


Abstract
Smarcal1 is a SWI/SNF-family protein with an ATPase domain involved in DNA-annealing activities and a binding site for the RPA single-strand-DNA-binding protein. Although the role played by Smarcal1 in the maintenance of replication forks has been established, it remains unknown whether Smarcal1 contributes to genomic DNA maintenance outside of the S phase. We disrupted the SMARCAL1 gene in both the chicken DT40 and the human TK6 B cell lines. The resulting SMARCAL1−/− clones exhibited sensitivity to chemotherapeutic topoisomerase 2 inhibitors, just as nonhomologous end-joining (NHEJ) null-deficient cells do. SMARCAL1−/− cells also exhibited an increase in radiosensitivity in the G1 phase. Moreover, the loss of Smarcal1 in NHEJ null-deficient cells does not further increase their radiosensitivity. These results demonstrate that Smarcal1 is required for efficient NHEJ-mediated DSB repair. Both inactivation of the ATPase domain and deletion of the RPA-binding site cause the same phenotype as does null-mutation of Smarcal1, suggesting that Smarcal1 enhances NHEJ, presumably by interacting with RPA at unwound single-strand sequences and then facilitating annealing at DSB ends. SMARCAL1−/−cells showed a poor accumulation of Ku70/DNA-PKcs and XRCC4 at DNA-damage sites. We propose that Smarcal1 maintains the duplex status of DSBs to ensure proper recruitment of NHEJ factors to DSB sites.
INTRODUCTION
Smarcal1 (a SWI/SNF-related, matrix associated, actin-dependent regulator of chromatin a-like 1) is a SWI/SNF family protein that carries an ATPase domain and the binding site for replication protein A (RPA), the single-strand-DNA-binding protein (1,2). Smarcal1 is an ATP-driven annealing helicase that catalyzes the formation of double-strand DNA from complementary single-strand DNA strands associated with RPA. Thus, Smarcal1 could counteract unwinding by DNA helicases at DSB sites. The annealing activity is stimulated by substrate DNA containing both single-strand and double-strand regions, such as a chicken foot structure (3). Mutations in the SMARCAL1 gene cause a rare autosomal recessive disease, Schimke immuno-osseous dysplasia (SIOD), which is characterized by short stature, kidney disease and a severely compromised immune system (4–7). Phenotypic analysis of Smarcal1-depleted cells suggests that Smarcal1 stabilizes replication forks when cells are exposed to aphidicolin, hydroxyurea and camptothecin (a topoisomerase 1 poison) (1,2,8–10).
The two major double-strand-break (DSB) repair pathways, homologous recombination (HR) and nonhomologous end-joining (NHEJ) (11–13) significantly contribute to cellular tolerance to anti-malignant therapies. First, both pathways contribute to cellular tolerance to radiotherapy, HR in the S to G2 phases and NHEJ throughout the cell cycle. Second, HR plays the dominant role in repairing DSBs generated during DNA replication by chemotherapeutic agents such as camptothecin and poly[ADP ribose]polymerase inhibitor (olaparib). These chemotherapeutic agents cause the accumulation of single-strand breaks, which are converted by DNA replication to DSBs called one-end breaks. These DSBs are repaired by HR but not by NHEJ (14–16). Third, NHEJ plays the dominant role in repairing DSBs caused by chemotherapeutic topoisomerase 2 inhibitors such as ICRF193 and etoposide (15,17). Measuring the sensitivity of gene-disrupted cells to various anti-malignant therapies allows us to define the role of the gene in HR, NHEJ or both. In addition to the above, the capability of canonical NHEJ is evaluated by examining the V(D)J recombination of Immunoglobulin (Ig) V genes, which requires a collaboration between NHEJ and V(D)J recombinase encoded by the recombination-activating-genes 1 and 2 (Rag1/Rag2) (18–20).
Canonical NHEJ is initiated by associating a Ku70/Ku80 heterodimer with DSB sites. Ku70/Ku80 associates preferentially with duplex DNA ends, rather than with DSBs carrying single-strand tails generated by exonucleases or DNA helicases (21–24). Ku70/Ku80 forms a complex with DNA-dependent-protein-kinase catalytic subunit (DNA-PKcs), leading to the activation of DNA-PKcs at DSB sites (25–27). DNA-PKcs phosphorylates a number of substrates, including itself (28–31). Ligase4 (Lig4) completes DSB repair in collaboration with the essential co-factors, XLF and XRCC4, which form clamp-like structures along duplex DNA (32–35). If canonical NHEJ does not perform DSB repair, non-canonical end-joining such as microhomology-mediated alternative end-joining (MMEJ) repairs DSBs, though less efficiently than canonical NHEJ, causing deletion near the DSB sites (36,37).
We disrupted the SMARCAL1 gene in the chicken DT40 and human B lymphoblastoid TK6 cell lines (38,39). The resulting SMARCAL1−/− clones exhibited sensitivity to camptothecin, suggesting that Smarcal1 plays a role in DNA replication, as indicated previously (9,10). Remarkably, Smarcal1 is also required for efficient NHEJ in human as well as in chicken cells. This conclusion is in agreement with the fact that SIOD patients exhibit reduced V(D)J recombination products in peripheral lymphocytes as well as increased chromosomal breakage (40,41). We propose that the decreased efficiency of NHEJ in V(D)J recombination as well as the compromised maintenance of replication fork progression result in severe lymphocytopenia in SIOD patients (4,40,41).
MATERIALS AND METHODS
Cell clones
All the clones used in this study are summarized in Table 1.
Table 1. Panel of cell lines used in this study.
| Genotype | Parental Cell Line | Markers genes | References |
|---|---|---|---|
| SMARCAL1−/− | DT40 | hisD, puroR | * |
| SMARCAL1Δ30/− | DT40 | hisD | * |
| KU70−/− | DT40 | bsrR, puroR | (55) |
| BRCA2−/− | DT40 | hygroR, hisD | (46) |
| KU70−/−/SMARCAL1Δ30/− | DT40 | hisD, bsrR, puroR | * |
| SMARCAL1−/− | TK6-derived TSCER2 and TSCE5 | puroR, hygroR | * T |
| LIG4−/−/− | TK6-derived TSCER2 and TSCE5 | puroR, neoR | * C |
| RAD54−/− | TK6-derived TSCER2 | puroR, neoR | * T |
| SMARCAL1−/−/LIG4−/−/− | TK6-derived TSCE5 | puroR, neoR, hygroR | * T/C |
| DNA-PKcs−/− | TK6-derived TSCE5 | neoR, hisD | * C |
| SMARCAL1−/−/DNA-PKcs−/− | TK6-derived TSCE5 | neoR, hisD | * T/C |
* = This study; T = TALEN; C = CRISPR.
Cell culture
DT40 and TK6 cells were cultured in the same manner as described previously (39,42).
Generation of SMARCAL1−/− DT40 cells
SMARCAL1 gene disruption constructs were generated from genomic polymerase chain reaction (PCR) products combined with histidinol dehydrogenase (hisD) and puromycin-resistance (puroR) marker genes. Genomic DNA from wild-type cells was amplified using the F1 and R1 primers for the 5′-arm and the F2 and R2 primers for the 3′-arm. The amplified 3′-arm PCR product was subcloned into pCR2.1-TOPO vector (Invitrogen, US). The 5′-arm PCR product harboring the SacI and BamHI sites at the 5′- and 3′-ends, respectively, was cloned into the SacI and BamHI sites of the pCR2.1-TOPO vector carrying the 3′-arm. The BamHI fragment containing either the hisD or puroR gene was cloned into the BamHI site between the 3′-arm and the 5′-arm in the pCR2.1-TOPO vector. To generate SMARCAL1−/− cells, the SMARCAL1 gene-disruption constructs carrying hisD and puroR were linearized, using the NotI restriction enzyme, and sequentially transfected by electroporation (Bio-Rad, US). A 0.5 kb probe was generated by PCR of genomic DNA using primers F3 and R3 for Southern blot analysis. The genomic DNA of the candidate clones was digested with SmaI and XhoI for Southern blot analysis. The gene disruption was confirmed by RT-PCR, using primers F4 and R4. When generating SMARCAL1+/− clones from wild-type cells the targeting efficiency was 20% (4/20), while that of generating SMARCAL1−/− cells from SMARCAL1+/− cells was 1.4% (1/71). All primers used here are shown in Supplemental Table S1.
Generation of SMARCAL1Δ30/− and KU70−/−/SMARCAL1Δ30/− DT40 cells
The intact allele of the SMARCAL1+/− cell was targeted for deleting the first 30 amino acids, which domain is responsible for RPA binding (10). To generate the Δ30 construct, the 5′-arm was amplified from genomic DNA using the NotI-tagged F5 and the BamHI-tagged R5, and the 3′-arm was amplified using the BamHI-tagged F6, which was designed from 90 bases downstream of start codon ATG in exon1, and the SalI-tagged R6. In the Zero Blunt TOPO vector (Invitrogen, US), the left and right arms were cloned at the site of NotI-BamHI and BamHI-SalI, respectively. The selection-marker gene, puromycin resistance (puroR) flanked by loxP sequences, was then inserted into the BamHI site between the left and right arms. The resulting Δ30-puroR construct was transfected into the SMARCAL1+/− and KU70−/−/SMARCAL1+/− cells. The puroR gene was popped out by transient expression of cre-recombinase, resulting in the generation of SMARCAL1Δ30/− and KU70−/−/SMARCAL1Δ30/− DT40 cells. To confirm the gene disruption by RT-PCR, the primer set F4 and R4 was used.
Generation of human SMARCAL1−/− and SMARCAL1−/−/LIG4−/−/− TK6 B cells
To generate a pair of TALEN expression plasmids against the SMARCAL1 gene, we used a Golden Gate TALEN kit and a TAL effector kit (Addgene, US) (43,44). The TALEN target sites are shown in Supplementary Figure S2. The gene-targeting constructs were generated from the genomic DNA of TK6 cells by amplifying with primers XhoI-flanked F7 and NheI-flanked R7 for the 5′-arm and NotI-flanked F8 and HindIII-flanked R8 for the 3′-arm. The 5′-arm PCR products were cloned into the XhoI and NheI sites found upstream of the puroR and hygroR marker genes of the DT-ApA/puro and DT-ApA/hygro vectors, respectively. The 3′-arm PCR products were cloned into the NotI and HindIII sites found downstream of the puroR and hygroR marker genes of the DT-ApA/puro and DT-ApA/hygro vectors, respectively. 6 μg TALEN-expression plasmids and 2 μg gene-targeting vectors were transfected into 4×106 TK6 cells using the Neon Transfection System (Life Technologies, US) with 3X pulse at 1350 V and with 10 ms pulse width. After electroporation, cells were released into 20 ml drug-free medium containing 10% horse serum. Forty-eight hours later, cells were seeded into 96-well plates with both hygromycin and puromycin antibiotics for two weeks. The genomic DNAs of the isolated clones resistant to both hygromycin and puromycin were digested with XbaI for Southern blot analysis. A 0.6 kb probe was generated by PCR of genomic DNA using primers F9 and R9. The loss of Smarcal1-protein expression was confirmed by western blot analysis (Supplementary Figure S2B). The efficiency of generating SMARCAL1−/−clones from wild-type cells was 100% (3/3). The method for generating LIG4−/−/− and RAD54−/− TK6 cells is described in the Supplemental Materials and Methods. SMARCAL1−/−/LIG4−/−/− clones were generated by disrupting the LIG4 gene in the SMARCAL1−/− cells. Gene-targeting efficiency was 10% (2/20).
Generation of human DNA-PKcs−/− and SMARCAL1−/−/ DNA-PKcs−/− TK6 B cells
To disrupt the DNA-PKcs gene, we designed a guide RNA targeting the 32nd exon using the Zhang CRISPR tool (45) and gene-targeting constructs. The CRISPR-target site is depicted in Supplementary Figure S3E. The gene-targeting constructs were generated using SLiCE (Seamless Ligation Cloning Extract). The genomic DNA was amplified with primers F19 and R19 from the DNA-PKcs-gene locus and the PCR product was used as template DNA for amplifying the 5′- and 3′-arms. The 5′-arm was amplified using primers F20 and R20 and the 3′-arm was amplified using primers F21 and R21, where each primer shared 20-base pair-end homology with the insertion site of the vector. Both vectors, DT-ApA/neo and DT-ApA/his, were linearized with AflII and ApaI. All the fragments of the vectors and inserts were purified using a qiaquick gel extraction kit (QIAGEN, Netherlands). The gene-targeting constructs were generated in a single reaction mixture containing DT-ApA/neo or DT-ApA/his vectors, 5′- and 3′-arms, and 2×SLiCE buffer (Invitrogen, US) and incubated for 30 min at room temperature. 6 μg of CRISPR and 2 μg of each gene-targeting vector were transfected into 4×106 TK6 cells using the Neon Transfection System (Life Technologies, US). After electroporation, cells were released into 20 ml drug-free medium containing 10% horse serum. Forty-eight hours later, cells were seeded into 96-well plates for selection with both neomycin and histidinol antibiotics for two weeks. The gene disruption was confirmed by RT-PCR using primers F22 and R22, and by western blot analysis with anti-DNA-PKcs antibody (Supplementary Figure S3F). The targeting efficiency of generating DNA-PKcs−/− clones from DNA-PKcs+/+ cells was 90% (9/10). The targeting efficiency of generating SMARCAL1−/−/DNA-PKcs−/− clones from SMARCAL1−/−/DNA-PKcs+/+ cells was 100% (2/2).
Generation of SMARCAL1−/− cells reconstituted with SMARCAL1WT, SMARCAL1R764Q or SMARCAL1Δ30 transgene
The SMARCAL1 cDNA was bought from the Kazusa DNA research institute (Chiba, Japan). The SMARCAL1R764Q cDNA was obtained by site-directed mutagenesis of SMARCAL1WT (wild-type SMARCAL1) cDNA using primers F18 and R18. The SMARCAL1Δ30 cDNA with the first 30 amino acids deleted was generated from SMARCAL1WT cDNA by PCR using primers F10 and R10. The SMARCAL1WT, SMARCAL1R764Q and SMARCAL1Δ30 transgenes were cloned into pMSCV-IRES-GFP retroviral expression vector (Clontech, US). The newly engineered retroviral expression vector was co-transfected into human 293T cells with a helper plasmid (pClampho, US) to produce a viral supernatant, which was collected after 24 hours and used to infect the SMARCAL1−/− cells. The efficiency of infection was assessed by quantifying the number of cells expressing GFP using flow-cytometric analysis (LSRFortessa, BD Biosciences, US). The cells expressing GFP were enriched using a cell sorter (FACSAria III, BD Biosciences, US) and seeded into 96 well plates to isolate single colonies. The expression level of the transgenes in the SMARCAL1−/− cells was measured by western blot (Figure 5D).
Figure 5.
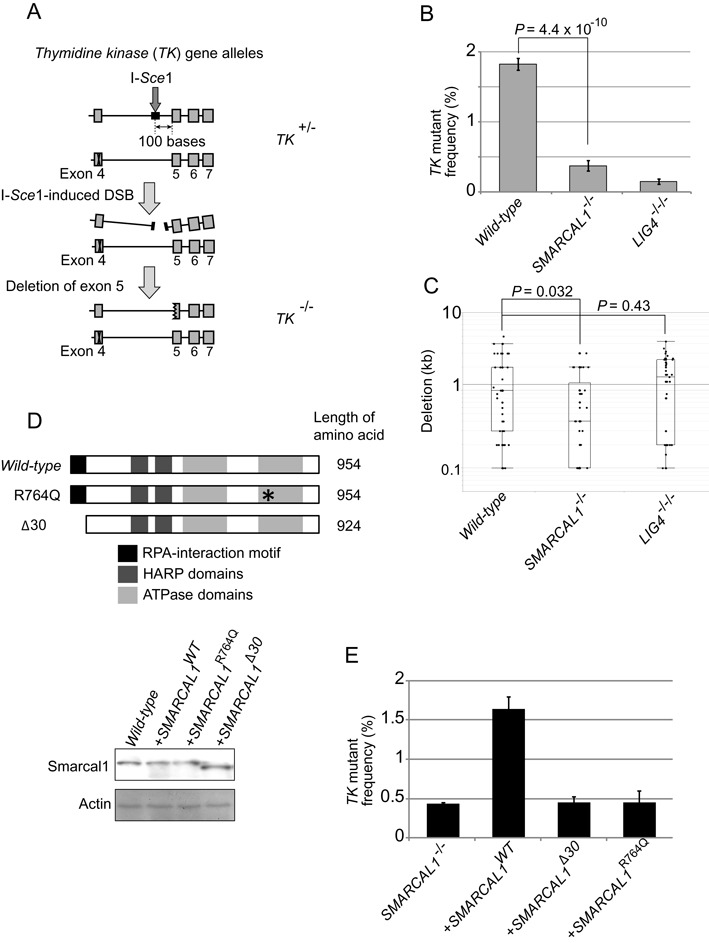
The fidelity of end-joining in SMARCAL1 mutant cells. (A) Schematic diagram showing DSB-repair events that repair I-Sce1-induced DSBs in the endogenous thymidine kinase (TK) locus. TK+/− cells carry an I-Sce1 site in intronic sequences of the wild-type TK allele and a mutation in exon4 of the mutant TK allele. DSB repair associated with deletion in exon5 coding sequences would yield TK−/− clones from TK+/− cells. The number of TK−/− clones was measured by counting the number of trifluorothymidine (TFT)-resistant colonies. (B) Histogram representing the frequency of DSB-repair events (y-axis) in the indicated genotypes (x-axis). Error bars indicate SD of more than three independent experiments. P-value was calculated by Student's t-test. (C) Box plot representing the length of nucleotide deletion (y-axis) in the indicated genotypes (x-axis). PCR was performed from genomic DNA isolated from at least 50 TFT-resistant clones of each genotype, as shown in Supplementary Figure S6. (D) Schematic representation of the structure of wild-type, R764Q and Δ30 Smarcal1 proteins. SMARCAL1−/− TK6 cells were reconstituted with SMARCAL1WT, SMARCAL1Δ30 or SMARCAL1R764Q transgene. Western blot analysis for the expression of individual transgenes in SMARCAL1−/− cells. β-actin was used as a loading control. (E) Histogram representing the frequency of TFT-resistant colonies (y-axis) in the indicated genotypes (x-axis). Error bars indicate SD of more than three independent experiments.
Colony-survival assay
To measure sensitivity, cells were treated with camptothecin (Topogen, Inc, US) and ICRF193 (Funakoshi, Japan) (17) and irradiated with ionizing radiation (137Cs). Cell sensitivity to these DNA-damaging agents was evaluated by counting colony formation in methylcellulose plates as described previously (46,47).
Cell-cycle synchronization
Cells were synchronized by centrifugal-counter-flow elutriation (Hitachi Koki, Japan). The cell suspension (∼5×107 TK6 cells) was loaded at a flow rate of 15 ml/min into an elutriation chamber rotating at 2000 rpm. Cell synchrony was confirmed by FACS analysis (LSRFortessa, BD Biosciences, US).
Immunostaining and microscopic analysis
Cells were fixed with 4% paraformaldehyde (Nacalai Tesque, Japan) for 10 min at room temperature and permeabilized with 0.5% TritonX-100 (Sigma, St. Louis, US) for 20 min. To exclude S-phase cells from the count shown in Figure 3C and D, a Click-iT EdU imaging kit (Alexa594, Invitrogen, US) was used. Images were taken with a confocal microscope (TCS SP8, Leica Microsystems, Germany) and a BX-61 microscope (Olympus, Japan).
Figure 3.
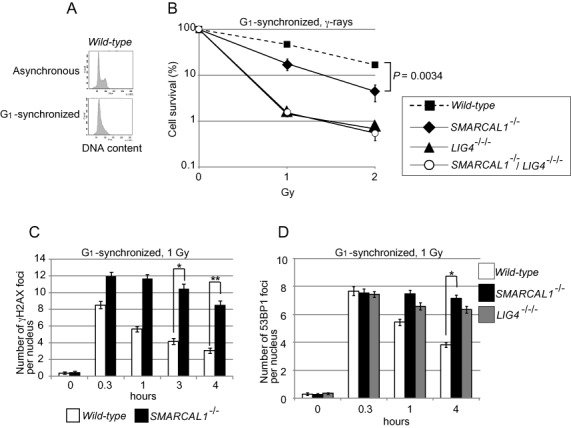
Repair of γ-ray induced DSBs at the G1 phase in TK6 cells. (A) DNA content of G1-synchronized TK6 cells. (B) Cellular sensitivity of G1-synchronized cells to γ-rays, shown as in Figure 1. Error bars indicate the SD of the mean for three independent assays. P-value was calculated by Student's t-test. (C) Histogram representing the γH2AX subnuclear foci of G1 cells after irradiation with 1 Gy γ-rays. The x-axis represents time after γ-irradiation (time zero); the y-axis represents the average number of γH2AX foci in individual cells. The nuclei of 100 morphologically intact cells were analyzed at each time point in individual experiments. The experiment was performed at least three times, with the averages presented with SD and P-values. Asterisks indicate statistical significance; *P = 0.0055 and **P = 0.00020. (D) Histogram representing the 53BP1 subnuclear foci of G1 cells, as shown in (C). The experiment was performed at least three times, with averages presented with SD and P-value. Asterisks indicate statistical significance; *p = 6.3×10−5.
V(D)J-recombination assay
The V(D)J-recombination assay was performed as described previously (48). Briefly, 4×106 TK6 cells were transfected (Neon, Life Technologies, US) with 600 ng of circular pJH200 or pJH290, 5.4 μg of RAG1- and 6.6 μg of RAG2-expression vector. The extrachromosomal plasmids were recovered from cells after 48 hours using a modified Hirt extraction method (49). The mixture of 15 μl of ElectroMAXTM DH10BTM (Life Technologies, US) competent bacteria and 300 ng (range of 100–500 ng) of recovered plasmids were added to an electroporation cuvette (0.1 cm gap) and incubated on ice for 10 min. The bacteria were then electroporated at 1.8 kV, 200 Ω and 25 uF for 2 seconds using a Gene PulserII (Bio-Rad, US). Pre-warmed SOC media was added to the bacteria and the reaction was incubated at 37°C for 2 hours. The reaction was plated on LB-agar plates containing 100 μg/ml ampicillin and 10 μg/ml chloramphenicol and incubated for 16–24 hours at 37°C. The ampicillin+chloramphenicol-resistant plasmids were isolated and subjected to ApaLI digestion to examine the fidelity of the signal joints. Digestion of original pJH200 plasmid yields a 4.3 kb band, while that of the correct recombination products yields 3.5 kb + 0.8 kb bands due to the newly generated ApaLI site. Signal-joint and coding-joint sequences were analyzed using the sequencing primer R17 (50).
NHEJ assay of I-Sce1-induced DSBs
A TK6-derived line that is heterozygous for point mutation in exon4 of the thymidine-kinase gene (TK+/−) was used to measure the frequency of NHEJ events as described previously (51,52). To measure the length of deletion in DSB-repair products formed in wild-type and SMARCAL1−/− cells, primers Fa and Ra were used. To measure the length of the deletion formed in the LIG4−/−/− cells, primers Fa and Rb were used.
Chromatin fractionation and chromatin immunoprecipitation
A Subcellular Protein Fractionation Kit from Thermo Scientific (78840) was used for chromatin fractionation. Expression plasmids for a TALEN and I-Sce1 were transfected into TK6 cells using the Neon Transfection System. After 20 hours, transfected cells were analyzed by western blotting and ChIP. ChIP was performed as described previously (53), with some modifications. Briefly, samples were sonicated to generate DNA fragments of <500 bp. The antibody was incubated with Dynabeads Protein G for 3 hours at 4°C. Sheared chromatin was centrifuged at 15000 rpm for 15 min at 4°C. After centrifugation, supernatants were incubated with antibody-protein G conjugates for 3 hours at 4°C. The conjugated beads were washed thoroughly with IP buffer-140, IP buffer-500, IP buffer-750, LiCl/detergent and TE. Real-time PCR was carried out as described previously (53). Sequences of primers are given in Supplemental Table S1.
Antibodies
Anti-γH2AX mouse monoclonal (1:1000, Millipore, US); anti-TP53BP1 rabbit polyclonal (1:100, Sigma, US); alexa fluor 488-conjugated anti-mouse IgG (1:1000, Molecular Probes); alexa fluor 488–conjugated anti-rabbit IgG (1:1000, Molecular Probes); anti-Smarcal1 rabbit polyclonal (ab154226, abcam, UK); anti-XRCC4 goat polyclonal (C-20, Santa Cruz, US); anti-Ku70 mouse polyclonal (#GTX70270, Gene Tex, US); anti-DNA-PKcs mouse monoclonal (ab1832, abcam, UK).
RESULTS
SMARCAL1-deficient cells are sensitive to DNA-damaging agents
To analyze the role of Smarcal1 in the DSB-repair pathway, we disrupted the SMARCAL1 gene and generated SMARCAL1−/− DT40 cells (Supplementary Figures S1A and S1C). Moreover, to selectively analyze the function of the Smarcal1-RPA interaction, we generated SMARCAL1Δ30/− DT40 mutant cells by deleting the N-terminal region encoding the RPA-binding site of the endogenous SMARCAL1 gene (9) (Supplementary Figures S1D and S1E). To define the role played by Smarcal1 in various DNA-repair processes, we measured cellular responses to exogenous DNA damages. SMARCAL1−/− and SMARCAL1Δ30/− DT40 cells exhibited increased sensitivities to various DNA-damaging agents, including camptothecin and ICRF193 (Figures 1A and B). The sensitivity profile of the SMARCAL1Δ30/− cells was very similar to that of the SMARCAL1−/− cells, suggesting that the physical association of Smarcal1 with RPA is essential for its DNA-damage response (9). The elevated sensitivity to camptothecin supports the idea that Smarcal1 helps to prevent replication forks from replication collapse at one-end breaks, as indicated previously (1,2,8–10,54). NHEJ-deficient KU70−/− cells (55), but not HR-deficient BRCA2−/−cells (46), showed a hypersensitivity to ICRF193 (Figure 1B). We further analyzed the functional relationship between Smarcal1 and Ku70 by generating KU70−/−/SMARCAL1Δ30/− double-mutant cells. The double-mutant cells showed virtually the same ICRF193 sensitivity as did the KU70−/− single mutant (Figure 1D), indicating that Smarcal1 is epistatic to Ku70.
Figure 1.
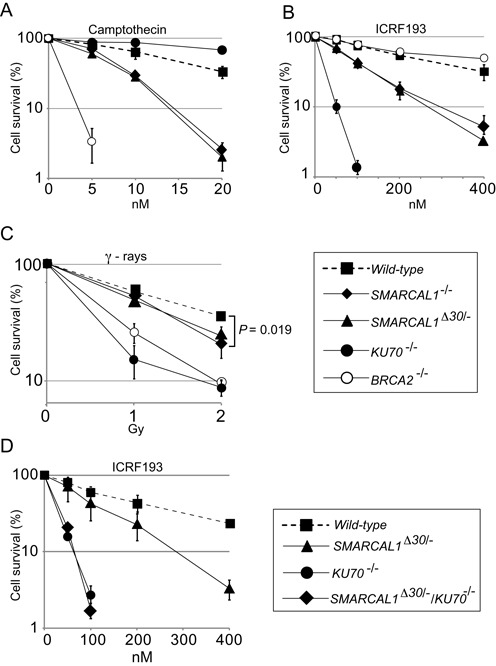
Smarcal1-deficient DT40 cells are sensitive to ICRF193 and camptothecin. Clonogenic-cell-survival assay following exposure of the indicated genotypes to DNA-damaging agents (A–D). The x-axis represents the dose of the indicated DNA-damaging agent on a linear scale; the y-axis represents the survival fraction on a logarithmic scale. Error bars show the SD of the mean for three independent assays. The P-value of (C) was calculated by Student's t-test or two-sample t-test for IR sensitivity at 2 Gy.
To investigate the role of Smarcal1 in human cells, we disrupted the SMARCAL1 gene in the human TK6 B cell line using a TALEN pair combined with gene-disruption constructs (Supplementary Figures S2A and S2B). We also generated both LIG4−/−/− (Supplementary Figures S3A and S3B) and RAD54−/− TK6 clones (Supplementary Figures S3C and S3D) as controls deficient in NHEJ and HR, respectively. In addition, we generated DNA-PKcs−/− TK6 clones (Supplementary Figures S3E and S3F). The TK6 cell line has been widely used by the governments of developed countries to detect environmental mutagens due to the very stable phenotype and karyotype of its cells (56,57). The SMARCAL1−/−cells proliferated with kinetics (15 h per cell cycle) and plating efficiency (58%) similar to that of wild-type TK6 cells. Like the DT40 mutants, human SMARCAL1−/− cells were moderately but also significantly sensitive to camptothecin (Figure 2A). Three SMARCAL1−/− clones and NHEJ-deficient LIG4−/−/− and DNA-PKcs−/−, but not HR-deficient RAD54−/−, were sensitive to ICRF193 (Figure 2B, Supplementary Figures S2C and S4B). Additionally, LIG4−/−/− and SMARCAL1−/−/LIG4−/−/− cells showed the same sensitivity to ICRF193 (Figure 2D). Likewise, DNA-PKcs−/− and SMARCAL1−/−/DNA-PKcs−/− cells showed the same sensitivity to ICRF193 (Supplementary Figure S4B). We thus conclude that Smarcal1 promotes the canonical NHEJ pathway in both DT40 and TK6 cell lines.
Figure 2.
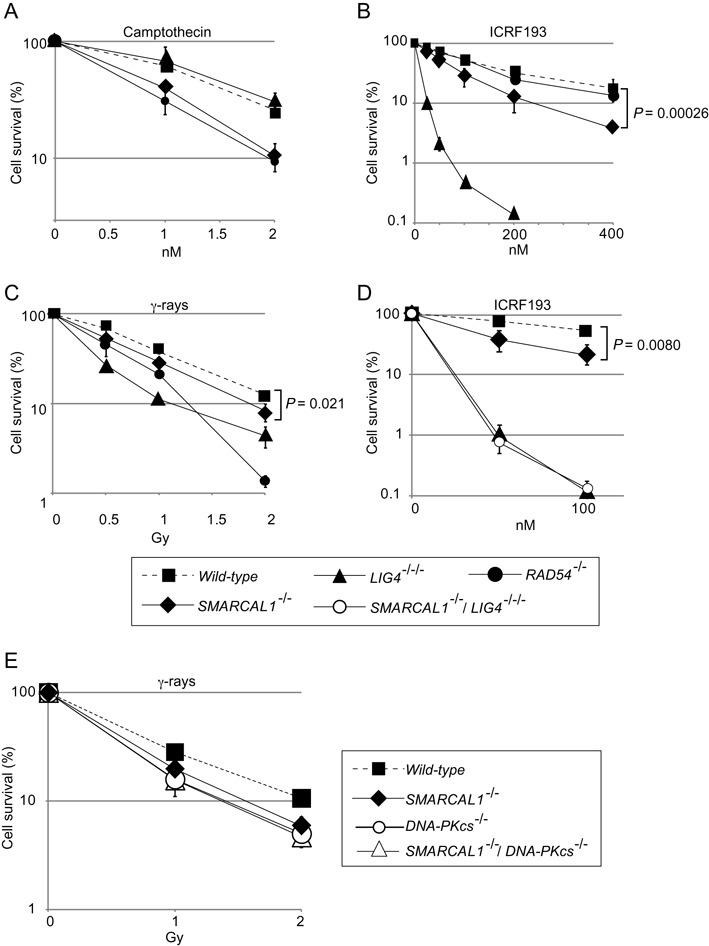
Sensitivity of human SMARCAL1−/−, SMARCAL1−/−/LIG4−/−/− and SMARCAL1−/−/DNA-PKcs−/− TK6 B cells to ICRF193 and γ-rays. (A–E) Cellular sensitivity is shown as in Figure 1. Error bars show the SD of the mean for three independent assays. P-values were calculated by Student's t-test.
Human SMARCAL1−/− cells are defective for DSB repair in the G1 phase
SMARCAL1−/− DT40 and TK6 cells were significantly radiosensitive (Figures 1C and 2C and Supplementary Figure S2C). Remarkably, SMARCAL1−/−, DNA-PKcs−/− and SMARCAL1−/−/DNA-PKcs−/− TK6 cells showed very similar radiosensitivity (Figure 2E and Supplementary Figure S4A). To further investigate the role of Smarcal1 in NHEJ, we measured sensitivity to ionizing radiation (IR) in the G1 phase (Figure 3A and Supplementary Figure S5A). SMARCAL1−/− as well as LIG4−/−/− cells showed a hypersensitivity to IR (Figure 3B). As expected, the SMARCAL1−/−/LIG4−/−/− cells showed the same hypersensitivity to IR as did the LIG4−/−/− cells in the G1 phase (Figure 3B). We then monitored DSB repair kinetics in the G1 phase by counting the number of γH2AX and 53BP1 foci over time after exposure to IR. The resolution of 53BP1 and γH2AX foci was significantly delayed in SMARCAL1−/− cells compared to wild-type cells (Figures 3C, D and Supplementary Figure S5B). These results indicate that SMARCAL1−/− cells are deficient in DSB repair in the G1 phase. We therefore conclude that Smarcal1 promotes DSB repair by NHEJ in both human and chicken DT40 cells.
Deletion of Smarcal1 does not compromise the fidelity of NHEJ
To address the accuracy of individual DSB-repair events, we performed two experiments using TK6 cells, (i) the analysis of V(D)J recombination (Figure 4) and (ii) the repair of I-Sce1 induced DSBs by NHEJ (Figure 5).
Figure 4.
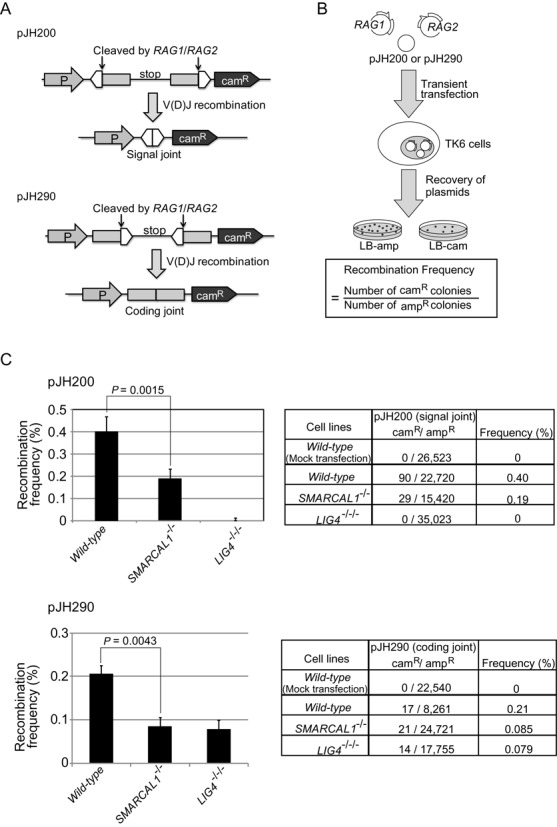
The loss of Smarcal1 reduces the efficiency of V(D)J recombination without compromising its fidelity. (A) The structure of two episomal V(D)J-recombination substrates, pJH200 and pJH290, and their recombination products. Open triangles and closed boxes represent recombination signals (RSs) and V(D)J-coding sequences, respectively. CamR = chloramphenicol-resistance gene; P = promoter. (B) Schematic representation of the experimental method for the V(D)J-recombination assay. Frequency of recombination was calculated by dividing the number of rearranged products (the number of camR colonies) by the number of recovered plasmids (the number of ampicillin-resistant [ampR] colonies). (C) Recombination frequency of TK6 cells carrying the indicated genotypes. Data shown are the means of more than three experiments. Error bars indicate SD of more than three independent experiments. P-value was calculated by Student's t-test. The total number of ampicillin- and chloramphenicol-resistant colonies is shown in the right panel. Supplemental Table S2 shows the nucleotide sequences of coding joints associated with deletion events.
V(D)J recombination is initiated by the Rag1 and Rag2 recombinase proteins, which introduce DSBs at the recombination signal (RS) (58,59) and complete recombination by collaborating with canonical NHEJ. We used two episomal V(D)J-recombination substrates, pJH200 and pJH290, where the recombinase generates the RS and coding-joint products, respectively (50) (Figure 4A). We transiently transfected expression plasmids encoding RAG1 and RAG2 along with either pJH200 or pJH290 into wild-type and SMARCAL1−/− and LIG4−/−/− cells (Figure 4B). We then recovered the transfected substrate plasmids from the TK6 cells, introduced the plasmids into bacterial cells, and plated them on LB agar plates containing either ampicillin or chloramphenicol. Ampicillin-resistant colonies contained recovered pJH200 or pJH290 plasmids, while colonies resistant to chloramphenicol contained only rearranged pJH200 or pJH290 plasmids. Thus, the frequency of rearrangement can be calculated as the gain of chloramphenicol-resistant (camR) colonies relative to the total number of ampicillin-resistant (ampR) colonies. The signal-joint ends are precisely ligated, whereas the coding ends are joined in a process that can involve nucleotide loss or gain in addition to simple ligation (60). We failed to recover any rearranged signal-joint products from LIG4−/−/− cells, which agrees with the essential role for DNA Ligase4 in signal-joint formation (61,62). The efficiency of recombination in the signal-joint and coding-joint plasmids was decreased 2.3 and 2.4 times, respectively, in SMARCAL1−/− cells, when compared with wild-type cells (Figure 4C). The nucleotide sequence analysis of signal-joint products indicated that only a single product among the 29-analyzed sequences contained one nucleotide deletion in SMARCAL1−/− cells (data not shown). Analysis of coding joints, on the other hand, showed more frequent deletion events in SMARCAL1−/− as well as in wild-type cells, with the extent and frequency of deletion being comparable between the two genotypes (Supplemental Table S2). Thus, the loss of Smarcal1 reduces the efficiency of canonical NHEJ without compromising its fidelity.
We next examined individual DSB-repair events in the human TK6 cell line, which is heterozygous (+/-) for the thymidine kinase (TK) gene and contains an I-Sce1 endonuclease recognition site in the fourth intron of the intact TK allele (51) (Figure 5A). If the repair of I-Sce1-induced DSBs causes the deletion of more than 100 nucleotides downstream from the I-Sce1 site, the deletion would inactivate the fifth exon, leading to the formation of TK−/− cells, which cells are able to form colonies in the negative-selection media containing trifluorothymidine (TFT) (Figure 5A). We found that the number of TFT-resistant clones was reduced by 8 and 5 times in the LIG4−/−/− and SMARCAL1−/− cells, respectively, when compared with wild-type cells (Figure 5B). To analyze deletion range and pattern, we isolated at least 50 individual TFT-resistant clones from each genotype. Genomic PCR amplification over the I-Sce1 site indicated that deletion was two times shorter in the SMARCAL1−/− cells and approximately 0.5 times longer in LIG4−/−/− cells, compared with wild-type cells (Figure 5C and Supplementary Figure S6). In summary, we conclude that Smrcal1 promotes NHEJ without affecting its accuracy.
The RPA-binding domain and the ATPase domains are both required for the promotion of NHEJ by Smarcal1
The phenotypic analysis of SMARCAL1Δ30/− DT40 cells indicated that the RPA-binding site is essential for Smarcal1 to function in NHEJ. To confirm the relevance of this finding to human cells, we reconstituted the SMARCAL1−/− TK6 cells with the RPA-binding-site-deficient (SMARCAL1Δ30) transgene as well as the wild-type (SMARCAL1WT) transgene. The expression level of the Smarcal1 protein in the reconstituted cells was similar to that in the wild-type cells (Figure 5D, lower panel). As expected, reconstitution of SMARCAL1−/− cells with the SMARCAL1WT transgene normalized the NHEJ-mediated repair of I-Sce1-induced DSBs (Figure 5E). The SMARCAL1Δ30 transgene failed to normalize NHEJ, indicating that the RPA-binding site plays a role in promoting NHEJ by Smarcal1 in human cells. Moreover, SMARCAL1−/− cells and SMARCAL1Δ30 transgene exhibited similar sensitivity to DNA-damaging agent (Supplementary Figure S4C), demonstrating that the RPA-binding site is critical for Smarcal1 DNA repair function. Next, to investigate the role of annealing helicase activity, we introduced a point mutation (Arginine 764 to Glutamine, R764Q) into the ATPase domain of the SMARCAL1WT transgene, which mutation has no detectable annealing helicase activity (2) and causes a severe form of Schimke immuno-osseous dysplasia (SIOD) (4). Reconstitution with the resulting SMARCAL1R764Q transgene did not restore the repair of I-Sce1-induced DSBs (Figure 5D and E). We therefore conclude that the annealing helicase activity as well as the physical association of Smarcal1 with RPA is required for the promotion of NHEJ by Smarcal1. One possible scenario is that Smarcal1 promotes NHEJ by interacting with unwound DSB ends associated with RPA and facilitate their annealing.
Annealing of double-strand DNA by purified Smarcal1 (2) suggests that Smarcal1 facilitates NHEJ by stabilizing the physical interaction between Ku70/Ku80/DNA-PKcs proteins and DSB ends, thereby activating DNA-PKcs. To test this hypothesis, we analyzed the phosphorylation status of threonine 2609 (63,64) after treatment with the topoisomerase 2 inhibitors ICRF193 (Supplementary Figures S7A and S7B) and etoposide (Supplementary Figure S7C). Strikingly, the phosphorylated threonine 2609 was detectable only in the wild-type and not in the SMARCAL1−/− cells (Supplementary Figures S7B and S7C). The compromised phosphorylation of DNA-PKcs may account for the decreased efficiency of NHEJ, as the substitution of the threonine 2609 site to alanine causes radio-sensitivity and reduced V(D)J recombination efficiency, which sensitivity is less prominent than that of DNA-PKcs null-mutant cells (64). We propose that Smarcal1 promotes NHEJ, presumably at an initial step, by facilitating the physical association of Ku70/Ku80/DNA-PKcs proteins and DSB ends.
Smarcal1 is required for the recruitment of XRCC4 to DNA-damage sites
To investigate early and late steps of NHEJ, we monitored the dynamics of Ku70 and XRCC4 proteins, respectively, in the chromatin-bound fraction, following exposure of cells to ICRF193 for one hour (Figure 6). This exposure did not affect the purification of the chromatin-bound or the nuclear-soluble fractions (Figure 6A). Exposure to ICRF193 caused a marked increase in the amounts of both Ku70 and XRCC4 in the chromatin-bound fraction of wild-type cells. In marked contrast, accumulations of Ku70 and XRCC4 in the SMARCAL1−/− as well as DNA-PKcs−/− cells were, significantly smaller compared with wild-type cells (Figure 6B and C).
Figure 6.
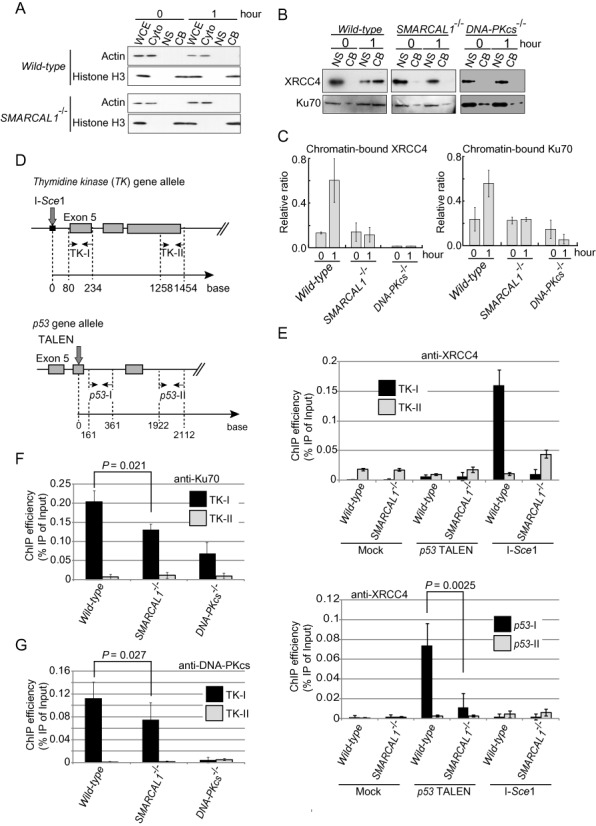
The loss of Smarcal1 results in compromised accumulation of Ku70, DNA-PKcs and XRCC4 at DSB sites. (A) Western blot data showing the validation of fractionation of the cytoplasmic (Cyto), nuclear soluble (NS) and chromatin-bound (CB) fractions isolated from the whole-cell extract (WCE). (B) Western blot data show the accumulation of XRCC4 (upper panel) and Ku70 (lower panel) in the chromatin-bound fraction after one-hour exposure of cells to ICRF193. (C) Histogram showing the quantification of XRCC4 and Ku70 in (B). The y-axis represents the amount of the chromatin-bound fraction relative to the total amount of the chromatin-bound fraction plus the nuclear soluble fraction. (D) Downward arrows represent the I-Sce1- (upper) and the TALEN- (lower) cutting sites in the TK and p53 genes, respectively. Pairs of horizontal opposing arrows indicate the sets of primers for quantitative real-time PCR. (E) Histograms represent the accumulation of XRCC4 near the I-Sce1-induced DSB in the TK locus (upper panel) and near the TALEN-induced DSB at the p53 locus (lower panel). (F) Histogram represents the accumulation of Ku70 near the I-Sce1-induced DSB in the TK locus. (G) Histogram represents the accumulation of DNA-PKcs near the I-Sce1-induced DSB in the TK locus.
To validate the significant reduction in the accumulation of XRCC4 near DSB sites, we conducted chromatin immunoprecipitation (ChIP) following transient transfection of empty vector and expression vectors encoding the I-Sce1 restriction enzyme and a TALEN towards the p53 locus (Figure 6D). XRCC4 is supposed to accumulate at the TK-I or p53-I sites, which are adjacent to the DSB sites, but not at the TK-II or p53-II sites, which are distant from the DSB sites (Figure 6D). Transient expression of I-Sce1 or TALEN caused the accumulation of XRCC4 near the DSB sites in wild-type cells, whereas the extent of the accumulation was significantly reduced in SMARCAL1−/− cells (Figure 6E and Supplementary Figure S7D). We checked the overall expression levels of XRCC4 (Supplementary Figure S7B) to confirm that the deletion of Smarcal1 did not alter the level of protein expression. Thus, Smarcal1 is required for recruitment of XRCC4 to DSB sites.
Like reduced accumulation of Ku70 in the chromatin fraction (Figures 6B and C), the amounts of Ku70 and DNA-PKcs near the I-Sce1 site were approximately 30% smaller in SMARCAL1−/− cells in comparison with wild-type cells (Figures 6F and G). In summary, Smarcal1 may promote annealing of DSB ends, which stabilizes complex formation of Ku/DNA-PKcs at DSB sites and fully activates DNA-PKcs. The activation may enhance the recruitment of XRCC4 for the completion of DSB repair by canonical NHEJ.
DISCUSSION
We herein show that Smarcal1 significantly contributes to canonical NHEJ. Although the role of Smarcal1 during DNA replication has been well established (1,3,8–10,65), it has remained unclear whether Smarcal1 plays a role outside the S phase. We here reveal that Smarcal1 contributes to DSB repair by NHEJ during the G1 phase. The role played by Smarcal1 in NHEJ is demonstrated by six points, as follows. First, the loss of Smarcal1 increases cellular sensitivity to ICRF193, which induces DSBs repair only by NHEJ and not by HR (17) (Figures 1B and 2B). Second, null-mutation of KU70 is epistatic to SMARCAL1Δ30 mutation in DT40 cells (Figure 1D) and null-mutation of LIG4 is epistatic to SMARCAL1 null-mutation in TK6 cells (Figure 2D), in terms of cellular tolerance to ICRF193. Third, the loss of Smarcal1 significantly reduces the efficiency of DSB repair in the G1 phase in TK6 cells (Figure 3). Fourth, SMARCAL1 null-mutation in TK6 cells significantly compromises V(D)J recombination (Figure 4) as well as I-Sce1-induced DSB repair by NHEJ (Figure 5). Fifth, SMARCAL1 null-mutation impairs the phosphorylation of DNA-PKcs at threonine 2609 (Supplementary Figures S7B and S7C). Lastly, SMARCAL1 null-mutation diminishes the accumulation of Ku70, DNA-PKcs and XRCC4 at DNA-damage-induced DSB sites (Figure 6). We therefore conclude that Smarcal1 plays a role in NHEJ.
The molecular mechanisms underlying the severe lymphocytopenia of SIOD patients remain unclear (4,40,41). The lymphocytopenia might result primarily from a severe defect in V(D)J recombination due to the reduced efficiency of NHEJ, according to the following studies. The analysis of V(D)J recombination in peripheral T lymphocytes indicates that the size of the T-cell-antigenic receptor (TCR) repertoire is extremely small in SIOD patients (40). Moreover, the peripheral T lymphocytes of SIOD patients have severely low copy number of episomal circular DNA generated as a consequence of V(D)J recombination and contain signal joints of the T cell receptor genes (40,41). These observations support the following scenario. A moderate defect in NHEJ can cause a very strong decrease in the efficiency of T-cell development in patients, since it requires productive D-J- and V-D-recombination events in both TCRα and TCRβ chain genes in individual thymocytes. The reduced T-cell production by the thymus may be compensated by enhanced proliferation of newly generated peripheral T lymphocytes (66) in SIOD patients, leading to the quick dilution of episomal circular DNA in individual peripheral T lymphocytes. This enhanced proliferation could cause a strong replication stress in SIOD patients, due to their attenuated stabilization of replication forks. In summary, we propose that the severe lymphocytopenia of SIOD patients (4,40,41) is attributable to the significantly reduced efficiency of V(D)J recombination, together with attenuated stabilization of replication forks.
A prominent question is, how does Smarcal1 facilitate the promotion of NHEJ? We have shown that both the loss of the RPA-binding site and the inactivation of ATPase activity in Smarcal1 completely abolished the promotion of NHEJ by Smarcal1 (Figure 5E). Thus, Smarcal1 plays a role in NHEJ by physically interacting with RPA on unwound DSB ends and then facilitating their annealing. The existence of unwound single-stranded sequences at the DSB sites is supported by the presence of RPA foci in the γ-ray irradiated G1 phase cells (67). The annealing by Smarcal1 may stabilize the interaction of DSB sites with DNA-PKcs/Ku70/Ku80, since Ku70/Ku80 associates with duplex DSB ends more stably than with DSBs carrying single-strand tails (21–24). The stabilization of DNA-PKcs/Ku70/Ku80 at DSB sites by Smarcal1 is verified by data shown in Figure 6. The following data suggest the important role of Smarcal1 in the functioning of DNA-PKcs. DNA-PKcs/Ku70/Ku80 also interacts with Smarcal1 in vivo (68,69). We here show that DNA-PKcs−/− and SMARCAL1−/− have an epistatic relationship in cellular tolerance to IR (Figure 2E). We also show that the loss of Smarcal1 inhibits the phosphorylation of DNA-PKcs at threonine 2609 following exposure of cells to the topoisomerase 2 inhibitors (Supplementary Figure S7). These observations suggest that Smarcal1 may be required for DNA-PKcs/Ku70/Ku80 to function appropriately. We also show that the loss of Smarcal1 reduces the recruitment of XRCC4 to DSB sites by several times (Figure 6). Previous studies indicate that DNA-PKcs is necessary for the stabilization of recruited XRCC4 (70,71), which is consistent with our data (Figure 6B). Thus, the effect of Smarcal1 on the recruitment of XRCC4 might be mediated by DNA-PKcs/Ku70/Ku80. We therefore propose that Smarcal1 maintains duplex DNA status at DSB ends by interacting with unwound single-strand DNA associated with RPA and facilitating their annealing. This annealing then stabilizes DNA-PKcs/Ku70/Ku80 at duplex DNA termini, which is essential for the proper accumulation and stabilization of XRCC4 at DNA damage sites. Future studies should clarify the molecular mechanism.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
We are grateful to Dr Susan P. Lees-Miller for her critical reading and comments and to the Laboratory for Animal Resources and Genetic Engineering, Center for Developmental Biology (CDB), RIKEN Kobe (http://www.cdb.riken.jp/arg/cassette.html). Thanks also go to Dr Miki Shinohara for providing us with the Ligase4 antibody and to Dr Junko Murai for giving us technical advice. Finally, we wish to thank A. Noguchi and A. Kobayashi for their technical assistance and the lab members for their stimulating discussions.
FUNDING
JSPS Core-to-Core Program, A. Advanced Research Networks [to S.T.]; Grant-in-Aid for Scientific Research on Innovative Areas; Core-to-Core Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan [to S.T. and H.S.]. Funding for open access charge: JSPS Core-to-Core Program, A. Advanced Research Networks; Grant-in-Aid for Scientific Research on Innovative Areas; Core-to-core Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan (KAKENHI, grant number 23221005 to T.S. and H.S., 26740018 to H.S.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Yusufzai T., Xiangduo K., Kyoko Y., Kadonaga J.T. The annealing helicase HARP is recruited to DNA repair sites via an interaction with RPA. Genes Dev. 2009;23:2400–2404. doi: 10.1101/gad.1831509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yusufzai T., Kadonaga J.T. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bétous R., Mason A.C., Rambo R.P., Bansbach C.E., Badu-Nkansah A., Sirbu B.M., Eichman B.F., Cortez D. SMARCAL1 catalyzes fork regression and holliday junction migration to maintain genome stability during DNA replication. Genes Dev. 2012;26:151–162. doi: 10.1101/gad.178459.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boerkoel C.F., Takashima H., John J., Yan J., Stankiewicz P., Rosenbarker L., André J.-L., Bogdanovic R., Burguet A., Cockfield S., et al. Mutant chromatin remodeling protein SMARCAL1 causes Schimke immuno-osseous dysplasia. Nat. Genet. 2002;30:215–220. doi: 10.1038/ng821. [DOI] [PubMed] [Google Scholar]
- 5.Deguchi K., Clewing J.M., Elizondo L.I., Hirano R., Huang C., Choi K., Sloan E.A., Lücke T., Marwedel K.M., Powell R.D., et al. Neurologic phenotype of Schimke immuno-osseous dysplasia and neurodevelopmental expression of SMARCAL1. J. Neuropath. Exp. Neurol. 2008;67:565–577. doi: 10.1097/NEN.0b013e3181772777. [DOI] [PubMed] [Google Scholar]
- 6.RN S., WA H., CR K. Chondroitin-6-sulphaturia, defective cellular immunity, and nephrotic syndrome. Lancet. 1971;2:1088–1089. doi: 10.1016/s0140-6736(71)90400-4. [DOI] [PubMed] [Google Scholar]
- 7.Spranger J., Hinkel G.K., Stöss H., Thoenes W., Wargowski D., Zepp F. Schimke immuno-osseous dysplasia: a newly recognized multisystem disease. J. Pediatr. 1991;119:64–72. doi: 10.1016/s0022-3476(05)81040-6. [DOI] [PubMed] [Google Scholar]
- 8.Yuan J., Ghosal G., Chen J. The annealing helicase HARP protects stalled replication forks. Genes Dev. 2009;23:2394–2399. doi: 10.1101/gad.1836409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ciccia A., Bredemeyer A.L., Sowa M.E., Terret M.E., Jallepalli P.V., Harper J.W., Elledge S.J. The SIOD disorder protein SMARCAL1 is an RPA-interacting protein involved in replication fork restart. Genes Dev. 2009;23:2415–2425. doi: 10.1101/gad.1832309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bansbach C.E., Bétous R., Lovejoy C.A., Glick G.G., Cortez D. The annealing helicase SMARCAL1 maintains genome integrity at stalled replication forks. Genes Dev. 2009;23:2405–2414. doi: 10.1101/gad.1839909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jackson S.P. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002;23:687–696. doi: 10.1093/carcin/23.5.687. [DOI] [PubMed] [Google Scholar]
- 12.Kanaar R., Hoeijmakers J.H., van Gent D.C. Molecular mechanisms of DNA double strand break repair. Trends Cell Biol. 1998;8:483–489. doi: 10.1016/s0962-8924(98)01383-x. [DOI] [PubMed] [Google Scholar]
- 13.Mehta A., Haber J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNARepair. Cold Spring Harb. Perspect. Biol. 2014;6:1–17. doi: 10.1101/cshperspect.a016428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi S., Kasaishi Y., Nakada S., Takagi T., Era S., Motegi A., Chiu R.K., Takeda S., Hirota K. Rad18 and Rnf8 facilitate homologous recombination by two distinct mechanisms, promoting Rad51 focus formation and suppressing the toxic effect of nonhomologous end joining. Oncogene. 2014;0:1–9. doi: 10.1038/onc.2014.371. [DOI] [PubMed] [Google Scholar]
- 15.Maede Y., Shimizu H., Fukushima T., Kogame T., Nakamura T., Miki T., Takeda S., Pommier Y., Murai J. Differential and common DNA repair pathways for topoisomerase I- and II-targeted drugs in a genetic DT40 repair cell screen panel. Mol. Cancer Therap. 2014;13:214–220. doi: 10.1158/1535-7163.MCT-13-0551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murai J., Huang S.Y.N., Das B.B., Renaud A., Zhang Y., Doroshow J.H., Ji J., Takeda S., Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adachi N., Suzuki H., Iiizumi S., Koyama H. Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: implications for the repair of topoisomerase II-mediated DNA damage. J. Biol. Chem. 2003;278:35897–35902. doi: 10.1074/jbc.M306500200. [DOI] [PubMed] [Google Scholar]
- 18.Boboila C., Alt F.W., Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- 19.Gellert M. V(D)J recombination: RAG proteins, repair factors, and regulation. Annu. Rev. Biochem. 2002;71:101–132. doi: 10.1146/annurev.biochem.71.090501.150203. [DOI] [PubMed] [Google Scholar]
- 20.Verkaik N.S., Esveldt-van Lange R.E.E., Van Heemst D., Brüggenwirth H.T., Hoeijmakers J.H.J., Zdzienicka M.Z., Van Gent D.C. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 2002;32:701–709. doi: 10.1002/1521-4141(200203)32:3<701::AID-IMMU701>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 21.Dynan W.S., Yoo S. Interaction of Ku protein and DNA-dependent protein kinase catalytic subunit with nucleic acids. Nucleic Acids Res. 1998;26:1551–1559. doi: 10.1093/nar/26.7.1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mimitou E.P., Symington L.S. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J. 2010;29:3358–3369. doi: 10.1038/emboj.2010.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mimori T., Hardin J.A. Mechanism of interaction between Ku protein and DNA. J. Biol. Chem. 1986;261:10375–10379. [PubMed] [Google Scholar]
- 24.Symington L.S., Gautier J. Double-Strand Break End Resection and Repair Pathway Choice. Ann. Rev. Genet. 2011;45:247–271. doi: 10.1146/annurev-genet-110410-132435. [DOI] [PubMed] [Google Scholar]
- 25.Lees-Miller S.P., Chen Y.R., Anderson C.W. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol. Cell. Biol. 1990;10:6472–6481. doi: 10.1128/mcb.10.12.6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carter T., Vancurova I., Sun I., Lou W., DeLeon S. A DNA-activated protein kinase from HeLa cell nuclei. Mol. Cell. Biol. 1990;10:6460–6471. doi: 10.1128/mcb.10.12.6460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blunt T., Gell D., Fox M., Taccioli G.E., Lehmann A.R., Jackson S.P., Jeggo P.A. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10285–10290. doi: 10.1073/pnas.93.19.10285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chan D.W., Chen B.P.-C., Prithivirajsingh S., Kurimasa A., Story M.D., Qin J., Chen D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev. 2002;16:2333–2338. doi: 10.1101/gad.1015202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cui X., Yu Y., Gupta S., Cho Y.-M., Lees-Miller S.P., Meek K. Autophosphorylation of DNA-dependent protein kinase regulates DNA end processing and may also alter double-strand break repair pathway choice. Mol. Cell. Biol. 2005;25:10842–10852. doi: 10.1128/MCB.25.24.10842-10852.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma Y., Pannicke U., Lu H., Niewolik D., Schwarz K., Lieber M.R. The DNA-dependent protein kinase catalytic subunit phosphorylation sites in human Artemis. J. Biol. Chem. 2005;280:33839–33846. doi: 10.1074/jbc.M507113200. [DOI] [PubMed] [Google Scholar]
- 31.Reddy Y.V.R., Ding Q., Lees-Miller S.P., Meek K., Ramsden D.A. Non-homologous end joining requires that the DNA-PK complex undergo an autophosphorylation-dependent rearrangement at DNA ends. J. Biol. Chem. 2004;279:39408–39413. doi: 10.1074/jbc.M406432200. [DOI] [PubMed] [Google Scholar]
- 32.Ahnesorg P., Smith P., Jackson S.P. XLF interacts with the XRCC4-DNA Ligase IV complex to promote DNA nonhomologous end-joining. Cell. 2006;124:301–313. doi: 10.1016/j.cell.2005.12.031. [DOI] [PubMed] [Google Scholar]
- 33.Buck D., Malivert L., De Chasseval R., Barraud A., Fondanèche M.C., Sanal O., Plebani A., Stéphan J.L., Hufnagel M., Le Deist F., et al. Cernunnos, a novel nonhomologous end-joining factor, is mutated in human immunodeficiency with microcephaly. Cell. 2006;124:287–299. doi: 10.1016/j.cell.2005.12.030. [DOI] [PubMed] [Google Scholar]
- 34.Critchlow S.E., Bowater R.P., Jackson S.P. Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Curr. Biol. 1997;7:588–598. doi: 10.1016/s0960-9822(06)00258-2. [DOI] [PubMed] [Google Scholar]
- 35.Grawunder U., Wilm M., Wu X., Kulesza P., Wilson T.E., Mann M., Lieber M.R. Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature. 1997;388:492–495. doi: 10.1038/41358. [DOI] [PubMed] [Google Scholar]
- 36.Lieber M.R. NHEJ and its backup pathways in chromosomal translocations. Nat. Struct. Mol. Biol. 2010;17:393–395. doi: 10.1038/nsmb0410-393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simsek D., Jasin M. Alternative end-joining is suppressed by the canonical NHEJ component Xrcc4-ligase IV during chromosomal translocation formation. Nat. Struct. Mol. Biol. 2010;17:410–416. doi: 10.1038/nsmb.1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buerstedde J.M., Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 39.Takashima Y., Sakuraba M., Koizumi T., Sakamoto H., Hayashi M., Honma M. Dependence of DNA double strand break repair pathways on cell cycle phase in human lymphoblastoid cells. Environ. Mol. Mutagen. 2009;50:815–822. doi: 10.1002/em.20481. [DOI] [PubMed] [Google Scholar]
- 40.Lev A., Amariglio N., Levy Y., Spirer Z., Anikster Y., Rechavi G., Dekel B., Somech R. Molecular assessment of thymic capacities in patients with Schimke immuno-osseous dysplasia. Clin. Immunol. 2009;133:375–381. doi: 10.1016/j.clim.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 41.Simon A.J., Lev A., Jeison M., Borochowitz Z.U., Korn D., Lerenthal Y., Somech R. Novel SMARCAL1 bi-allelic mutations associated with a chromosomal breakage phenotype in a severe SIOD patient. J. Clin. Immunol. 2014;34:76–83. doi: 10.1007/s10875-013-9957-3. [DOI] [PubMed] [Google Scholar]
- 42.Sonoda E., Sasaki M.S., Buerstedde J.M., Bezzubova O., Shinohara A., Ogawa H., Takata M., Yamaguchi-Iwai Y., Takeda S. Rad51-deficient vertebrate cells accumulate chromosomal breaks prior to cell death. EMBO J. 1998;17:598–608. doi: 10.1093/emboj/17.2.598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cermak T., Doyle E., Christian M. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sakuma T., Ochiai H., Kaneko T., Mashimo T., Tokumasu D., Sakane Y., Suzuki K.-i., Miyamoto T., Sakamoto N., Matsuura S., et al. Repeating pattern of non-RVD variations in DNA-binding modules enhances TALEN activity. Sci. Rep. 2013;3:3379. doi: 10.1038/srep03379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., et al. Double nicking by RNA-guided CRISPR cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qing Y., Yamazoe M., Hirota K., Dejsuphong D., Sakai W., Yamamoto K.N., Bishop D.K., Wu X., Takeda S. The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet. 2011;7:1002148. doi: 10.1371/journal.pgen.1002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao G.Y., Sonoda E., Barber L.J., Oka H., Murakawa Y., Yamada K., Ikura T., Wang X., Kobayashi M., Yamamoto K., et al. A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol. Cell. 2007;25:663–675. doi: 10.1016/j.molcel.2007.01.029. [DOI] [PubMed] [Google Scholar]
- 48.Hesse J.E., Lieber M.R., Gellert M., Mizuuchi K. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell. 1987;49:775–783. doi: 10.1016/0092-8674(87)90615-5. [DOI] [PubMed] [Google Scholar]
- 49.Arad U. Modified Hirt procedure for rapid purification of extrachromosomal DNA from mammalian cells. Biotechniques. 1998;24:760–762. doi: 10.2144/98245bm14. [DOI] [PubMed] [Google Scholar]
- 50.Taccioli G.E., Rathbun G., Oltz E., Stamato T., Jeggo P.A., Alt F.W. Impairment of V(D)J recombination in double-strand break repair mutants. Science. 1993;260:207–210. doi: 10.1126/science.8469973. [DOI] [PubMed] [Google Scholar]
- 51.Honma M., Izumi M., Sakuraba M., Tadokoro S., Sakamoto H., Wang W., Yatagai F., Hayashi M. Deletion, rearrangement, and gene conversion; genetic consequences of chromosomal double-strand breaks in human cells. Environ. Mol. Mutagen. 2003;42:288–298. doi: 10.1002/em.10201. [DOI] [PubMed] [Google Scholar]
- 52.Honma M., Sakuraba M., Koizumi T., Takashima Y., Sakamoto H., Hayashi M. Non-homologous end-joining for repairing I-SceI-induced DNA double strand breaks in human cells. DNA Repair. 2007;6:781–788. doi: 10.1016/j.dnarep.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Sasanuma H., Murakami H., Fukuda T., Shibata T., Nicolas A., Ohta K. Meiotic association between Spo11 regulated by Rec102, Rec104 and Rec114. Nucleic Acids Res. 2007;35:1119–1133. doi: 10.1093/nar/gkl1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Postow L., Woo E.M., Chait B.T., Funabiki H. Identification of SMARCAL1 as a component of the DNA damage response. J. Biol. Chem. 2009;284:35951–35961. doi: 10.1074/jbc.M109.048330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Takata M., Sasaki M.S., Tachiiri S., Fukushima T., Sonoda E., Schild D., Thompson L.H., Takeda S. Chromosome instability and defective recombinational repair in knockout mutants of the five Rad51 paralogs. Mol. Cell. Biol. 2001;21:2858–2866. doi: 10.1128/MCB.21.8.2858-2866.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kirkland D., Pfuhler S., Tweats D., Aardema M., Corvi R., Darroudi F., Elhajouji A., Glatt H., Hastwell P., Hayashi M., et al. How to reduce false positive results when undertaking in vitro genotoxicity testing and thus avoid unnecessary follow-up animal tests: Report of an ECVAM Workshop. Mut. Res. 2007;628:31–55. doi: 10.1016/j.mrgentox.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 57.Zhang L.S., Honma M., Hayashi M., Suzuki T., Matsuoka A., Sofuni T. A comparative study of TK6 human lymphoblastoid and L5178Y mouse lymphoma cell lines in the in vitro micronucleus test. Mut. Res. 1995;347:105–115. doi: 10.1016/0165-7992(95)00027-5. [DOI] [PubMed] [Google Scholar]
- 58.Oettinger M.A., Schatz D.G., Gorka C., Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 59.Schatz D.G., Oettinger M.A., Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 60.Schatz D.G. V(D)J recombination moves in vitro. Semin. Immunol. 1997;9:149–159. doi: 10.1006/smim.1997.0068. [DOI] [PubMed] [Google Scholar]
- 61.Frank K.M., Sekiguchi J.M., Seidl K.J., Swat W., Rathbun G.A., Cheng H.L., Davidson L., Kangaloo L., Alt F.W. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature. 1998;396:173–177. doi: 10.1038/24172. [DOI] [PubMed] [Google Scholar]
- 62.Grawunder U., Zimmer D., Fugmann S., Schwarz K., Lieber M.R. DNA ligase IV is essential for V(D)J recombination and DNA double-strand break repair in human precursor lymphocytes. Mol. Cell. 1998;2:477–484. doi: 10.1016/s1097-2765(00)80147-1. [DOI] [PubMed] [Google Scholar]
- 63.Chen B.P.C., Uematsu N., Kobayashi J., Lerenthal Y., Krempler A., Yajima H., Löbrich M., Shiloh Y., Chen D.J. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J. Biol. Chem. 2007;282:6582–6587. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- 64.Ding Q., Reddy Y.V., Wang W., Woods T., Douglas P., Ramsden D.A., Lees-Miller S.P., Meek K. Autophosphorylation of the catalytic subunit of the DNA-dependent protein kinase is required for efficient end processing during DNA double-strand break repair. Mol. Cell. Biol. 2003;23:5836–5848. doi: 10.1128/MCB.23.16.5836-5848.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bansbach C.E., Boerkoel C.F., Cortez D. SMARCAL1 and replication stress: An explanationfor SIOD. Nucleus. 2010;1:245–248. doi: 10.4161/nucl.1.3.11739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Takeda S., Rodewald H.R., Arakawa H., Bluethmann H., Shimizu T. MHC class II molecules are not required for survival of newly generated CD4+ T cells, but affect their long-term life span. Immunity. 1996;5:217–228. doi: 10.1016/s1074-7613(00)80317-9. [DOI] [PubMed] [Google Scholar]
- 67.Barton O., Naumann S.C., Diemer-Biehs R., Kunzel J., Steinlage M., Conrad S., Makharashvili N., Wang J., Feng L., Lopez B.S., et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell Biol. 2014;206:877–894. doi: 10.1083/jcb.201401146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Betous R., Glick G.G., Zhao R., Cortez D. Identification and characterization of SMARCAL1 protein complexes. PloS One. 2013;8:e63149. doi: 10.1371/journal.pone.0063149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quan J., Yusufzai T. HARP preferentially co-purifies with RPA bound to DNA-PK and blocks RPA phosphorylation. Epigenetics. 2014;9:693–697. doi: 10.4161/epi.28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Drouet J., Delteil C., Lefrancois J., Concannon P., Salles B., Calsou P. DNA-dependent protein kinase and XRCC4-DNAligase IV mobilization in the cell in response to DNA double strand breaks. J. Biol. Chem. 2005;280:7060–7069. doi: 10.1074/jbc.M410746200. [DOI] [PubMed] [Google Scholar]
- 71.Yano K.I., Chen D.J. Live cell imaging of XLF and XRCC4 reveals anovel view of protein assembly in the non-homologous end-joining pathway. Cell Cycle. 2008;7:1321–1325. doi: 10.4161/cc.7.10.5898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.