


Abstract
N6-methyladenosine (m6A) is the most abundant internal modification in eukaryotic messenger RNA (mRNA). Recent discoveries of demethylases and specific binding proteins of m6A as well as m6A methylomes obtained in mammals, yeast and plants have revealed regulatory functions of this RNA modification. Although m6A is present in the ribosomal RNA of bacteria, its occurrence in mRNA still remains elusive. Here, we have employed ultra-high pressure liquid chromatography coupled with triple-quadrupole tandem mass spectrometry (UHPLC-QQQ-MS/MS) to calculate the m6A/A ratio in mRNA from a wide range of bacterial species, which demonstrates that m6A is an abundant mRNA modification in tested bacteria. Subsequent transcriptome-wide m6A profiling in Escherichia coli and Pseudomonas aeruginosa revealed a conserved m6A pattern that is distinct from those in eukaryotes. Most m6A peaks are located inside open reading frames and carry a unique consensus motif of GCCAU. Functional enrichment analysis of bacterial m6A peaks indicates that the majority of m6A-modified genes are associated with respiration, amino acids metabolism, stress response and small RNAs, suggesting potential functional roles of m6A in these pathways.
INTRODUCTION
N6-methyladenosine (m6A) is the most frequent internal mRNA modification in a wide range of eukaryotes and certain viral RNAs (1–6). Modification of m6A is mediated by an N6-adenosine methyltransferase complex including a 70-kD SAM (S-adenosylmethionine)-binding subunit methyltransferase like 3 (METTL3, also called MT-A70), methyltransferase like 14 (METTL14) and Wilms tumor 1 associated protein (WTAP) (7). METTL3 and METTL14 form a heterodimer that catalyzes m6A RNA methylation, while WTAP interacts with the complex and affects the mRNA methylation (8). These methyltransferases play important roles in mouse embryonic stem cell differentiation and circadian rhythms (9–11) Fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) are m6A RNA demethylases, which are involved in mammalian development, RNA metabolism and fertility (12,13). The human YTH-domain family 2 (YTHDF2) has recently been shown to specifically bind m6A-modified mRNA and to promote the decay of the bound mRNA (14,15). These discoveries present the first examples of reversible RNA modification and reveal the unique regulatory functions of reversible m6A methylation on mRNA and non-coding RNAs.
Transcriptome-wide profiling of m6A distributions in mammals and yeast further characterize the dynamic nature of m6A modification, which is enriched around the stop codon and at 3′ UTRs, as well as in long internal exons and at the transcription start site (11,15,16). m6A in Arabidopsis thaliana is also enriched around the stop codon, 3′ UTRs and around the start codon (17). The majority of m6A-peaks in these organisms harbor a consensus motif of RRACU (R = A/G).
Although m6A has been well documented in the rRNA in bacteria, its presence on mRNA is still elusive. In Escherichia coli, A1618 and A2030 of 23S rRNA are methylated by methyltransferases RlmF and RlmJ, respectively (18,19). Both deletion and overexpression of rlmF result in a loss of cell fitness and growth defect (18), while an rlmJ mutant shows mild phenotypes under various growth conditions (19). Interestingly, the modifications of m2A or m8A on A1607, A2503 and A2508 play important roles in antibiotic resistance, an extensively studied subject in microbiology during the last 10 years (20).
In order to investigate the potential presence and functions of m6A in bacterial mRNA, we calculated the m6A/A ratio in mRNA from seven diverse bacterial species, which reveal that m6A is an abundant mRNA modification in Gram-negative bacteria. High-resolution transcriptome-wide m6A profiling in two model bacteria E. coli and Pseudomonas aeruginosa reveal a conserved and distinct m6A distribution pattern. Most m6A-modified genes are involved in energy metabolism and small RNAs, suggesting potential functional roles of m6A in these processes.
MATERIALS AND METHODS
Bacterial strains and mRNA purification
The strains and culture conditions used in this study are listed in Table 1. Total RNA was purified from bacterial pellets of 2-ml culture by using an RNeasy Mini Kit (Qiagen) that removes tRNA. Two micrograms of total RNA were applied to a MICROExpressTM Bacterial mRNA Enrichment Kit (Life technologies). A Ribo-Zero™ rRNA Removal Kit (Bacteria) (Epicentre) was used in order to further remove remaining rRNA. All procedures in the manufacturer's protocols were strictly followed. In order to verify the removal of rRNA, a qPCR (7300 Real-Time PCR System, Applied Biosystems) was done against the rRNA background in order to check relative enrichment levels. One nanogram of either total RNA or purified mRNA from E. coli was used per qPCR reaction (Power SYBR Real-Time PCR mater mix, Life technologies). The primers used were 5′-CTCCTACGGGAGGCAGCAG-3′ and 5′-GTATTACCGCGGCGGCTG-3′. The P. aeruginosa MPAO1 strain were cultured overnight at different temperatures (37, 40, 42 or 45°C) and then subjected to the mRNA purification protocol described above in the temperature variation studies.
Table 1. Strains and growth conditions.
| Strain | Growth condition |
|---|---|
| Escherichia coli K-12 (CGSC) | LB, 37°C overnight |
| E. coli K-12 rlmJ mutant (CGSC) | LB, 37°C overnight |
| E. coli K-12 rlmF mutant (CGSC) | LB, 37°C overnight |
| E. coli K-12 ksgA mutant (CGSC) | LB, 37°C overnight |
| E. coli 5α | LB, 37°C overnight |
| E. coli XL-blue | LB, 37°C overnight |
| Pseudomonas aeruginosa MPAO1 | LB, 37°C overnight |
| P. aeruginosa PA14 | LB, 37°C overnight |
| Pseudomonas syringae pv. tomato DC3000 | King's B medium, 28°C for 2 d |
| Staphylococcus aureus Newman | TSB medium, 37°C overnight |
| S. aureus USA100 | TSB medium, 37°C overnight |
| S. aureus USA400 | TSB medium, 37°C overnight |
| S. aureus USA700 | TSB medium, 37°C overnight |
| S. aureus COL | TSB medium, 37°C overnight |
| S. aureus RN4220 | TSB medium, 37°C overnight |
| Bacillus subtilis | LB, 37°C overnight |
| Anabaena sp. PCC 7120 | Z8 medium, 25°C overnight |
| Synechocystis sp. PCC 6803 | Z8 medium, 25°C overnight |
Ultra-high pressure liquid chromatography coupled with triple-quadrupole tandem mass spectrometry (UHPLC-QQQ-MS/MS) analysis for m6A/A ratio
The highly purified bacterial mRNA was subjected to an UHPLC-QQQ-MS/MS (Agilent) analysis. Two hundred ng of mRNA or rRNA (on the beads of the mRNA Enrichment Kit) were digested by nuclease P1 (2 U) in 40 μl of nuclease buffer (25 mM of NaCl and 2.5 mM of ZnCl2) at 37°C for 2 h, followed by the addition of NH4HCO3 (1 M, 2 μl) and alkaline phosphatase (0.5 U) at 37°C for 2 h. The nucleosides were separated by reverse phase ultra-performance liquid chromatography by a C18 column on an Agilent 6410 QQQ triple-quadrupole LC mass spectrometer in positive electrospray ionization mode. The nucleosides were quantified using the nucleoside-to-base ion mass transitions of 282 to 150 (m6A), 294 to 164 (m62A) and 268 to 136 (A). Quantification was performed by comparison with the standard curve obtained from pure nucleoside standards. Three biological repeats have been performed for all bacterial strains.
High-throughput and high-resolution m6A sequencing
Procedures were slightly modified from previously described protocols (21). In a 0.5-ml IP reaction, 5 μg purified bacterial mRNA and 15 μl of 0.5 mg/ml rabbit anti-m6A antibody (202003; Synaptic Systems) were incubated for 2 h at 4°C in IPP buffer (150 mM NaCl, 0.1% NP-40, 10 mM Tris–HCl, pH 7.4, 1 U/μl RNasin). The mixture was exposed to UV irradiation at 254 nm 3× (90 s each time), before RNase T1 (0.1 U/μl) digestion for 15 min at 22°C. After the digestion reaction was quenched on ice for 5 min, 200 μl pre-blocked protein A bead slurry was added into reaction for 1 h at 4°C. After washing thrice with IP wash buffer (50 mM HEPES-KOH, pH 7.5, 300 mM KCl, 0.05% NP-40, with proteinase inhibitor and RNasin), the beads were treated by a second round of RNase T1 digestion (15 U/μl) at 22°C for 15 min. The beads were cooled down on ice for 5 min and then thrice washed with high salt wash buffer (50 mM HEPES-KOH, pH 7.5, 500 mM KCl, 0.05% NP-40, with proteinase inhibitor and RNasin). The beads were then treated with Antarctic phosphatase (0.5 U/μl) for 20 min at 37°C. After dephosphorylation, beads were washed twice with phosphatase wash buffer (50 mM Tris–HCl, pH 7.5, 20 mM EGTA, 0.5% NP-40) and twice with PNK buffer without DTT (50 mM Tris–HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2). Polynucleotide kinase (1 U/μl) and 200 μM adenosine triphosphate was then added to the beads at 37°C for 15 min. The RNA fragments were further purified by proteinase K digestion and TRIzol extraction. For IP samples, small RNA libraries were made by using NEBNext® Small RNA Library Prep Set for Illumina® (NEB). The input samples followed the above procedures without anti-m6A antibody pull-down and RNase T1 digestion. Libraries for input samples were made by using TruSeq RNA Sample Preparation Kits (Illumina, non-strand specific). Six libraries were constructed, containing one control sample and two duplicate IP samples for each strain.
Data analyses
All samples were sequenced using the HiSeq 2000 system (Illumina, with 50-bp and single end mode) at the Genomics Core Facility at the University of Chicago. FastQC was done to check the quality of each dataset; all datasets were obtained in high quality and can afford further reliable analyses. Two E. coli IP libraries obtained 1,718,364 mapped reads, and two P. aeruginosa libraries obtained 3,720,658 mapped reads. Sequence data were analyzed by following the procedures described previously (17). Briefly, Tophat (version 2.0.0, with the parameter: -p 8 –read-mismatches 2 –max-multihits 1) with Bowtie was run in order to align the input and IP-sequenced samples to the E. coli K-12 substr. MG1655 (ASM584v2, NC_000913.3) and P. aeruginosa PAO1 (ASM676v1, NC_002516.2) genomes and annotation files (22,23). In TopHat each read was only mapped to the genome once. The enriched peaks were identified using MACS software (version 2.0.0, with the parameter: callpeak -t ip.bam -c ck.bam -f BAM -g 6000000 –nomodel -n -p 1e-5) (24). Consensus sequence motifs were identified by using HOMER (version 4.7, with the parameter: -p 3 -rna -len 6) (25). A scrambled sequence was used as the background. Gene function analysis (GO enrichment) was performed with the online DAVID (version 6.7) tool (http://david.abcc.ncifcrf.gov/) (26). The m6A peaks were divided into three categories based on their relative positions in their corresponding genes: Overlap Start (±100 nucleotides around the start codon), Overlap End (±100 nucleotides around the stop codon) and Inside (other locations inside a coding region). The functional association with each gene was determined by NCBI annotation.
RESULTS
m6A is presented in mRNA of a wide range of bacterial species
Although m6A is the most abundant internal mRNA modification in eukaryotes, its potential presence in the kingdom of bacteria has yet to be investigated. To this end, we selected seven diverse model bacterial species (E. coli, P. aeruginosa, Pseudomonas syringae, Staphylococcus aureus, Bacillus subtilis, Anabaena sp. PCC 7120 and Synechocystis sp. PCC 6803) to grow in a common laboratory environment and measured their m6A/A ratios in purified mRNA. Unlike eukaryotes, bacterial mRNA lacks a poly(A) tail, which makes it challenging to purify mRNA. By following the protocols described in the Method section, we were able to remove >90% rRNA in the purified mRNA sample (Supplementary Figure S1).
We then tried to use an UHPLC-QQQ-MS/MS approach in order to quantify the m6A/A level in the bacterial mRNA samples that contain residual rRNA (<10%). Given that two m6A (catalyzed by RlmF and RlmJ) and two N6,N6-dimethladenosine (m62A, catalyzed by KsgA) are known to be present in rRNA of E. coli and other related bacterial species (18,19,27), the values of m62A levels can be used as an internal reference for the m6A level from the residue of rRNA presented in the purified mRNA. We first determined the m62A/m6A ratio of rRNA as 1.30 in the wild-type strain and 2.04 in either an rlmF mutant or an rlmJ mutant. As a negative control, the m62A modification was not detectable in a ksgA mutant (Supplementary Figure S2). Based on the m62A/m6A ratio in rRNA, we were able to accurately calculate the real m6A/A level as ([m6A-m62A/1.30]/A) in the purified mRNA samples.
We observed the presence of m6A in all of the tested bacterial species, whose m6A/A ratio varied within the range of 0.02–0.28% (Figure 1A). We obtained the m6A/A ratios in mRNA from three Gram-negative bacteria (E. coli, P. aeruginosa and P. syringae) (>0.2%) and from two Gram-positive bacteria (S. aureus and B. subtilis) (<0.08%). Unlike E. coli and Pseudomonas spp., two other Gram-negative cyanobacteria (Anabaena sp. PCC 7120 and Synechocystis sp. PCC 6803) showed low m6A/A ratios (<0.04%). In order to test if m6A/A ratios also vary among different strains in the same species, three strains of E. coli (K-12, 5α and XL-blue), two strains of P. aeruginosa (MPAO1 and PA14) and six strains of S. aureus (Newman, USA100, USA400, USA700, RN4220 and COL) were tested, all of which revealed a constant ratio in the same species (Figure 1B). These results clearly demonstrate the widespread occurrence of m6A in bacterial mRNA. Gram-negative bacteria tend to have higher m6A/A ratios in mRNA than Gram-positive bacteria. The high m6A/A ratio (>0.2%) in mRNA from E. coli and Pseudomonas spp. is comparable to that from eukaryotes (1), suggesting that m6A could be an important mRNA modification playing functional roles in these and other bacteria.
Figure 1.

Presence of m6A in bacterial mRNA. (A) The m6A/A ratios of mRNA isolated from seven bacterial species. (B) The m6A/A ratios of mRNA isolated from different strains of Escherichia coli and Pseudomonas aeruginosa. Error bars represent standard deviations, which were calculated from three replicates.
m6A distribution exhibits a distinct topology in E. coli
To obtain the transcriptome-wide m6A map of E. coli, we employed an m6A-specific antibody for pull-down coupled with high-throughput sequencing (15,16). In order to obtain a high-resolution m6A-map, bacterial mRNAs were subjected to a modified photo-crosslinking-assisted m6A-seq approach (PA-m6A-seq), which significantly improves the m6A peak resolution from ∼200 nt to around 23 nt (21). In total, we identified 265 m6A peaks representing the transcripts of 213 genes in E. coli (Supplementary Table S1).
We next analyzed the distribution of m6A in the whole transcriptome of E. coli. We determined the distribution of m6A reads along transcripts in both the m6A-IP and non-IP (input) samples. Intriguingly, we found that reads from m6A-IP tend to be equally distributed throughout a gene, with a peak in the middle of open reading frames (ORFs) (Figure 2A). The prevalence is quite different from that observed in mammals, which accumulates around the stop codon and within 3′ UTRs (15,16). To further confirm the preferential locations of m6A on transcripts, we investigated the metagene profiles of m6A peaks. Consistent with the distribution of reads, m6A peaks are abundant inside ORF (72%), followed by regions at the start of gene (15%) and then the end of gene (13%) (Figure 2B).
Figure 2.

Overview of m6A methylome in Escherichia coli. (A) The m6A peak distribution within different gene contexts. The y-axis represents (number of reads/length unit)/(number of total reads), which is an indicator of the extent to which sequencing reads are enriched in different segments across the entire transcript. (B) Accumulation of m6A reads along transcripts. Each transcript is divided into three parts: Overlap Start, Inside and Overlap End. (C) The UGCCAG conserved sequence motif for m6A-containing peak regions. (D) Pie chart displaying the percentage of genes containing m6A peaks with functional categories. (E) GO-enrichment analysis of all the genes with m6A peaks. The effect size (number of enriched genes/total genes in the GO category) for each category is 15/45 (aerobic respiration), 15/122 (amino acids biosynthesis), 6/138 (response to abotic stimulus), 5/61 (cell wall biosynthesis) and 6/39 (anaerobic respiration), respectively. The statistical test (P-value) used by DAVID was the Fisher Exact test.
We then used the HOMER tool to identify a leading m6A consensus sequence (UGCCAG, P < 1e-14), which could be found in more than 41.2% of all m6A peaks (Figure 2C and Supplementary Table S1) (25). This motif is different from the conserved one (RRACU, R = A/G) found in eukaryotes. The unique feature of the m6A distribution suggests a likely unique role of m6A in perhaps bacteria-specific pathways.
m6A-containing mRNAs in important biological pathways in E. coli
Diverse functions are encoded by m6A-containing genes, which include metabolism (52%), transportation (12%), gene regulation (11%), cell envelope (5%), ribosome (3%), nucleic acids (1%), stress response (5%), genes with unknown function/annotation (6%) and pseudogenes (2%) (Figure 2D). To further uncover potential functional insights on m6A in E. coli, we selected 213 m6A-containing transcripts and identified the enriched gene ontology (GO) terms using the DAVID tool (26). We found that these genes are highly enriched in aerobic respiration, amino acids biosynthesis, response to abiotic stresses, cell wall biosynthesis and anaerobic respiration (Figure 2E). The first two classes belong to housekeeping genes that are involved in central energy production and metabolism, while the latter three classes are bacteria-specific categories. The m6A distribution pattern suggests that m6A may play roles in these important biological pathways. For instance, hydrogenase 1 mediates hydrogen uptake and transport in the process of anaerobic respiration. Four (hyaA, hyaB, hyaC and hyaD) genes of the six-gene-operon encoding hydrogenase 1 contain multiple m6A peaks inside the transcript, suggesting a clustering of m6A in this operon (Figure 3A) (28). We also observed concentrated m6A peaks in gabD and gabT genes, which encode succinate-semialdehyde dehydrogenase and 4-aminobutyrate aminotransferase in the pathway of amino acid metabolism (Figure 3B) (29).
Figure 3.

Accumulation of m6A reads in hyaABCD genes (A), gabDT (B) and lacZI(C) in Escherichia coli transcriptome. CK represents the control sample and IP represents ChIP-seq sample.
We next sought to determine if the unique m6A position patterns are related to bacteria-specific GO categories. As aforementioned, we classified genes into three subgroups according to the location of m6A peaks on a gene: Overlap Start (m6A peaks within 100-bp from the start codon), Overlap End (m6A peaks within 100-bp from the stop codon) and Inside (m6A peaks inside the coding region) (Figure 2B). We then performed GO-enrichment analysis for each subgroup. As expected, the same five GO categories were enriched in the Inside subgroup that consists of 72% of all m6A peaks. Two GO categories (aerobic respiration and stress responses) were enriched in the Overlap Start subgroup, while amino acids biosynthesis and response to stimulus were enriched in the Overlap End subgroup (Supplementary Figure S3).
Beside these five GO categories, we identified high m6A peaks in a group of functionally important genes, such as lacZ and lacI (Figure 3C). LacI negative regulates the classic lac operon (lacZYA) that is required for transport and metabolism of lactose (30). Interestingly, we also noticed 15 small RNAs carrying m6A modification (Supplementary Table S1). These newly found m6A marks in these transcripts could open a new angle to study novel regulatory roles in well-established pathways.
Unique patterns of P. aeruginosa methylome
Given that P. aeruginosa, a widely-spread human opportunistic pathogen, also possesses a high m6A/A ratio in mRNA, we applied the same modified photo-crosslinking-assisted m6A-seq approach to obtain a high-resolution map of its m6A methylome. We identified 109 m6A peaks representing the transcripts of 68 genes in P. aeruginosa (Supplementary Table S2). The m6A-modified transcripts identified are around half of those in E. coli.
We next determined the distribution of m6A reads along transcripts in both the m6A-IP and non-IP (input) samples. Like in E. coli, we found that reads from m6A-IP are equally distributed throughout a gene, with two peaks in the middle of ORFs as well as in the beginning of genes (Figure 4A). m6A peaks are abundant inside ORF (77%), followed by the start of gene regions (15%) and end of gene regions (8%) (Figure 4B). We were also able to identify an m6A consensus sequence (GGYCAG, Y = C/U P < 1e-16), which were found in >70% of all m6A peaks (Figure 4C and Supplementary Table S2). This motif is almost identical to the one in E. coli (UGCCAG). A similar feature of the m6A distribution in both E. coli and P. aeruginosa indicates that m6A possesses functions unique to these bacteria.
Figure 4.

Overview of m6A methylome in Pseudomonas aeruginosa. (A) The m6A peak distribution within different gene contexts. The y-axis represents (number of reads/length unit)/(number of total reads), which is an indicator of the extent to which sequencing reads are enriched in different segments across the entire transcript. (B) Accumulation of m6A reads along transcripts. Each transcript is divided into three parts: Overlap Start, Inside and Overlap End. (C) The GGCCAG conserved sequence motif for m6A-containing peak regions. (D) Pie chart displaying the percentage of genes containing m6A peaks with functional categories. (E) KEGG-enrichment analysis of all the genes with m6A peaks. The effect size (number of enriched genes/total genes in the KEGG category) for each category is 7/148 (amino acids metabolism), 4/37 (glycolysis) and 3/56 (TCA cycle), respectively.
The m6A-containing genes cover different gene categories in P. aeruginosa, including metabolism (40%), gene regulation (8%), transportation (1%), virulence (4%), ribosome (12%), stress response (4%) and genes with unknown function/annotation (31%) (Figure 4D). DAVID analysis revealed significant GO enrichments in amino acids metabolism, glycolysis, and tricarboxylic acid (TCA) cycle (Figure 4E), all of which belong to housekeeping genes that are involved in central energy production and metabolism. The m6A pattern suggests that m6A may play roles in these essential pathways in P. aeruginosa. For instance, three adjacent genes (PA3415-PA3417 encoding branched-chain alpha-keto acid dehydrogenase, pyruvate dehydrogenase β and α subunit, respectively) involved in glycolysis and TCA carry numerous m6A peaks (Figure 5A) (31). We also observed multiple m6A peaks in the next downstream gene ldh, which encodes leucine dehydrogenase in the pathway of amino acid metabolism (Figure 5A). Beside these housekeeping genes, we identified high m6A peaks in two important small RNAs, namely RsmY and RsmZ, as well as in two key virulence genes rhlA and rhlB (Figure 5B).
Figure 5.

Accumulation of m6A reads in PA3415–3417 and ldh (A), rsmY (B) and rsmZ (C) in Pseudomonas aeruginosa transcriptome. CK represents control sample and IP represents ChIP-seq sample.
Temperature tunes m6A level in P. aeruginosa
In humans and mice, dynamic changes of certain m6A peaks have been observed under different stress conditions, indicating a link between m6A and stress response (15). In order to test if this trend also exists in bacteria, we measured the m6A/A ratios under a variety of growth environments or stress conditions (such as varying temperatures, different growth media, exposure to different antibiotics and oxidative stresses) for both E. coli and P. aeruginosa. For most tested conditions, we did not observe a significant difference in the m6A/A level compared to the normal condition (LB, mid-log phase, 37°C) for both bacteria. Interestingly, we noticed that increasing the culture temperature (from 37–45°C) led to a clear decrease in the m6A/A ratio in P. aeruginosa (Figure 6). Although P. aeruginosa still slowly grew, particularly at 45°C, the m6A modification was almost abolished.
Figure 6.
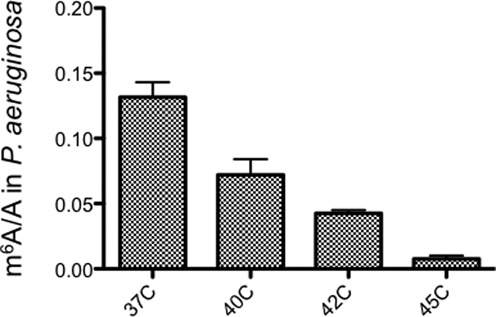
Growth temperature significantly affects the m6A/A ratio in Pseudomonas aeruginosa. Error bars are calculated from three replicates.
DISCUSSION
Recent discoveries and characterization of m6A erasers (demethylase), binders (m6A-specific binding protein) and writers (methyltransferase) as well as advances in profiling the m6A methylomes in eukaryotic systems reveal that m6A is a reversible and dynamic modification with important regulatory functions (1). On the other hand, the m6A methylomes in bacterial mRNA remain poorly studied. Here, we report the presence of m6A in a wide range of bacterial species and the ratio of m6A/A in mRNA from diverse bacterial strains vary within the range of 0.02–0.28%. We noticed that S. aureus and B. subtilis showed very low m6A/A ratios, which suggests that they may not possess an m6A methylase that could be present in the Gram-negative bacteria. Based on the genome annotation in NCBI, there are at least 43 proteins containing a S-adenosyl methionine (SAM)-binding domain in E. coli K-12, but only 24 in S. aureus USA300. We further present the high-resolution, transcriptome-wide m6A distributions in E. coli and human pathogen P. aeruginosa, which contain 265 and 109 peaks, respectively.
In order to provide additional insights into the overall m6A patterns in the bacterial kingdom, we compared these two newly identified methylomes. E. coli and P. aeruginosa share many similarities in their m6A distributions that are distinct from those of mammals: (i) a similar motif GCCAG instead of RRACU motif in mammals; (ii) most peaks are in the middle of coding region, while mammalian m6A peaks enrich around the stop codon and at 3′ UTRs; (iii) enrichment of GO categories of energy and amino acids metabolism; (iv) many small noncoding RNAs were found to carry m6A for both organisms. These shared characteristics suggest that other bacterial species, especially Gram-negative bacteria, may have similar m6A characteristics in mRNA. The new consensus sequence (GCCAG) is different from known rRNA methylation sites, including the two m6A sites on rRNA (CACA*GGU for RlmF and GUGA*AGA for RlmJ) and one on tRNAval (UACA*AGG for YfiC). Given that our recent study demonstrates that m6A and its specific binding protein, YTHDF2, affect the translation status and lifetime of mRNA in eukaryotes (14), m6A may play a similar role in bacteria.
On the other hand, there are clear species-specific features of the m6A distribution between these two bacteria. Although the two species share major GO categories (energy and protein metabolism), we observed a very low rate of overlapping genes. Besides rRNA genes that are previously known to carry m6A modification, only one gene (aceA encoding isocitrate lyase) was shared between E. coli and P. aeruginosa. Each bacterium has distinct functional categories of mRNAs that carry m6A. For example, genes involved in cell wall biosynthesis and anaerobic respiration are enriched in the E. coli methylome only, while a group of virulence genes (RsmYZ and rhlAB) are enriched in the P. aeruginosa methylome (Figure 5B and C). RsmYZ binds to RsmA and dissociates RsmA away from its mRNA targets, which in turn tunes a group of important virulence pathways including Type III secretion system (T3SS) and biofilm formation (32,33). rhlAB encodes rhamnosyltransferase, producing rhamnolipid biosurfactants that are involved in uptake of hydrophobic substrates, virulence, biofilm and antibiotic resistance (34). m6A marks in these virulence genes could connect RNA modification to bacterial pathogenesis. Our result also suggests a relationship between m6A and P. aeruginosa adaption to temperature changes. Alternatively, the putative m6A methylase in P. aeruginosa may be inactive at high temperature.
Given that multiple known rRNA or tRNA adenine methylases have been characterized in E. coli, we measured the m6A/A ratios in two methylase mutants, rlmF and rlmJ. However, the ratios were not significantly lower than the wild-type strain, suggesting that they are not mRNA methyltransferases (Supplementary Figure S4). As a negative control, the ksgA mutant lost the m62A modification and showed a higher m6A/A ratio than the other strains, suggesting a small content of rRNA in the mRNA sample. We also tried to look for bacterial homologs of mammalian m6A methyltransferases (METTL3, METTL14 and WTAP), but could not identify one, reminiscent of a distinct bacterial m6A motif that is different from that of mammals. These results suggest that bacterial m6A modification in mRNA is possessing of a mechanism that differs from eukaryotes.
Recent m6A profiling in yeast revealed dynamic changes in methylation during meiosis, which led us to test if bacterial m6A patterns also vary in different growth stage (11). To this end, we measured the m6A/A ratios during lag phase, log phase, stationary phase and death phase for both E. coli and P. aeruginosa. No significant difference was recorded throughout the bacterial growth curve, which indicates that m6A is a stable modification during bacterial growth.
The first bacterial m6A maps presented here provide a starting roadmap for uncovering bacterial distinct m6A functions in the future. Recent breakthroughs in the characterization of m6A-associated proteins as well as in the development of high-throughput assays in mammals present a very useful toolbox for us to study m6A in bacteria. Given the high abundance of m6A in numerous bacterial species, we foresee unique functions of m6A modification in mRNA in the wide bacteria kingdom.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACCESSION NUMBER
m6A-seq data are available in the National Center for Biotechnology Information Gene Expression Omnibus under series GSE65347.
FUNDING
US National Institutes of Health [AI074658 to C.H.]. Project of Tianjin, China (13TXSYJC40100). Funding for open access charge: US National Institutes of Health [AI074658 to C.H.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Fu Y., Dominissini D., Rechavi G., He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat. Rev. Genet. 2014;15:293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 2.Machnicka M.A., Milanowska K., Osman Oglou O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M., et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyer K.D., Jaffrey S.R. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat. Rev. Mol. Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhong S., Li H., Bodi Z., Button J., Vespa L., Herzog M., Fray R.G. MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell. 2008;20:1278–1288. doi: 10.1105/tpc.108.058883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clancy M.J., Shambaugh M.E., Timpte C.S., Bokar J.A. Induction of sporulation in Saccharomyces cerevisiae leads to the formation of N6-methyladenosine in mRNA: a potential mechanism for the activity of the IME4 gene. Nucleic Acids Res. 2002;30:4509–4518. doi: 10.1093/nar/gkf573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krug R.M., Morgan M.A., Shatkin A.J. Influenza viral mRNA contains internal N6-methyladenosine and 5′-terminal 7-methylguanosine in cap structures. J. Virol. 1976;20:45–53. doi: 10.1128/jvi.20.1.45-53.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., et al. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 2014;10:93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li R., Yang Y.G., Gao Y., Wang Z.Q., Tong W.M. A distinct response to endogenous DNA damage in the development of Nbs1-deficient cortical neurons. Cell Res. 2012;22:859–872. doi: 10.1038/cr.2012.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y., Li Y., Toth J.I., Petroski M.D., Zhang Z., Zhao J.C. N6-methyladenosine modification destabilizes developmental regulators in embryonic stem cells. Nat. Cell Biol. 2014;16:191–198. doi: 10.1038/ncb2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Batista P.J., Molinie B., Wang J., Qu K., Zhang J., Li L., Bouley D.M., Lujan E., Haddad B., Daneshvar K., et al. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz S., Agarwala S.D., Mumbach M.R., Jovanovic M., Mertins P., Shishkin A., Tabach Y., Mikkelsen T.S., Satija R., Ruvkun G., et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng G., Dahl J.A., Niu Y., Fedorcsak P., Huang C.M., Li C.J., Vagbo C.B., Shi Y., Wang W.L., Song S.H., et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang X., Lu Z., Gomez A., Hon G.C., Yue Y., Han D., Fu Y., Parisien M., Dai Q., Jia G., et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 16.Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo G.Z., MacQueen A., Zheng G., Duan H., Dore L.C., Lu Z., Liu J., Chen K., Jia G., Bergelson J., et al. Unique features of the m(6)A methylome in Arabidopsis thaliana. Nat. Commun. 2014;5:5630. doi: 10.1038/ncomms6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sergiev P.V., Serebryakova M.V., Bogdanov A.A., Dontsova O.A. The ybiN gene of Escherichia coli encodes adenine-N6 methyltransferase specific for modification of A1618 of 23 S ribosomal RNA, a methylated residue located close to the ribosomal exit tunnel. J. Mol. Biol. 2008;375:291–300. doi: 10.1016/j.jmb.2007.10.051. [DOI] [PubMed] [Google Scholar]
- 19.Golovina A.Y., Dzama M.M., Osterman I.A., Sergiev P.V., Serebryakova M.V., Bogdanov A.A., Dontsova O.A. The last rRNA methyltransferase of E. coli revealed: the yhiR gene encodes adenine-N6 methyltransferase specific for modification of A2030 of 23S ribosomal RNA. RNA. 2012;18:1725–1734. doi: 10.1261/rna.034207.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poehlsgaard J., Douthwaite S. The bacterial ribosome as a target for antibiotics. Nat. Rev. Microbiol. 2005;3:870–881. doi: 10.1038/nrmicro1265. [DOI] [PubMed] [Google Scholar]
- 21.Chen K., Lu Z., Wang X., Fu Y., Luo G.Z., Liu N., Han D., Dominissini D., Dai Q., Pan T., et al. High resolution N6-Methyladenosine (m6A) map using photo-crosslinking-assisted m6A-Seq. Angew. Chem. Int. Ed. Engl. 2015;127:1607–1610. doi: 10.1002/anie.201410647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8:R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham P.R., Weitzmann C.J., Nurse K., Masurel R., Van Knippenberg P.H., Ofengand J. Site-specific mutation of the conserved m6(2)A m6(2)A residues of E. coli 16S ribosomal RNA. Effects on ribosome function and activity of the ksgA methyltransferase. Biochim. Biophys. Acta. 1990;1050:18–26. doi: 10.1016/0167-4781(90)90135-o. [DOI] [PubMed] [Google Scholar]
- 28.Menon N.K., Robbins J., Wendt J.C., Shanmugam K.T., Przybyla A.E. Mutational analysis and characterization of the Escherichia coli hya operon, which encodes [NiFe] hydrogenase 1. J. Bacteriol. 1991;173:4851–4861. doi: 10.1128/jb.173.15.4851-4861.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartsch K., von Johnn-Marteville A., Schulz A. Molecular analysis of two genes of the Escherichia coli gab cluster: nucleotide sequence of the glutamate:succinic semialdehyde transaminase gene (gabT) and characterization of the succinic semialdehyde dehydrogenase gene (gabD) J. Bacteriol. 1990;172:7035–7042. doi: 10.1128/jb.172.12.7035-7042.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Beckwith J.R. Regulation of the lac operon. Recent studies on the regulation of lactose metabolism in Escherichia coli support the operon model. Science. 1967;156:597–604. doi: 10.1126/science.156.3775.597. [DOI] [PubMed] [Google Scholar]
- 31.Winsor G.L., Lam D.K., Fleming L., Lo R., Whiteside M.D., Yu N.Y., Hancock R.E., Brinkman F.S. Pseudomonas Genome Database: improved comparative analysis and population genomics capability for Pseudomonas genomes. Nucleic Acids Res. 2011;39:D596–D600. doi: 10.1093/nar/gkq869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valverde C., Heeb S., Keel C., Haas D. RsmY, a small regulatory RNA, is required in concert with RsmZ for GacA-dependent expression of biocontrol traits in Pseudomonas fluorescens CHA0. Mol. Microbiol. 2003;50:1361–1379. doi: 10.1046/j.1365-2958.2003.03774.x. [DOI] [PubMed] [Google Scholar]
- 33.Intile P.J., Diaz M.R., Urbanowski M.L., Wolfgang M.C., Yahr T.L. The AlgZR two-component system recalibrates the RsmAYZ posttranscriptional regulatory system to inhibit expression of the Pseudomonas aeruginosa type III secretion system. J. Bacteriol. 2014;196:357–366. doi: 10.1128/JB.01199-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ochsner U.A., Fiechter A., Reiser J. Isolation, characterization, and expression in Escherichia coli of the Pseudomonas aeruginosa rhlAB genes encoding a rhamnosyltransferase involved in rhamnolipid biosurfactant synthesis. J. Biol. Chem. 1994;269:19787–19795. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.