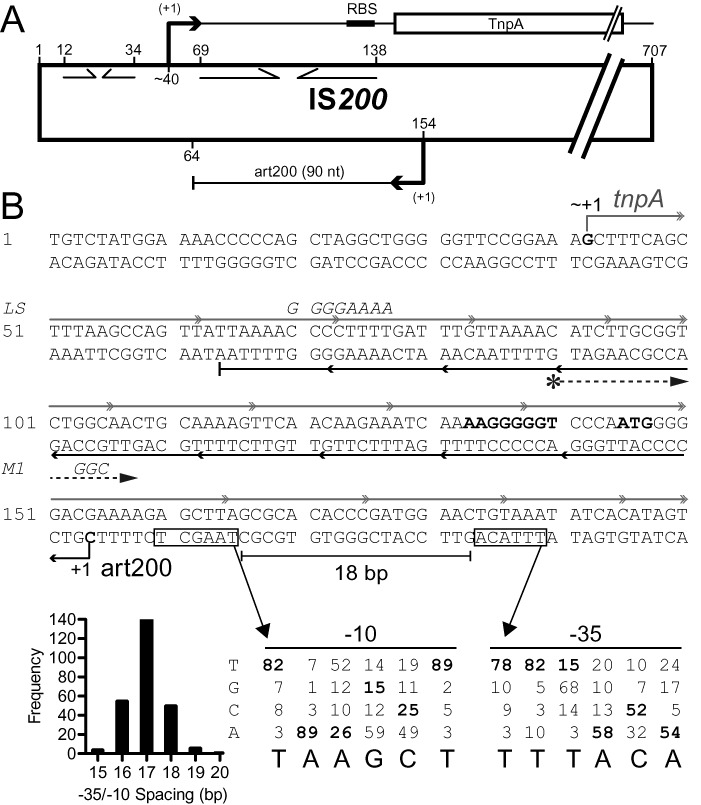
Schematic of IS200. (A) IS200 is 707 basepairs in length. It contains a single protein coding gene (transposase; tnpA), transcription of which originates at about nt 40 (23,35); tnpA promoter elements have not been defined. The ‘left end’ contains two internal inverted repeats (opposing arrows), one of which acts as a transcription terminator (nts 12–34) and the other (nts 69–138) was predicted to encode a stem-loop structure in the 5'UTR of the tnpA mRNA that sequesters the Shine-Dalgarno sequence (35). IS200 in Salmonella also expresses a 90 nt sRNA (art200, previously STnc490), which is perfectly complementary to the 5'UTR and the first three codons of tnpA. The transcription start site and 3’ end for art200 in Salmonella (derived from RNA-Seq experiments) are shown but promoter elements were not previously defined (19). (B) The DNA sequence of the first 200 nucleotides of IS200 is shown. The tnpA and art200 transcripts are shown in gray and black, respectively. Putative promoter elements for art200 are boxed and the Salmonella transcription start site (+1) is shown. The former were predicted using a position weight matrix (showing nucleotide identity of −10 and −35 promoter elements for E. coli) and the optimal spacing between −10 and −35 elements in E. coli (histogram) (data from (41)). The SD sequence and start codon for tnpA are shown in bold. Mutations introduced into tnpA/art200 in this work (LS and M1) are indicated in italics. A DNA primer used to map the 5' end of art200 in E. coli (Figure 2) is depicted with an asterisk followed by a dashed arrow.
In the current work we asked if IS200 transposase expression is down-regulated by the cis-encoded sRNA in E. coli. We show that transposase expression is strongly repressed by STnc490, which we renamed art200 (antisense regulator of transposase IS200). Hfq does not play a role in art200-tnpA pairing, although it does repress tnpA expression in the absence of art200. This repression appears to be the result of Hfq binding to the 5'UTR of tnpA immediately upstream of the SD. We also show that the tnpA SD sequence is sequestered in secondary structure and that this inhibits 30S ribosome subunit binding. Finally, we demonstrate that IS200 transposition increases in E. coli upon disruption of translational control mechanisms. Implications of these results are considered in the context of tight regulation of IS200 transposition, a possible role for art200 in the control of host gene expression and the potential application of the IS200 sRNA system in synthetic biology/metabolic engineering.
MATERIALS AND METHODS
Bacterial strains, plasmids and oligonucleotides
All Miller assays and related RNA analyses were performed in E. coli K-12 derivatives DBH323 (MC4100 ΔrecA774::kan) or DBH326 (DBH323 hfq-1::cat) (36). Salmonella enterica subsp. enterica serovar Typhimurium str. LT2 was used as a source of IS200. For mating out experiments, DBH33 was lysogenized with λDBH881 to create the donor strain DBH291 (DBH33 Mini IS200-kan) and DBH13 was used as the recipient strain (see Supporting Information for details of strain construction). DH5α was used for routine cloning and plasmid propagation. Strains and plasmids used in the main text are listed in Table 1; all other plasmids and oligonucleotides are listed in Supplementary Tables S1 and S2 respectively.
Table 1. Strains and plasmids.
| Name | Description | Notes |
|---|---|---|
| E. coli | ||
| DBH13 | HB101 [F- leu- pro-]; SmR | Recipient strain for mating out experiments |
| DBH33 | NK5830 [recA- arg -ΔlacproXIII nalR rifR/ F’ lacpro+] | Parent strain for mating out donor |
| DBH291 | DBH33 λDBH881 (Mini IS200-kan); KanR | Donor strain for mating out experiments |
| DBH323 | MC4100 ΔrecA774::kan; SmRKanR | Miller assays (hfq+) (36) |
| DBH326 | DBH323 hfq-1::cat; SmRKanRCmR | Miller assays (hfq−) (36) |
| Plasmids | ||
| pDH857 | pBAD24-SDBAD24-tnpAWT; ApR | TnpA expression for mating out, pBAD24 regulatory elements |
| pDH860 | pBAD24-SDBAD24-tnpAY125F; ApR | TnpAY125F (cat. dead) expression for mating out, pBAD24 regulatory elements |
| pDH861 | pGEM-T Easy derived, tnpA-lacZ TLF; ApR | tnpAWT-lacZ TLF |
| pDH862 | pDH861 with lower-stem mutations, tnpALS-lacZ; ApR | TLF with disrupted stem-loop structure |
| pDH880 | pDH861 with PA-6 mutation; ApR | tnpA-lacZ, no cis-art200 |
| pDH896 | pBAD24-tnpAWT; ApR | TnpA expression for mating out, WT IS200 5'UTR |
| pDH897 | pBAD24-tnpALS; ApR | TnpA expression for mating out, LS IS200 5'UTR |
| pDH898 | pDH900 with T7 PA1-tnpA45–298; TetR | High-copy titrator, no cis-art200 |
| pDH899 | pDH900 with PTet-tnpA45–298; TetR | Low-copy titrator, no cis-art200 |
| pDH900 | pACYC184 derivative; TetR CmS | Vector control |
| pDH902 | pDH900 with IS20045–298; TetR | trans-art200 |
| pDH912 | pDH902 with M1 mutations; TetR | trans-art200M1 |
| pDH914 | pDH899 with M1 mutations; TetR | Low-copy titratorM1’ |
| pDH916 | pDH861 with M1 mutations; ApR | tnpAM1’-lacZ TLF |
| pDH918 | pDH880 with M1 mutations; ApR | tnpAM1’-lacZ TLF, no cis-art200 |
The IS200-lacZ translational fusion (TLF; pDH861) and mutant derivatives consist of the first 323 nt of IS200 fused to codon 10 of the lacZ gene cloned into pGEM-T easy (Promega). The art200 titrator plasmids (pDH898 and pDH899; Figure 3A) consist of nt 45–298 of IS200 (no cis-art200) transcribed from either the T7 phage PA1 or PTet. The sgrS transcriptional terminator was inserted immediately downstream of IS200, and the entire construct was cloned into pACYC184. The plasmids expressing art200 in trans (pDH902 and pDH912) consist of nt 45–298 of IS200 cloned into pACYC184. Transposase expression in mating out experiments (pDH857, pDH860, pDH896 and pDH897) was from pBAD24 derivatives (37) where TnpA was expressed from native or exogenous regulatory elements. Further details of constructing these plasmids are provided in Supporting Material.
Figure 3.
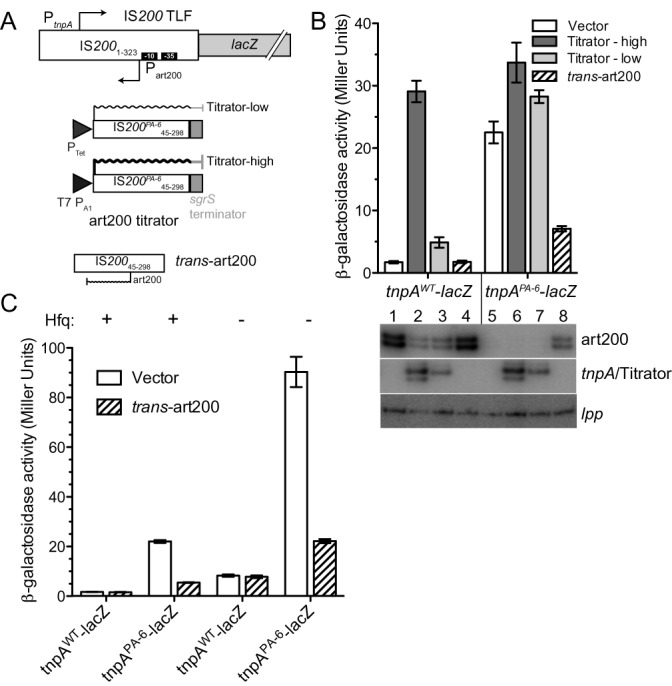
Impact of art200 and Hfq on tnpA expression. (A) An IS200-lacZ translational fusion (TLF) was constructed to measure tnpA expression in E. coli. art200 levels were manipulated by: (i) introducing the PA-6 mutations into the TLF (down-regulated); (ii) co-expressing an art200 titrator RNA (IS200 45–298) with the TLF (down-regulated) or (iii) co-expressing art200 in trans relative to the TLF (up-regulated). tnpA expression was entirely under the control of native (IS200) regulatory elements. Titrator RNAs were constitutively expressed from promoters PTet (moderate strength) or T7 PA1 (strong).
RNA footprinting and toeprinting
In vitro transcription templates were generated by polymerase chain reaction (PCR) using plasmids pDH861 (WT tnpA1–173 and art200), pDH862 (LS tnpA1–173 and art200), and pDH916 (M1 tnpA1–173 and art200) and primers oDH450 and oDH394 (tnpA1–173) or oDH500 and oDH501 (art200). RNAs were generated by in vitro transcription and 5' labeled with [γ32P]-ATP as previously described (38). Wild-type Hfq was purified by heat treatment and poly(A) affinity purification (39). RNase, Pb2+ and hydroxyl radical footprinting were performed essentially as previously described (7,38,40). For footprinting reactions studying art200-tnpA pairing, each RNA was denatured at 95°C for 2 min and snap-cooled on ice for 3 min. Ambion 10X RNA Structure Buffer was added to a final concentration of 1X (10 mM Tris-HCl (pH 7), 100 mM KCl, 10 mM MgCl2), and the RNAs were incubated separately at 37°C for 5 min to fold before mixing. For the control reactions in Figure 4B where the RNAs were folded together (FT; lanes 6, 11, 16 and 21) 5'32P-labeled tnpA and art200 were mixed before the denaturing step.
Figure 4.
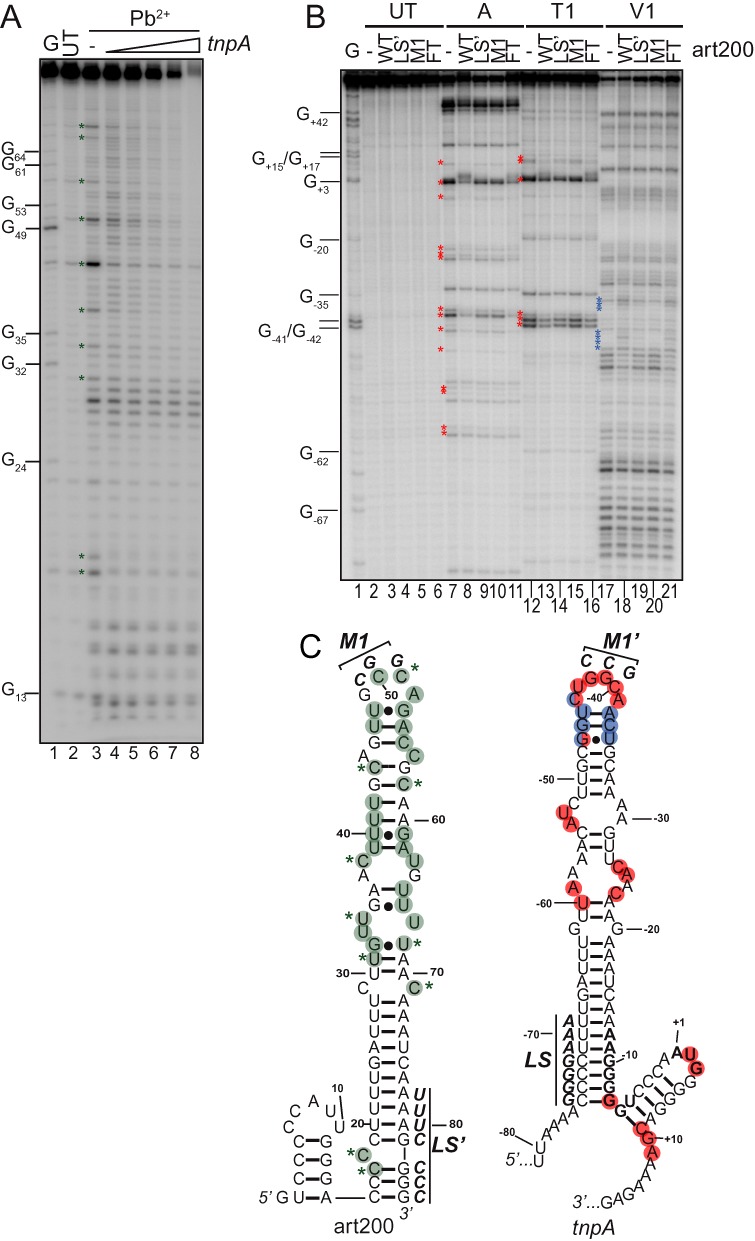
Pb2+ and RNase footprinting of art200, tnpA1–173 and an art200-tnpA1–173 complex. (A) 5'32P-labeled art200 (69 nM) was incubated in the absence or presence of increasing concentrations tnpA1–173 (69, 138, 276, 460 or 1380 nM) before limited treatment with Pb2+. Note that each RNA was denatured and allowed to fold before mixing. Positions that were most strongly protected from Pb2+ cleavage in a tnpA-concentration dependent manner are indicated with a green asterisk. UT is untreated art200 RNA and G is an RNase T1 sequencing lane. (B) 5'32P-labeled tnpA1–173 (40 nM) was incubated with wild-type and mutant variants (LS’ and M1 – see Figure 1B) of art200 (600 nM) or folding buffer (-) before treatment with RNase A, T1 or V1. tnpA1–173 and art200 RNA were denatured and allowed to fold independently before mixing, except for a control reaction with WT art200 where RNAs were mixed, denatured and allowed to fold together (FT; lanes 6, 11, 16 and 21). Nucleotide number is relative to the AUG start codon in tnpA. Nucleotides that were most strongly protected from single-strand specific RNase (A/T1) in the presence of art200 are indicated with a red asterisk and positions that showed an increased sensitivity to RNase V1 (double-strand specific) are indicated with a blue asterisk. (C) Structural constraints derived from footprinting were input into mFold to produce structures for art200 and tnpA1–173 (see also Supplementary Figures S2 and S3). Residues in art200 that showed either weak (green circle) or strong (green circle plus asterisk) decreases in Pb2+ reactivity upon mixing with tnpA1–173 are highlighted. Residues in tnpA1–173 that showed strong (red circles) decreases in RNase A or T1, or strong increases (blue circles) in V1 reactivity upon art200 addition are highlighted. Two residues (−44 and −47) showed increased V1 sensitivity and decreased A1/T1 sensitivity (blue-red circles). Nucleotide changes present in M1 and M1’ versions of art200 and tnpA1–173, respectively, are shown in bold.
Toeprinting was performed essentially as previously described (40). Briefly, unlabeled tnpA (2 pmol) was annealed to 5'32P-labeled oDH394 before incubation with purified Hfq (0–8 pmol hexamer) or art200 (30 pmol) at 37°C. This was followed by addition of the 30S ribosomal subunit (3.6 pmol) and then initiator fmet-tRNA (10 pmol; Sigma-Aldrich) for a final volume of 10 μl. Reverse transcription reactions were carried out at 37°C for 10 min with 200U of SuperScript II (Invitrogen).
Following ethanol precipitation, samples were resuspended in denaturing load dye (95% [v/v] formamide, 0.5X TBE, 3% [w/v] xylene cyanol) and resolved on a 10% polyacrylamide gel containing 7M urea. Dried gels were exposed to a phosphorimager storage screen, imaged with a Storm imager and quantitated with ImageQuant (GE Healthcare).
β-galactosidase assays
Cells were grown in LB supplemented (where necessary for plasmid selection) with ampicillin (100 μg/ml) and tetracycline (10 μg/ml). Saturated overnight cultures were used to seed subcultures (1:40 dilution), which were grown to mid-log phase (OD600 = 0.4–0.6). The Miller assay was performed as previously described (39).
RNA extraction, primer extension
Cells were grown in LB supplemented (where appropriate for plasmid maintenance) with ampicillin (100 μg/ml) and/or tetracycline (10 μg/ml) to OD600 = 0.6 at which time total RNA was extracted with acid phenol as previously described (40). 10 μg of total RNA was subject to primer extension analysis with 5'32P-labeled oDH427 (Figures 2 and 3B) or oDH537 (Figure 6) (art200), oDH428 (tnpA), and oDH390 (lpp) and SuperScript III (Invitrogen) according to the manufacturer's instructions. Following ethanol precipitation, samples were resuspended in denaturing load dye and resolved on a 10% polyacrylamide gel containing 7M urea. Dried gels were exposed to a phosphorimager storage screen, imaged with a Storm imager and quantitated with ImageQuant (GE Healthcare).
Figure 2.
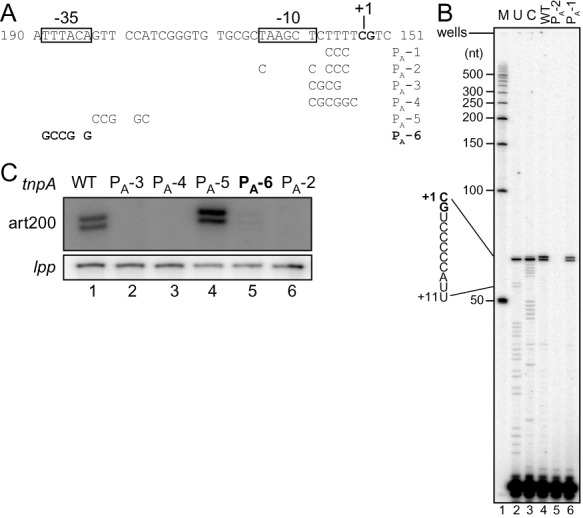
Primer extension analysis of art200 in E. coli. (A) The DNA sequence of the 5' end of the art200 gene plus mutations introduced to deduce the −35/−10 promoter elements are shown. (B, C) Primer extension reactions performed to detect art200 from WT and the indicated mutant forms of tnpA-lacZ are shown. Total RNA was isolated from exponential phase cells grown in rich media (LB) and the primer (5'-end labeled with 32P) shown in Figure 1B was used to make cDNA. The cells contained IS200 on a multicopy plasmid. cDNAs were analyzed on a 10% denaturing polyacrylamide (sequencing) gel. The image in (B) shows a full gel; M = markers, U and C are RNA sequencing reactions. The image in (C) shows only the portion of a gel that includes the art200 primer extension products. For the latter, primer extension was multiplexed to include lpp as a loading control. The mutant PA-6 (bold letters) was used in subsequent experiments in this work to knock-down art200 levels in E. coli.
Figure 6.
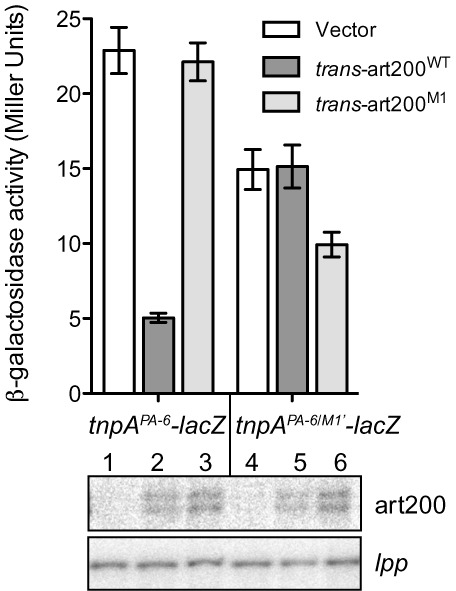
Impact of terminal loop mutations on translational repression in vivo. A plasmid encoding tnpAPA-6-lacZ or tnpAPA-6/M1’-lacZ was co-transformed into DBH323 with a compatible plasmid expressing art200 (WT or M1) in trans to tnpA-lacZ or an empty vector control. β-galactosidase activity was measured in cells grown to mid-exponential phase in LB media. Bars show the mean expression from two independent experiments and error bars indicate standard error on the mean (n = 6). Thebottom panel shows primer extension analysis using RNA extracted from cells grown in the Miller assay (top panel). lpp was used as a loading control.
Conjugal mating out assay
The conjugal mating out assay was performed as previously described (39,40); see Supplementary Figure S5 for schematic. Briefly, DBH291 was transformed with pDH857, pDH860, pDH896, or pDH897 and grown on LB agar plates containing ampicillin (100 μg/ml), kanamycin (25 μg/ml) and 0.05% arabinose (w/v). For the experiments presented in Figure 9C, DBH291 was transformed with pDH897 and pDH900 (vector) or pDH898 (titrator) and grown on LB agar plates containing ampicillin, tetracycline (10 μg/ml) and 0.05% arabinose (w/v). Individual colonies (‘donors’) were grown to saturation in LB containing (where appropriate for plasmid selection) ampicillin and tetracycline with 0.05% arabinose (w/v) and were subcultured 1:20 into LB containing 0.2% arabinose. Following mating with the recipient strain (DBH13), cells were plated on M9 glucose plates supplemented with thiamine, leucine and streptomycin (150 μg/ml) (‘exconjugants’) or streptomycin and kanamycin (‘hops’). Transposition frequency was determined by dividing ‘hops’ by ‘exconjugants’.
Figure 9.
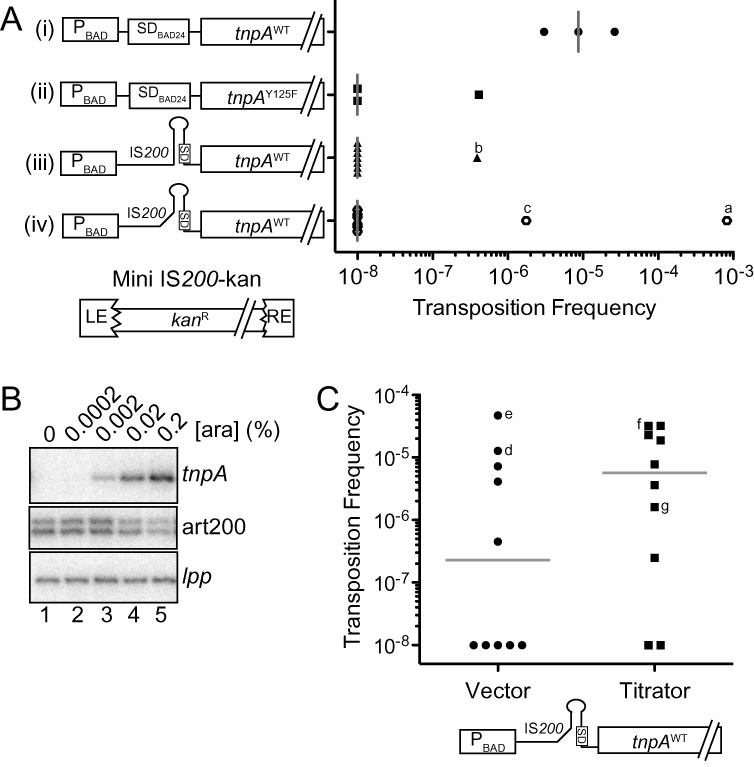
IS200 transposition assays. (A) IS200 transposition frequency was measured using the conjugal mating out assay. Briefly, E. coli (F+; DBH291) containing a single chromosomal copy of a marked IS200 element (mini IS200-kan) was transformed with a plasmid expressing TnpA under the control of various regulatory elements, including the PBAD promoter, the 5'UTR from pBAD24 (includes an optimized SD) and the IS200 5'UTR (constructs i–iv). These ‘donor’ cells were grown in the presence of arabinose (0.2%) to induce tnpA transcription, mixed with an F- recipient strain (DBH13) and then the mating mixes were plated on selective media for measuring mating efficiency (exconjugants) and transposition events (hops). Transposition frequency is the ratio of hop to exconjugant colonies. Transposition frequencies for individual donor clones are presented in scattergram form for each TnpA construct; gray bars show the median transposition frequency for one (constructs i and ii) or three (constructs iii and iv) independent experiments. Clones that did yield hops and were analyzed by Southern blot analysis are indicated (a–c). LE = left end (bp 1–163), RE = right end (bp 566–707) and kanR = kanamycin resistance gene. (B) Primer extension analysis of DBH291 donor cells transformed with construct (iii) and grown to mid-log phase in the presence of arabinose. Primer extension reactions were multiplexed to detect tnpA, art200 and lpp (loading control). (C) Mating out assay with donor strains containing construct (iv) and either the low expression art200 titrator plasmid or an empty vector control. Gray bars show the median transposition frequency for each donor strain from three independent experiments; d, e, f and g are hop colonies subjected to Southern blot analysis (Supplementary Figure S6). In (A) and (C) the transposition frequency for donor clones that did not produce hop colonies was set at 1 × 10−8.
RESULTS
Characterization of the IS200 antisense RNA gene in E. coli
Salmonella Typhimurium LT2 contains 6 copies of IS200 and expresses STnc490 (an RNA that is antisense to the transposase RNA) at high levels under standard laboratory growth conditions (15,19). However the gene encoding this transcript was not previously fully characterized. We show below that the IS200 asRNA is expressed in E. coli and characterized components of the gene encoding this transcript. The putative transcription start site for the IS200 asRNA (predicted based on RNA-Seq experiments in Salmonella (19)) is shown in Figure 1B. We scanned upstream of this position for possible promoter elements and based on consensus sequences for −10 and −35 elements and the optimal spacing between these elements in E. coli (41), we defined putative −10 and −35 promoter elements for the IS200 asRNA (Figure 1B). We then introduced various mutations into the promoter (PA) of the asRNA (PA-1 to PA-6), which were designed to affect the predicted −10/−35 elements or the surrounding sequences (in the context of a multi-copy plasmid encoding IS200) and performed primer extension analysis (Figure 2). The results from our ‘WT’ construct revealed two 5' ends for the IS200 asRNA (nts 153 and 154), one of which matches the transcription start site previously identified in Salmonella (19). Based on the signal intensity it is evident that the IS200 asRNA is abundantly expressed and the two start sites are used at roughly an equal frequency. Variants, including PA-2, PA-3, PA-4 and PA-6, showing the greatest reduction in asRNA expression (Figure 2B and C) all had one or more mutations in either the predicted −35 or −10 region, supporting our assignment of these promoter elements. We note that relative to other E. coli genes the predicted −35 element of the antisense gene is a better match to the consensus than is the −10 element, the latter of which was almost unrecognizable. In addition to defining fundamental information regarding the IS200 antisense gene in E. coli, results from this analysis also provided us with a means of knocking down the levels of the IS200 asRNA in E. coli.
The IS200 antisense RNA inhibits IS200 transposase expression
To measure the impact of the IS200 asRNA on tnpA expression we constructed in a multi-copy plasmid an IS200-lacZ translational fusion (TLF) in which codon 60 of the tnpA gene was fused to codon 10 of the lacZ gene (Figure 3A). In this construct tnpA-lacZ expression was under the control of native IS200 regulatory elements. We also made a version of the TLF plasmid in which the PA-6 mutations (see Figure 2A) had been incorporated. These TLFs were introduced into a Δlac strain of E. coli (MC4100 derivative) and tnpA-lacZ expression was measured using the Miller assay. We show in Figure 3B (compare columns 1 and 5) that knocking down IS200 asRNA expression increased tnpA-lacZ expression about 13-fold. We also performed primer extension on cells used in the Miller assay and this confirmed that asRNA levels were extremely low in the strain containing the PA-6 TLF (compare lanes 1 and 5 in Figure 3B).
A potential drawback of using the PA-6 mutations to knock down IS200 asRNA levels was that the tnpA RNA sequence is altered by these mutations and this could affect the stability of the tnpA transcript and therefore its expression in the Miller assay. Accordingly, we developed an alternative means of knocking down the antisense RNA that did not alter the sequence of the tnpA transcript. In this approach we expressed a segment of the tnpA mRNA that included a region of the mRNA that was fully complementary to the antisense RNA (nt 45–298 of IS200). Pairing of the two RNAs would potentially promote the degradation of one or both RNA molecules through the action of double-strand specific ribonucleases. This ‘RNA titration’ approach has previously been used in the IS10 system to decrease levels of the IS10 encoded asRNA and was also used to knock-down endogenous levels of the MicA sRNA in E. coli (13,42). For our purposes we prepared two titrator constructs differing only in the strength of the promoters used to drive titrator expression (Figure 3A; Titrator-high and Titrator-low). We show in Figure 3B that titrator RNA expression from both constructs increased tnpA expression and the fold increase correlated well with the amount of titrator RNA expressed (compare Miller units in columns 1–3 and the corresponding lanes in the image from primer extension analysis).
We also asked if the IS200 antisense RNA could function in trans to repress tnpA expression. For this experiment we cloned the antisense RNA gene into a plasmid compatible with our TLF plasmids and co-transformed these plasmids into E. coli cells. In the situation where tnpA expression was expected to be relatively high because of the PA-6 mutations in the TLF (knock-down antisense RNA expression in cis), expression of the antisense RNA in trans reduced tnpA-lacZ expression about 3.5-fold (compare columns 5 and 8).
Based on the results presented in this section we conclude that the IS200 antisense RNA does function in vivo to down-regulate IS200 tnpA expression. Accordingly, we have renamed this RNA art200 for antisense regulator of transposase IS200.
Hfq negatively regulates tnpA expression but independent of art200
Based on previous work in the Tn10 system where we demonstrated that Hfq promotes antisense RNA pairing with the transposase RNA, potentially through restructuring of both RNAs (7), we wanted to test the possibility that Hfq might play a similar role in the IS200 system. Toward this end we repeated the experiment described in Figure 3B in isogenic hfq+ and hfq− strains of E. coli. We show in Figure 3C that tnpA-lacZ expression increased approximately 5-fold in the hfq− relative to the hfq+ strain in the context of the WT TLF (compare columns 1 and 5). This showed that Hfq does repress tnpA expression. Additionally, the PA-6 and hfq mutations acted synergistically to de-repress tnpA expression (compare column 1 to 3 and 7), and art200 provided in trans was able to repress expression of the PA-6 TLF regardless of Hfq status (compare columns 3 and 4 to columns 7 and 8). Finally, we performed primer extension analysis to measure tnpA and art200 levels in hfq+ and hfq− cells and found that tnpA levels decreased in the absence of Hfq while art200 levels were unaffected (Supplementary Figure S1). Thus we conclude that Hfq and art200 represent two distinct regulatory mechanisms that down-regulate tnpA expression independent of one another.
art200 and tnpA mRNA interact in vitro
Given that art200 and tnpA are complementary over 90 nt it seemed likely that art200 would inhibit tnpA expression through complementary basepairing. However, based on structure probing analysis of art200 and the first 173 nt of tnpA mRNA (tnpA1–173) along with secondary structure predictions, it is apparent that both RNAs are highly structured and this could limit their ability to pair (Supplementary Figures S2 and S3). An alternative possibility is that art200 acts via a protein titration mechanism. We tested for RNA pairing by performing lead(II) acetate (Pb2+) footprinting on a mixture of 5'32P-labeled art200 and unlabeled tnpA1–173. Both RNAs were generated by in vitro transcription and allowed to fold before mixing. Pairing would convert single to double stranded regions and consequently there would be a loss of Pb2+ reactivity at these positions. We show in Figure 4A that a cluster of residues (marked with a green asterisk) in the upper portion of the predicted stem-loop of art200 exhibited reduced Pb2+ reactivity upon addition of tnpA1–173. Note that most of these residues were in parts of art200 predicted by our model to be single stranded. The complementary nucleotides in tnpA1–173 include positions −23 to −62.
We also performed the complementary experiment with 5'32P-labeled tnpA1–173, but in this case used RNases (A, T1 and V1) as structure probes (Figure 4B). Both RNase A and T1 are single strand specific and accordingly reduced reactivity with these enzymes in the presence of unlabeled art200 would provide evidence of basepairing. Comparison of lanes 7 and 8 (RNase A) and lanes 12 and 13 (RNase T1) revealed two areas containing the most prominent reactivity decreases including residues −60 to −23 and −7 to +11 (indicated by red asterisks). The former region encompasses the upper stem-loop of tnpA1–173, thus supporting results from the Pb2+ footprinting that were consistent with the upper stem-loop region of art200 participating in base-pairing with the upper stem-loop of tnpA1–173. Also consistent with this interpretation, there were several examples of nucleotides in this region showing increased reactivity to RNase V1 a double-strand specific ribonuclease (blue asterisks). A summary of the footprinting data is presented in Figure 4C (Pb2+, green; RNase A/T1, red; RNase V1, blue).
The terminal loops of art200 and tnpA1–173 include four and six unpaired residues respectively, and have the highest G-C content of any of the single stranded regions in the two RNA molecules. This led us to predict that art200-tnpA1–173 pairing might initiate with a kissing loop interaction involving these two loops. Accordingly, we mutated three residues in the terminal loop of art200 (art200M1) and asked if this form of art200 could still pair with tnpA1–173 using RNase footprinting. For all of the residues that showed decreased RNase A or T1 reactivity in the presence of art200WT (red asterisks), we observed reduced protection in the presence of art200M1. Similarly, all of the residues that showed increased V1 reactivity in the presence of art200WT showed decreased reactivity in the presence of art200M1. We also introduced mutations to the lower stem region of art200 (nts 78–84, LS’) and observed an intermediate effect on pairing relative to WT and M1 suggesting this region is less important for pairing.
The above results show that art200 and tnpA do indeed interact in vitro; however pairing is limited to loosely structured regions of both RNAs. In particular, the terminal loop region of each RNA is important for pairing, which may indicate that pairing initiates with these sequences through a kissing loop interaction.
Basepairing between art200 and tnpA blocks 30S ribosome binding in vitro and inhibits transposase expression in vivo
Based on our Pb2+ and RNase footprinting experiments we thought it likely that art200 pairing with the 5'UTR of tnpA would inhibit translation initiation. We tested this possibility by performing toeprinting analysis. In this assay purified 30S ribosomal subunit plus initiator tRNA (fMet-tRNA) was mixed with tnpA1–173 either in the presence or absence of art200. Primer extension with a 5'32P-labeled primer complementary to nucleotides +51 to +70 on tnpA1–173 was then performed and reactions were analyzed on a sequencing gel. Typically the 30S ribosome leaves a footprint of ∼30 nucleotides spanning the SD sequence and first five codons such that a strong stop is produced in the primer extension reaction about 15 nt downstream of the start codon. We show that in the absence of art200, a relatively weak stop signal was observed at position +16 when 30S ribosome and fMet-tRNA were incubated with tnpA1–173 (Figure 5A, lane 6); the weak toeprint signal is consistent with previous work suggesting that the tnpA SD sequence is sequestered in a secondary structure element (35) (see also Supplementary Figure S3). Addition of art200 (15:1 molar excess) inhibited formation of the toeprint signal by approximately 95% (lane 7 and Figure 5B). However, when we used art200M1 instead of art200WT inhibition of the toeprint signal was greatly reduced to approximately 50%. Given the evidence presented in Figure 4 that art200M1 fails to basepair with tnpA, the inability of art200M1 to suppress the toeprint signal to the same degree as art200WT is consistent with art200 inhibiting 30S ribosome binding through a basepairing interaction with tnpA. We also show that when tnpA is mutated to restore complementarity with the terminal loop region of art200M1, ribosome binding is blocked by art200M1 but not art200WT (compare lanes 12–14 in Figure 5A).
Figure 5.

Impact of antisense pairing on ribosome binding to tnpA1–173 in vitro. (A) 30S ribosome binding to WT and M1’ tnpA1–173 was measured in a toeprint assay. Where indicated WT or M1 art200 (3 μM) was added to tnpA RNA (200 nM) prior to addition of the 30S ribosomal subunit and initiator tRNA. Strong pauses in reverse transcription (G+15/G+16) produced upon incubating the above mix with reverse transcriptase, dNTPs and a 5'32P-labeled DNA primer (anneals downstream of the tnpA start codon) define the toeprint signal. Positions of prominent art200-dependent pauses in reverse transcription that occur independent of 30S ribosome addition are also indicated. G, A and C are sequencing lanes and nucleotide numbering is relative to the start codon of tnpA. (B) Toeprint signal band intensities (G+15 and G+16) from (A) were quantified. The toeprint signal for tnpAWT in the absence of art200 was set at 100%.
Finally, the toeprint analysis also provided further details of the tnpA-art200 pairing interaction. In all of the reactions that included a form of art200 that was fully complementary to tnpA (lanes 5, 7, 11 and 13) there were a series of prominent primer extension pauses upstream of the SD. These strong pauses can be explained by art200 pairing with tnpA and thus the experiment reveals that position −25 in tnpA defines a ‘downstream’ boundary of antisense pairing. This fits well with our structure probe data, which were consistent with position −23 being the downstream boundary.
We also looked at the impact of terminal loop mutations (art200M1 and tnpAM1’) on tnpA-lacZ expression in vivo. We show in Figure 6 that when art200 was provided in trans (in the PA-6 TLF background) strong repression of tnpA was only achieved when the terminal loops of art200 and tnpA were perfectly complementary (compare columns 2 and 3). Also, trans-art200M1 was capable of repressing expression of tnpAPA-6/M1’-lacZ but not tnpAPA-6-lacZ (compare columns 3 and 6). Primer extension analysis on RNA prepared from the strains in Figure 6 showed that both forms of trans-art200 were expressed at similar levels in these experiments (Figure 6, lower panel).
Taken together the results from experiments in Figures 5 and 6 show that despite 90 nt of perfect complementarity, the primary determinant for antisense repression in the IS200 system is complementarity between the upper stem-loop regions of tnpA and art200 and that basepairing between residues in the terminal loops is critically important for antisense repression. Although we haven't investigated the effect of other mutations in single-stranded regions of either RNA (e.g. nt 62 to 65 in art200 and −53 to −56 in tnpA) it seems likely that pairing initiates with the 3 G/C base-pairs affected by the M1 mutation and then propagates roughly half-way down the respective stems. An initial kissing-loop interaction has been shown in many other antisense systems to be important for pairing (43–45). Further pairing might be inhibited by the absence of bulges in the lower portions of the respective stems, as such discontinuities in intramolecular base-pairing have been shown in other studies to be important in destabilizing stem structures and allowing intermolecular basepairing (11,46–47).
tnpA translation is also repressed by mRNA secondary structure
Previous work in the IS200 system revealed that deleting the 5' portion of tnpA mRNA (nts −32 to −103) resulted in a ∼10-fold increase in tnpA-lacZ expression (35). The authors from this study concluded that the increased expression resulted from the loss of an inhibitory stem-loop structure; however their deletion also removed half of art200. To determine if RNA secondary structure plays an important role in inhibiting tnpA expression, we introduced mutations to the lower stem (nts −69 to −75) and evaluated the impact of these mutations on ribosome binding in vitro and on tnpA expression in vivo. We show in the toeprinting assay in Figure 7A that tnpA1–173 with the lower stem mutations (tnpALS) gave a much higher toeprint signal (20-fold increase) than tnpAWT (compare lanes 6 and 11; also see Figure 7B). This indicates that nucleotides comprising the lower stem are important determinants for tnpA translation (as previously suggested). We also asked if art200 could still repress ribosome binding in the tnpALS background. Both art200WT and art200LS’ strongly repressed ribosome binding (compare lanes 12 and 13 with lane 11) but a mutant form of art200 (art200M1) lacking full terminal loop complementarity with tnpALS failed to fully block ribosome binding (lane 14). These results indicate that the nucleotides comprising the lower stem of tnpA are not critical for antisense repression and thus contribute to a distinct mode of tnpA translational regulation.
Figure 7.
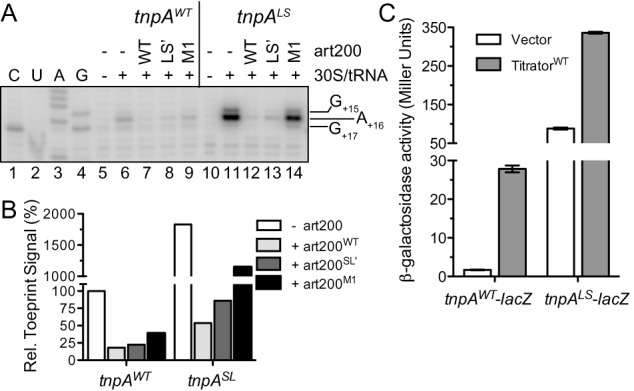
Impact of tnpA lower stem (LS) mutations on transposase expression. (A) The toeprinting assay was performed on tnpA RNA (WT and LS; 200 nM) in the presence or absence of art200 (WT, LS’ or M1; 3 μM) as described in Figure 5. The toeprint signal spans nucleotides 15–17. C, U, A and G are sequencing lanes. (B) Toeprint signal band intensities (G+15, A+16 and G+16) from (A) were quantified. The toeprint signal for tnpAWT in the absence of art200 was set at 100%. (C) A plasmid encoding tnpAWT-lacZ or tnpALS-lacZ was co-transformed into DBH323 with a compatible plasmid expressing art200 titrator RNA (WT or M1) or an empty vector control. β-galactosidase activity was measured in cells grown to mid-exponential phase in LB media. Bars show the mean expression from two independent experiments and error bars indicate standard error on the mean (n = 6).
We also determined the impact of the LS mutations on tnpA expression in vivo. Consistent with the toeprinting assay, the LS mutations increased tnpA-lacZ expression 50-fold relative to that observed for WT tnpA-lacZ (compare columns 1 and 3 in Figure 7C). Titration of art200LS’ in this system with the high copy titrator further increased expression 4-fold (column 4) indicating that the two regulatory systems can act independent of each other to repress tnpA expression.
We therefore conclude that in addition to a cis-encoded sRNA, translation of the IS200 transposase is strongly repressed by an mRNA secondary structure that can directly sequester the SD.
Hfq blocks ribosome binding to tnpA in vitro
Although Hfq is not required for antisense pairing, tnpA-lacZ expression increased 5-fold in an hfq− versus hfq+ strain of E. coli. In addition, we have shown that this up-regulation in the absence of Hfq did not require the production of art200 (Figure 3C). This indicates that Hfq represses transposase expression in an antisense-independent manner. Additionally, tnpA levels do not increase in hfq− which indicates Hfq acts at the level of tnpA translation (Supplementary Figure S1). We have recently shown that in the IS10 system, Hfq binding to the ribosome binding site of transposase mRNA was sufficient for repressing translation initiation (40). We therefore considered the possibility that Hfq might be acting directly on tnpA to inhibit translation.
We show in Figure 8A and B that Hfq inhibited formation of the tnpA1–173 toeprint in a concentration dependent manner (see also Supplementary Figure S4). At a 1:1 molar ratio of Hfq:tnpA, the toeprint signal was reduced 40–50% compared to no Hfq addition and at a 4:1 ratio of Hfq:tnpA the toeprint signal was reduced 80%. Thus, Hfq can block 30S ribosomal subunit binding to tnpA1–173 in vitro independent of an sRNA. We note that the strength of the Hfq block on ribosome binding in the IS200 system is weaker than previously seen with the IS10 transposase mRNA (RNA-IN) but slightly stronger than observed with a control mRNA, usg (Figure 8B) (40,48). For example, when Hfq is limiting (1:2 ratio of Hfq:mRNA), the toeprint signal was reduced 30% for tnpA and only 12% for usg mRNA. However, at higher concentrations of Hfq the toeprint signal was reduced a comparable amount for both tnpA and usg.
Figure 8.
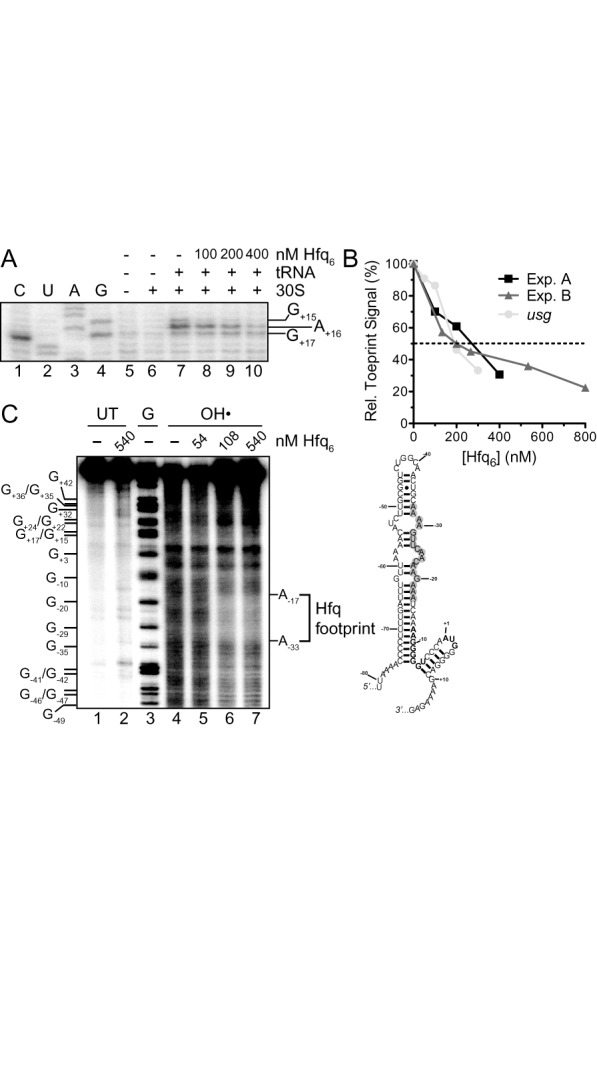
Hfq inhibits 30S ribosomal subunit binding to tnpA and binds upstream of the SD. (A) Toeprint assay showing the effect of Hfq on 30S ribosomal subunit binding to tnpA. Hfq (100–400 nM; hexamer concentration) was added to tnpA (200 nM) prior to addition of 30S ribosomal subunit and initiator tRNA. A section of the gel image including the toeprint signal is shown. (B) The percent inhibition of toeprint signal upon incubating Hfq with tnpA or an mRNA that does not interact with Hfq (usg, (48)) is shown; the usg data come from ref (40). For both mRNAs the toeprint signal in the absence of Hfq was set at 100%. Experiment A refers to part (A) of this figure while Experiment B refers to Supplementary Figure S4. (C) Hydroxyl radical footprinting experiment with 5'32P-labeled tnpA1–173 (68 nM) and the indicated concentrations of Hfq. Subsequent to mixing tnpA and Hfq limited RNA cleavage by hydroxyl radical treatment was carried out as previously described (7). UT is untreated RNA and G is an RNA cleavage ladder produced by RNase T1 cleavage. The Hfq footprint between residues −17 and −33 defines an Hfq binding site in tnpA and the position of this site is highlighted (gray circles) in our model for tnpA1–173; the tnpA SD and start codon are in bold. Note that a version of this gel image was previously published in Methods in Molecular Biology (38).
We further analyzed the Hfq-tnpA interaction by performing hydroxyl radical footprinting on 5'32P-labeled tnpA1–173 mixed with various concentrations of Hfq (Figure 8C). The results of the footprinting were consistent with Hfq binding tnpA in an interval extending from position −33 to −17. Together, the above results suggest that Hfq binding immediately upstream of the tnpA SD sequence represses tnpA translation by preventing ribosome binding.
IS200 transposition is limited by translational control
Typically for bacterial transposons, transposition frequency correlates strongly with transposase expression (32–34). We measured IS200 transposition by constructing a mini-IS200 element (IS200-kan) and using this marked element in mating out experiments (see Supplementary Figure S5 for schematic of the mating out assay). IS200 transposase was provided in trans from a plasmid in which the tnpA gene was under the control of different regulatory elements (Figure 9A).
We did not detect transposition events when tnpA expression was under the control of the fully native regulatory elements. We did detect transposition events when tnpA was fused to PBAD and SDBAD24 and arabinose (0.2%) was present during growth (construct (i)). Notably, the number of events was considerably higher than in a control where the tnpA gene contained a mutation in the catalytic tyrosine (construct (ii)). We confirmed that these were authentic transposition events by mapping two independent hops from construct (i) using ST-PCR (36,49) (Supplementary Figure S6A). We therefore conclude that the IS200 TnpA protein from Salmonella is active for transposition in E. coli.
When we replaced SDBAD24 with the native 5'UTR (construct (iii)), the transposition frequency dropped considerably; a single transposition event was observed in one of three experiments. This construct produced a large amount of tnpA mRNA suggesting that translational control strongly limits transposition (Figure 9B). We then introduced mutations into the native 5'UTR to disrupt the tnpA stem-loop (construct (iv)). This increased the frequency of transposition events, although the occurrence of these events was still sporadic.
We next measured transposition from construct (iv) in the presence of the art200 titrator plasmid or a vector control. Our expectation was that disrupting two regulatory pathways (mRNA structure and antisense control) would further increase transposition. In the presence of the titrator plasmid, 8/10 donor isolates produced measurable transposition while only 5/10 donors produced hops in the presence of the vector control (Figure 9C). This coincided with a 25-fold increase in the median value of transposition when art200 was depleted. Together these data show that (1) TnpA expression is in fact limiting for IS200 transposition, and (2) disrupting translational regulation (mRNA secondary structure and antisense control) of tnpA leads to an increase in IS200 transposition.
DISCUSSION
Translation of the IS200 transposase is repressed by a cis-encoded sRNA, Hfq and RNA secondary structure
IS200 is a very unusual transposable element in that it is widespread in Eubacteria and in some species has attained a very high copy number (see below), yet its ability to transpose is exceedingly poor. This correlates with very weak expression of the IS200 transposase protein. In the current work we have expanded our understanding of how IS200 transposase expression is suppressed to include two new levels of post-transcriptional regulation and further characterization of a predicted cis-regulatory element. First, we show that the recently identified sRNA art200 (previously STnc490 (15)) encoded opposite the transposase 5'UTR represses transposase translation by base-pairing with tnpA mRNA and blocking 30S ribosome binding. Additionally, we expand on previous work that suggested RNA secondary structure in the 5'UTR of tnpA inhibits translation by sequestering the SD in a stable stem loop structure (35). Finally, we show that the chaperone protein Hfq is also a negative regulator of tnpA expression. Footprinting revealed that Hfq binds immediately upstream to the tnpA SD raising the possibility that Hfq could block 30S subunit binding to tnpA. Support for this came from toeprinting studies where at low concentrations of Hfq (100 nM) ribosome binding to tnpA was reduced 30%. It is not clear at this point if this reduction is significant as the level of toeprint inhibition was only marginally higher than that detected in a control reaction; 15% with usg mRNA. By comparison in another system (cirA mRNA) where Hfq was reported to directly interfere with 30S subunit binding a similarly small reduction in toeprint signal (20%) was reported at low Hfq concentrations (50). Based on these data we suggest that this moderate effect on ribosome binding could account for at least a portion of the 5-fold repression Hfq has on tnpA-lacZ expression in vivo. Notably, this represents the first example of a bacterial ‘host’ protein suppressing IS200 (22) and the second example of Hfq directly repressing translation of a transposase protein (40).
An interesting aspect of the regulatory mechanisms described here is that all three are capable of acting independently to interfere with 30S subunit binding to tnpA. This conclusion comes from the following observations: (1) art200 repressed tnpA expression and ribosome binding in the absence of Hfq (Figures 3C and 5); (2) art200 suppressed 30S subunit binding to tnpA under conditions where the inhibitory stem-loop structure is destabilized by mutations in tnpA (Figure 7A); and (3) the effect of disrupting hfq on tnpAWT expression was almost 20-fold less than inhibiting formation of the stem-loop structure (compare Figures 3C and 7C). If Hfq acted to stabilize the stem-loop structure one might have expected tnpA expression to be comparable in hfq− and tnpALS situations.
What might be the explanation for this level of functional redundancy? IS200 tnpA contains an almost perfect Shine-Dalgarno sequence (tnpA, AAGGGGGU; E. coli consensus, AAGGAGGU) (51). However, this sequence is sequestered in secondary structure (this work and (35)). Interestingly, upstream of the SD there is a single-stranded C/A-rich sequence (nts −26 to −21) that potentially could act as a translational enhancer. Such sequences can provide an initial toehold for the 30S ribosomal subunit through a direct interaction between the S1 protein component of the 30S complex and the C/A-rich RNA sequence (52–55). S1 could tether the ribosome to tnpA and expose the downstream SD sequence for 30S subunit binding by altering the local RNA structure (56–59). As we have shown that art200 pairs with the C/A-rich containing portion of the tnpA transcript and Hfq binds this same region, it is possible that both art200 and Hfq repress the function of this putative translational enhancer sequence by sterically occluding S1 binding. The combination of sequestration of the SD and interference of translational enhancer function would be expected to provide a very strong block (synergistic or at least additive) to translation, which we observed here. We have some evidence of the C/A-rich region playing a regulatory role in translation as mutations in this region reduced tnpA expression almost 200-fold (Supplementary Figure S7), although as we were unable to measure steady-state tnpA RNA levels because of the extremely low abundance of this transcript we can't rule out the possibility that this decrease resulted from the mutations destabilizing the transcript.
Notably, there are several examples in the literature of sRNAs interfering with translational enhancer function. The sRNA GcvB represses initiation of translation for multiple mRNA transcripts by pairing with C/A-rich translational enhancers (60,61). In addition, other sequences upstream of the SD have been shown to influence 30S subunit binding. In the case of the tisAB transcript, which has its SD sequence sequestered in a highly structured region, a genetic element distinct from a C/A-rich translational enhancer was shown to provide a ‘standby’ site for 30S binding. It was inferred that 30S binding to this sequence opened up the downstream structure for subsequent 30S binding to the SD. The sRNA IstR-1 acts as a negative regulator of translation in this system by competing with the 30S subunit for the standby site (62). Art200 and/or Hfq could act in a similar manner in the IS200 system (Figure 10, (i)).
Figure 10.
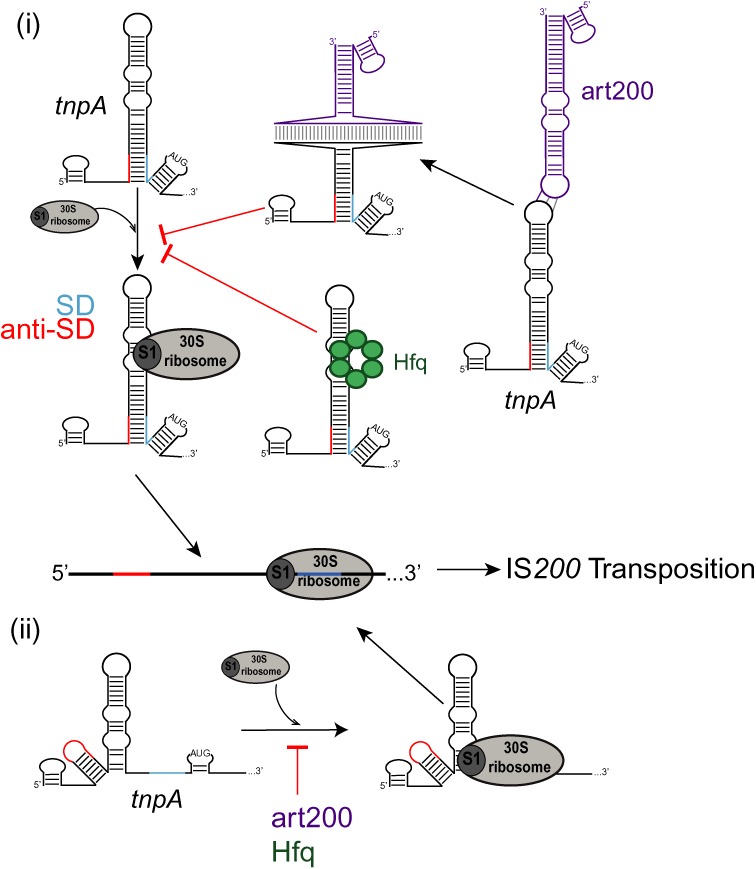
Model for translational repression of IS200 tnpA. 30S ribosomal subunit binding to tnpA is inhibited by art200 (purple) and Hfq (green) as well as RNA secondary structure which sequesters the Shine-Dalgarno sequence (SD, light blue line). Art200 and Hfq may act to (i) block ribosomal protein S1 binding to a translational enhancer or (ii) simply prevent the 30S-SD interaction. An alternative secondary structure of tnpA where the anti-Shine-Dalgarno (anti-SD, red line) is not paired to the SD is derived from secondary structure predictions (see Supplementary Figure S8). See discussion for more details.
Finally, it is also possible that art200 and/or Hfq exert their negative regulatory effects on the IS200 system by binding close enough to the SD to directly block 30S subunit binding. It has been reported that the maximal ribosome-binding region can include nucleotides as far as 39 residues upstream of the start codon (63) and the art200 pairing site and the Hfq binding site fall within this window. If this latter mechanism were in play in the IS200 system, then all three negative regulatory systems would be acting at the same step in translation and accordingly the reason for this level of redundancy would be less clear. Although one possibility could be that there are some circumstances where SD sequestration would be suboptimal. For example, under conditions where transcription rates are reduced it is possible that the anti-SD sequence in tnpA could pair with an alternative sequence to the SD (one such possible structure is shown in Supplementary Figure S8). In this case art200 and/or Hfq could provide important back-up functions for limiting 30S binding to tnpA (Figure 10, (ii)).
Given the current work, it is not surprising that IS200 transposition is exceptionally rare. In addition to weak transcription of the transposase gene, tnpA translation is suppressed by three independent mechanisms. As we have shown that TnpA expression is in fact limiting for transposition (Figure 9), we speculate that translation initiation represents the main point of regulation for IS200 transposition.
Might art200 function as both a cis and trans acting sRNA?
We have previously shown that Hfq represses IS10 transposase expression by facilitating the pairing between transposase mRNA (RNA-IN) and a cis-encoded sRNA (RNA-OUT) (7) and this led us to ask if similar regulation would occur in the IS200 system with art200. However, the current work shows that Hfq is not required for art200-mediated repression of tnpA expression. There are a large number of Hfq-binding RNAs in vivo and we and others have provided evidence that Hfq is in fact limiting for RNA binding (40,64–66). Since Hfq binding in vivo must therefore be selective, it seems likely that the Hfq-art200 interaction is biologically important (67), although for gram positive bacteria only a subset of Hfq-binding sRNAs seem to rely on Hfq for stability and/or riboregulation (68–72). It is possible that art200 also is a trans-acting sRNA, and that this secondary function requires Hfq.
In addition to its Hfq binding properties, art200 expression increases during stationary phase and under conditions that induce the Salmonella pathogenicity islands (15). There is no a priori expectation that expression of an RNA involved in repressing transposition would fluctuate in response to external stimuli or growth phase. Art200 is also expressed at a level far greater than that required to repress the poorly expressed tnpA mRNA (see Figure 3B).
One paradox of IS200 elements is that while these transposons are essentially dormant many genomes containing IS200 elements have multiple copies. For example, natural isolates of Salmonella and Shigella contain up to 25 and 4 copies of IS200, respectively (25) and the Y. pestis 6/69M genome contains at least 30 copies of the closely related IS1541 (73) In fact, a cursory BLAST search for IS200 elements in Salmonella revealed an average of 9.6 (n = 33) copies of IS200 per genome, while a similar search in Yersinia averaged 39.8 (n = 30) copies per genome. In contrast, E. coli contains an average of 2.9 (n = 31) IS200 elements per genome. The high copy number of IS200 elements in certain species may simply reflect host-specific adaptation by the transposon (74). Alternatively, IS200 might have been domesticated by certain host bacteria, in which case IS200 expansion (and utilization of art200 as a trans regulator) could be a response to selective pressure. There are several examples of Hfq integrating horizontally acquired genes into host regulatory networks (4,15,75–78) and art200 may represent one such case.
IS200 5'UTR as a platform for designing novel riboregulators of translation initiation
A major goal in the field of synthetic biology is to create tightly controlled gene regulatory networks to coordinate the expression of a range of desired protein products. The ultimate goal of this field is to produce microorganisms capable of producing biomaterials, pharmaceuticals and biofuels and acting as biosensors for a range of applications (79). Since these biosynthetic pathways must be tightly regulated yet easily manipulated, a great deal of work has been done to design riboregulators of transcription and translation. As it is advantageous to adapt naturally occurring regulators rather than de novo design, well-studied systems such as the pT181 transcriptional attenuator and IS10 antisense system have been modified and combined for synthetic biology applications (80–85).
We propose that the IS200 5'UTR will serve as a convenient platform for modular design of orthogonal regulators of protein synthesis. First, we show here that the cis-encoded antisense system can be easily re-programmed by altering 3-nt in the terminal loop region of each RNA. We have not investigated the impact of more extensive changes but predict that this could provide greater specificity. Additionally, our work shows that antisense regulation can be exploited for negative regulation (i.e. providing art200 in trans) or positive regulation (i.e. eliminating art200 through titration). In principle, the tnpA 5'UTR could be fused to a gene of interest and translation of this downstream gene could be modified by an art200 derivative provided in trans. Translation could be further regulated by selectively disrupting the secondary structure that naturally occludes the SD sequence on tnpA. An RNA which basepairs with the linear region of tnpA immediately 5' to the lower-stem as well as the ‘anti-SD’ sequence could reduce secondary structure in a manner analogous to the LS mutation we described here. This synthetic RNA would be similar to the recently described ‘trigger RNA’ which can activate expression of de novo designed toehold switches (86).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to J. Ross for productive discussions throughout this work and T. Tessier for assistance with initial IS200 plasmid constructions. We thank M. Valvano for providing Salmonella Typhimurium LT2, G. Storz for providing an E. coli hfq-1::Cm strain and N. Kleckner for providing pNK81 and λNK1039. Finally, we thank L. Haniford and F. Laditi for help in preparing genomic DNA and other reagents.
FUNDING
Canadian Institutes of Health Research [MOP 11281 to D.B.H.]; Ontario Graduate Scholarship and Western Graduate Research Scholarship to M.J.E. Funding for open access charge: Canadian Institutes of Health Research.
Conflict of interest statement. None declared.
REFERENCES
- 1.Waters L.S., Storz G. Regulatory RNAs in bacteria. Cell. 2009;136:615–628. doi: 10.1016/j.cell.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Storz G., Vogel J., Wassarman K.M. Regulation by small RNAs in bacteria: expanding frontiers. Mol. Cell. 2011;43:880–891. doi: 10.1016/j.molcel.2011.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Papenfort K., Vogel J. Small RNA functions in carbon metabolism and virulence of enteric pathogens. Front. Cell. Infect. Microbiol. 2014;4 doi: 10.3389/fcimb.2014.00091. doi:10.3389/fcimb.2014.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Papenfort K., Vogel J. Regulatory RNA in Bacterial Pathogens. Cell. Host Microbe. 2010;8:116–127. doi: 10.1016/j.chom.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Thomason M.K., Storz G. Bacterial antisense RNAs: how many are there, and what are they doing. Annu. Rev. Genet. 2010;44:167–188. doi: 10.1146/annurev-genet-102209-163523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Georg J., Hess W.R. cis-antisense RNA, another level of gene regulation in bacteria. Microbiol. Mol. Biol. Rev. 2011;75:286–300. doi: 10.1128/MMBR.00032-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ross J.A., Ellis M.J., Hossain S., Haniford D.B. Hfq restructures RNA-IN and RNA-OUT and facilitates antisense pairing in the Tn10/IS10 system. RNA. 2013;19:670–684. doi: 10.1261/rna.037747.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Opdyke J.A., Kang J.-G., Storz G. GadY, a small-RNA regulator of acid response genes in Escherichia coli. J. Bacteriol. 2004;186:6698–6705. doi: 10.1128/JB.186.20.6698-6705.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomizawa J., Itoh T., Selzer G., Som T. Inhibition of ColE1 RNA primer formation by a plasmid-specified small RNA. Proc. Natl. Acad. Sci. U.S.A. 1981;78:1421–1425. doi: 10.1073/pnas.78.3.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simons R.W., Hoopes B.C., McClure W.R., Kleckner N. Three promoters near the termini of IS10: pIN, pOUT, and pIII. Cell. 1983;34:673–682. doi: 10.1016/0092-8674(83)90400-2. [DOI] [PubMed] [Google Scholar]
- 11.Kittle J.D., Simons R.W., Lee J., Kleckner N. Insertion sequence IS10 anti-sense pairing initiates by an interaction between the 5’ end of the target RNA and a loop in the anti-sense RNA. J. Mol. Biol. 1989;210:561–572. doi: 10.1016/0022-2836(89)90132-0. [DOI] [PubMed] [Google Scholar]
- 12.Ma C., Simons R.W. The IS10 antisense RNA blocks ribosome binding at the transposase translation initiation site. EMBO J. 1990;9:1267–1274. doi: 10.1002/j.1460-2075.1990.tb08235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simons R.W., Kleckner N. Translational control of IS10 transposition. Cell. 1983;34:683–691. doi: 10.1016/0092-8674(83)90401-4. [DOI] [PubMed] [Google Scholar]
- 14.Sharma C.M., Vogel J. Experimental approaches for the discovery and characterization of regulatory small RNA. Curr. Opin. Microbiol. 2009;12:536–546. doi: 10.1016/j.mib.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 15.Sittka A., Lucchini S., Papenfort K., Sharma C.M., Rolle K., Binnewies T.T., Hinton J.C., Vogel J. Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 2008;4:e1000163. doi: 10.1371/journal.pgen.1000163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li S.K., Ng P.K., Qin H., Lau J.K., Lau J.P., Tsui S.K., Chan T.F., Lau T.C. Identification of small RNAs in Mycobacterium smegmatis using heterologous Hfq. RNA. 2013;19:74–84. doi: 10.1261/rna.034116.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lybecker M., Zimmermann B., Bilusic I., Tukhtubaeva N., Schroeder R. The double-stranded transcriptome of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2014;111:3134–3139. doi: 10.1073/pnas.1315974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilusic I., Popitsch N., Rescheneder P., Schroeder R., Lybecker M. Revisiting the coding potential of the E. coli genome through Hfq co-immunoprecipitation. RNA Biol. 2014;11:641–654. doi: 10.4161/rna.29299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kröger C., Dillon S.C., Cameron A.D.S., Papenfort K., Sivasankaran S.K., Hokamp K., Chao Y., Sittka A., Hébrard M., Händler K., et al. The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E1277–E1286. doi: 10.1073/pnas.1201061109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan Y., Su S., Meng X., Ji X., Qu Y., Liu Z., Wang X., Cui Y., Deng Z., Zhou D., et al. Determination of sRNA expressions by RNA-seq in Yersinia pestis grown in vitro and during infection. PLoS One. 2013;8:e74495. doi: 10.1371/journal.pone.0074495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raghavan R., Sloan D.B., Ochman H. Antisense transcription is pervasive but rarely conserved in enteric bacteria. mBio. 2012;3 doi: 10.1128/mBio.00156-12. doi:10.1128/mBio.00156-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beuzon C.R., Chessa D., Casadesus J. IS200: an old and still bacterial transposon. Int. Microbiol. 2004;7:3–12. [PubMed] [Google Scholar]
- 23.Beuzon C.R., Casadesus J. Conserved structure of IS200 elements in Salmonella. Nucleic Acids Res. 1997;25:1355–1361. doi: 10.1093/nar/25.7.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filée J., Siguier P., Chandler M. Insertion sequence diversity in archaea. Microbiol. Mol. Biol. Rev. 2007;71:121–157. doi: 10.1128/MMBR.00031-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gibert I., Barbé J., Casadesús J. Distribution of insertion sequence IS200 in Salmonella and Shigella. J. Gen. Microbiol. 1990;136:2555–2560. doi: 10.1099/00221287-136-12-2555. [DOI] [PubMed] [Google Scholar]
- 26.Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lam S., Roth J.R. IS200: A Salmonella-specific insertion sequence. Cell. 1983;34:951–960. doi: 10.1016/0092-8674(83)90552-4. [DOI] [PubMed] [Google Scholar]
- 28.Casadesus J., Roth J. Absence of insertions among spontaneous mutants of Salmonella typhimurium. Mol. Gen. Genet. 1989;216:210–216. doi: 10.1007/BF00334358. [DOI] [PubMed] [Google Scholar]
- 29.Schiaffino A., Beuzón C.R., Uzzau S., Leori G., Cappuccinelli P., Casadesús J., Rubino S. Strain typing with IS200 fingerprints in Salmonella abortusovis. Appl. Environ. Microbiol. 1996;62:2375–2380. doi: 10.1128/aem.62.7.2375-2380.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornelius C.A., Quenee L.E., Elli D., Ciletti N.A., Schneewind O. Yersinia pestis IS1541 transposition provides for escape from plague immunity. Infect. Immun. 2009;77:1807–1816. doi: 10.1128/IAI.01162-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bisercić M., Ochman H. The ancestry of insertion sequences common to Escherichia coli and Salmonella typhimurium. J. Bacteriol. 1993;175:7863–7868. doi: 10.1128/jb.175.24.7863-7868.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morisato D., Way J.C., Kim H.J., Kleckner N. Tn10 transposase acts preferentially on nearby transposon ends in vivo. Cell. 1983;32:799–807. doi: 10.1016/0092-8674(83)90066-1. [DOI] [PubMed] [Google Scholar]
- 33.Nagy Z., Chandler M. Regulation of transposition in bacteria. Res. Microbiol. 2004;155:387–398. doi: 10.1016/j.resmic.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Kleckner N. Regulation of transposition in bacteria. Annu. Rev. Cell Biol. 1990;6:297–327. doi: 10.1146/annurev.cb.06.110190.001501. [DOI] [PubMed] [Google Scholar]
- 35.Beuzon C.R., Marques S., Casadesus J. Repression of IS200 transposase synthesis by RNA secondary structures. Nucleic Acids Res. 1999;27:3690–3695. doi: 10.1093/nar/27.18.3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross J.A., Trussler R.S., Black M.D., McLellan C.R., Haniford D.B. Tn5 transposition in Escherichia coli is repressed by Hfq and activated by over-expression of the small non-coding RNA SgrS. Mobile DNA. 2014;5:27. doi: 10.1186/s13100-014-0027-z. doi:10.1186/s13100-014-0027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guzman L.M., Belin D., Carson M.J., Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ellis M., Trussler R., Ross J., Haniford D. Probing Hfq:RNA interactions with hydroxyl radical and RNase footprinting. Methods Mol. Biol. 2015;1259:403–415. doi: 10.1007/978-1-4939-2214-7_24. [DOI] [PubMed] [Google Scholar]
- 39.Ross J.A., Wardle S.J., Haniford D.B. Tn10/IS10 transposition is downregulated at the level of transposase expression by the RNA-binding protein Hfq. Mol. Microbiol. 2010;78:607–621. doi: 10.1111/j.1365-2958.2010.07359.x. [DOI] [PubMed] [Google Scholar]
- 40.Ellis M.J., Trussler R.S., Haniford D.B. Hfq binds directly to the ribosome-binding site of IS10 transposase mRNA to inhibit translation. Mol. Microbiol. 2015;96:633–650. doi: 10.1111/mmi.12961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harley C.B., Reynolds R.P. Analysis of E. coli Promoter sequences. Nucleic Acids Res. 1987;15:2343–2361. doi: 10.1093/nar/15.5.2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Udekwu K.I., Darfeuille F., Vogel J., Reimegård J., Holmqvist E., Wagner E.G.H. Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev. 2005;19:2355–2366. doi: 10.1101/gad.354405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brantl S. Antisense-RNA regulation and RNA interference. Biochim. Biophys. Acta. 2002;1575:15–25. doi: 10.1016/s0167-4781(02)00280-4. [DOI] [PubMed] [Google Scholar]
- 44.Franch T., Petersen M., Wagner E.G., Jacobsen J.P., Gerdes K. Antisense RNA regulation in prokaryotes: rapid RNA/RNA interaction facilitated by a general U-turn loop structure. J. Mol. Biol. 1999;294:1115–1125. doi: 10.1006/jmbi.1999.3306. [DOI] [PubMed] [Google Scholar]
- 45.Hjalt T., Gerhart E., Wagner H. The effect of loop size in antisense and target RNAs on the efficiency of antisense RNA control. Nucleic Acids Res. 1992;20:6723–6732. doi: 10.1093/nar/20.24.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brantl S. Regulatory mechanisms employed by cis-encoded antisense RNAs. Curr. Opin. Microbiol. 2007;10:102–109. doi: 10.1016/j.mib.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 47.Hjalt T.Å.H., Gerhart E., Wagner H. Bulged-out nucleotides in an antisense RNA are required for rapid target RNA binding in vitro and inhibition in vivo. Nucleic Acids Res. 1995;23:580–587. doi: 10.1093/nar/23.4.580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beisel C.L., Updegrove T.B., Janson B.J., Storz G. Multiple factors dictate target selection by Hfq-binding small RNAs. EMBO J. 2012;31:1961–1974. doi: 10.1038/emboj.2012.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chun K.T., Edenberg H.J., Kelley M.R., Goebl M.G. Rapid amplification of uncharacterized transposon-tagged DNA sequences from genomic DNA. Yeast. 1997;13:233–240. doi: 10.1002/(SICI)1097-0061(19970315)13:3<233::AID-YEA88>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 50.Salvail H., Caron M.P., Belanger J., Masse E. Antagonistic functions between the RNA chaperone Hfq and an sRNA regulate sensitivity to the antibiotic colicin. EMBO J. 2013;32:2764–2778. doi: 10.1038/emboj.2013.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shine J., Dalgarno L. Determinant of cistron specificity in bacterial ribosomes. Nature. 1975;254:34–38. doi: 10.1038/254034a0. [DOI] [PubMed] [Google Scholar]
- 52.Ringquist S., Jones T., Snyder E.E., Gibson T., Boni I., Gold L. High-affinity RNA ligands to Escherichia coli ribosomes and ribosomal protein s1: comparison of natural and unnatural binding sites. Biochemistry. 1995;34:3640–3648. doi: 10.1021/bi00011a019. [DOI] [PubMed] [Google Scholar]
- 53.Mogridge J., Greenblatt J. Specific binding of Escherichia coli ribosomal protein S1 to boxA transcriptional antiterminator RNA. J. Bacteriol. 1998;180:2248–2252. doi: 10.1128/jb.180.8.2248-2252.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sengupta J., Agrawal R.K., Frank J. Visualization of protein S1 within the 30S ribosomal subunit and its interaction with messenger RNA. Proc. Natl. Acad. Sci. U.S.A. 2001;98:11991–11996. doi: 10.1073/pnas.211266898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komarova A.V., Tchufistova L.S., Supina E.V., Boni I.V. Protein S1 counteracts the inhibitory effect of the extended Shine-Dalgarno sequence on translation. RNA. 2002;8:1137–1147. doi: 10.1017/s1355838202029990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Studer S.M., Joseph S. Unfolding of mRNA secondary structure by the bacterial translation initiation complex. Mol. Cell. 2006;22:105–115. doi: 10.1016/j.molcel.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 57.Rajkowitsch L., Schroeder R. Dissecting RNA chaperone activity. RNA. 2007;13:2053–2060. doi: 10.1261/rna.671807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bear D.G., Ng R., Van Derveer D., Johnson N.P., Thomas G., Schleich T., Noller H.F. Alteration of polynucleotide secondary structure by ribosomal protein S1. Proc. Natl. Acad. Sci. U.S.A. 1976;73:1824–1828. doi: 10.1073/pnas.73.6.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qu X., Lancaster L., Noller H.F., Bustamante C., Tinoco I. Ribosomal protein S1 unwinds double-stranded RNA in multiple steps. Proc. Natl. Acad. Sci. U.S.A. 2012;109:14458–14463. doi: 10.1073/pnas.1208950109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yang Q., Figueroa-Bossi N., Bossi L. Translation enhancing ACA motifs and their silencing by a bacterial small regulatory RNA. PLoS Genet. 2014;10:e1004026. doi: 10.1371/journal.pgen.1004026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma C.M., Darfeuille F., Plantinga T.H., Vogel J. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007;21:2804–2817. doi: 10.1101/gad.447207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Darfeuille F., Unoson C., Vogel J., Wagner E.G.H. An antisense RNA inhibits translation by competing with standby ribosomes. Mol. Cell. 2007;26:381–392. doi: 10.1016/j.molcel.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 63.Hüttenhofer A., Noller H.F. Footprinting mRNA-ribosome complexes with chemical probes. EMBO J. 1994;13:3892–3901. doi: 10.1002/j.1460-2075.1994.tb06700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hussein R., Lim H.N. Disruption of small RNA signaling caused by competition for Hfq. Proc. Natl. Acad. Sci. U.S.A. 2011;108:1110–1115. doi: 10.1073/pnas.1010082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Moon K., Gottesman S. Competition among Hfq-binding small RNAs in Escherichia coli. Mol. Microbiol. 2011;82:1545–1562. doi: 10.1111/j.1365-2958.2011.07907.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Papenfort K., Said N., Welsink T., Lucchini S., Hinton J.C.D., Vogel J. Specific and pleiotropic patterns of mRNA regulation by ArcZ, a conserved, Hfq-dependent small RNA. Mol. Microbiol. 2009;74:139–158. doi: 10.1111/j.1365-2958.2009.06857.x. [DOI] [PubMed] [Google Scholar]
- 67.Miyakoshi M., Chao Y., Vogel J. Regulatory small RNAs from the 3′ regions of bacterial mRNAs. Curr. Opin. Microbiol. 2015;24:132–139. doi: 10.1016/j.mib.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 68.Sievers S., Sternkopf Lillebæk E.M., Jacobsen K., Lund A., Mollerup M.S., Nielsen P.K., Kallipolitis B.H. A multicopy sRNA of Listeria monocytogenes regulates expression of the virulence adhesin LapB. Nucleic Acids Res. 2014;42:9383–9398. doi: 10.1093/nar/gku630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nielsen J.S., Lei L.K., Ebersbach T., Olsen A.S., Klitgaard J.K., Valentin-Hansen P., Kallipolitis B.H. Defining a role for Hfq in Gram-positive bacteria: evidence for Hfq-dependent antisense regulation in Listeria monocytogenes. Nucleic Acids Res. 2010;38:907–919. doi: 10.1093/nar/gkp1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dambach M., Irnov I., Winkler W.C. Association of RNAs with Bacillus subtilis Hfq. PLoS One. 2013;8:e55156. doi: 10.1371/journal.pone.0055156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Christiansen J.K., Nielsen J.S., Ebersbach T., Valentin-Hansen P., Søgaard-Andersen L., Kallipolitis B.H. Identification of small Hfq-binding RNAs in Listeria monocytogenes. RNA. 2006;12:1383–1396. doi: 10.1261/rna.49706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jousselin A., Metzinger L., Felden B. On the facultative requirement of the bacterial RNA chaperone, Hfq. Trends Microbiol. 2009;17:399–405. doi: 10.1016/j.tim.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 73.Odaert M., Devalckenaere A., Trieu-Cuot P., Simonet M. Molecular characterization of IS1541 insertions in the genome of Yersinia pestis. J. Bacteriol. 1998;180:178–181. doi: 10.1128/jb.180.1.178-181.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Siguier P., Gourbeyre E., Chandler M. Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 2014;38:895–891. doi: 10.1111/1574-6976.12067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vogt S.L., Raivio T.L. Hfq reduces envelope stress by controlling expression of envelope-localized proteins and protein complexes in enteropathogenic Escherichia coli. Mol Microbiol. 2014;92:681–697. doi: 10.1111/mmi.12581. [DOI] [PubMed] [Google Scholar]
- 76.Shakhnovich E.A., Davis B.M., Waldor M.K. Hfq negatively regulates type III secretion in EHEC and several other pathogens. Mol. Microbiol. 2009;74:347–363. doi: 10.1111/j.1365-2958.2009.06856.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Papenfort K., Podkaminski D., Hinton J.C.D., Vogel J. The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc. Natl. Acad. Sci. U.S.A. 2012;109:E757–E764. doi: 10.1073/pnas.1119414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pfeiffer V., Sittka A., Tomer R., Tedin K., Brinkmann V., Vogel J. A small non-coding RNA of the invasion gene island (SPI-1) represses outer membrane protein synthesis from the Salmonella core genome. Mol. Microbiol. 2007;66:1174–1191. doi: 10.1111/j.1365-2958.2007.05991.x. [DOI] [PubMed] [Google Scholar]
- 79.Khalil A.S., Collins J.J. Synthetic biology: applications come of age. Nat. Rev. Genet. 2010;11:367–379. doi: 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chappell J., Takahashi M.K., Meyer S., Loughrey D., Watters K.E., Lucks J. The centrality of RNA for engineering gene expression. Biotechnol. J. 2013;8:1379–1395. doi: 10.1002/biot.201300018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mutalik V.K., Qi L., Guimaraes J.C., Lucks J.B., Arkin A.P. Rationally designed families of orthogonal RNA regulators of translation. Nat. Chem. Biol. 2012;8:447–454. doi: 10.1038/nchembio.919. [DOI] [PubMed] [Google Scholar]
- 82.Qi L., Lucks J.B., Liu C.C., Mutalik V.K., Arkin A.P. Engineering naturally occurring trans-acting non-coding RNAs to sense molecular signals. Nucleic Acids Res. 2012;40:5775–5786. doi: 10.1093/nar/gks168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu C.C., Qi L., Lucks J.B., Segall-Shapiro T.H., Wang D., Mutalik V.K., Arkin A.P. An adaptor from translational to transcriptional control enables predictable assembly of complex regulation. Nat. Methods. 2012;9:1088–1094. doi: 10.1038/nmeth.2184. [DOI] [PubMed] [Google Scholar]
- 84.Takahashi M.K., Lucks J.B. A modular strategy for engineering orthogonal chimeric RNA transcription regulators. Nucleic Acids Res. 2013;41:7577–7588. doi: 10.1093/nar/gkt452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qi L., Haurwitz R.E., Shao W., Doudna J.A., Arkin A.P. RNA processing enables predictable programming of gene expression. Nat. Biotech. 2012;30:1002–1006. doi: 10.1038/nbt.2355. [DOI] [PubMed] [Google Scholar]
- 86.Green Alexander A., Silver Pamela A., Collins James J., Yin P. Toehold switches: de-novo-designed regulators of gene expression. Cell. 2014;159:925–939. doi: 10.1016/j.cell.2014.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.