


Abstract
The HIV-1 restriction factor SAMHD1 is a tetrameric enzyme activated by guanine nucleotides with dNTP triphosphate hydrolase activity (dNTPase). In addition to this established activity, there have been a series of conflicting reports as to whether the enzyme also possesses single-stranded DNA and/or RNA 3′-5′ exonuclease activity. SAMHD1 was purified using three chromatography steps, over which the DNase activity was largely separated from the dNTPase activity, but the RNase activity persisted. Surprisingly, we found that catalytic and nucleotide activator site mutants of SAMHD1 with no dNTPase activity retained the exonuclease activities. Thus, the exonuclease activity cannot be associated with any known dNTP binding site. Monomeric SAMHD1 was found to bind preferentially to single-stranded RNA, while the tetrameric form required for dNTPase action bound weakly. ssRNA binding, but not ssDNA, induces higher-order oligomeric states that are distinct from the tetrameric form that binds dNTPs. We conclude that the trace exonuclease activities detected in SAMHD1 preparations arise from persistent contaminants that co-purify with SAMHD1 and not from the HD active site. An in vivo model is suggested where SAMHD1 alternates between the mutually exclusive functions of ssRNA binding and dNTP hydrolysis depending on dNTP pool levels and the presence of viral ssRNA.
It is quite common that enzymes with one major activity also catalyze other minor reactions that take advantage of the same active site environment and catalytic residues (1,2). One classic example is the major DNA phosphodiester hydrolysis activity of DNase I and its minor activity of 3′-phosphate monoester hydrolysis from DNA ends (3,4). Both reactions occur in the same active site and likely take advantage of the similar active site elements, even though the transition states and catalytic requirements for hydrolysis of phosphate diesters and monoesters are quite different (5,6). Thus, in general it is not unanticipated that dNTP hydrolases might also possess other phosphate ester hydrolyzing activities and that these additional activities might be of biological significance.
Sterile Alpha Motif and Histidine-Aspartate Domain 1 protein (SAMHD1) is a Mg2+-dependent homotetrameric enzyme that indiscriminately hydrolyzes all dNTPs to deoxynucleoside and tripolyphosphate products (7,8). The enzyme plays a key role in an innate immunity pathway that restricts HIV-1 infection of resting immune cells by preventing efficient completion of reverse transcription. The mechanism may involve depletion of the dNTP substrates of reverse transcriptase and/or other activities of SAMHD1 (9). SAMHD1 also contributes to the stability of the genome by restricting the replication of mutagenic retroelements, although this function does not appear to require its dNTPase activity (10). Mutations at the SAMHD1 locus are associated with the inherited inflammatory autoimmune disease Aicardi-Goutières syndrome that mimics chronic viral infection (11).
SAMHD1 has a complex activation mechanism involving nucleotide binding to two classes of activator sites (A1 and A2) as well as four catalytic sites on the HD domain of each tetramer. Nucleotide binding energy is used to induce oligomerization of the enzyme from its inactive monomer and dimer forms that predominate in the absence of nucleotide activation (12,13). Nucleotide activation has been shown to follow an ordered-essential mechanism (14): preferential binding of GTP to the four A1 sites promotes dimerization, which is followed by binding of any dNTP to each of four A2 sites, and substrate dNTPs to each of the four catalytic sites. Occupation of all the sites is required to drive the dimer to tetramer transition. Thus, remarkably, the SAMHD1 tetramer binds a total of twelve nucleotides in its activated state, and the tetramer is stable for many hours after nucleotides have been depleted (14). The enzyme is evolutionarily related to a large superfamily of HD-domain proteins that contain a characteristic H...HD…D sequence motif required for divalent metal ion binding (15). Although the vast majority of the known HD family members have phosphohydrolase activities, recent studies have uncovered a clade that possesses mixed-valent diirion-dependent oxygenase activity (16,17). Thus, the architecture of this protein fold and its metal binding properties are capable of yielding diverse enzymatic catalytic properties.
Although several groups have qualitatively confirmed that SAMHD1 binds ssDNA and ssRNA, but not duplex DNA or duplex RNA/DNA hybrids (18–20), there have been multiple and conflicting reports as to whether SAMHD1 has 3′-5′ exonuclease activity (7,18,19,21). Notably, Beloglazova et al. reported that SAMHD1 exhibited a major 3′-5′ ssDNA exonuclease activity and also a minor ssRNA exonuclease activity if Ca2+ was used as the catalytic metal (19). This group assigned these activities to SAMHD1 rather than contaminating exonucleases on the basis that mutation of key active site groups that ablated dNTPase activity (H167A, H206A, D207A, D311A) also reduced exonuclease activity (19). In contrast, Ryoo et al. recently reported that SAMHD1 had no detectable DNase activity, but that its ssRNase activity was robust in buffers containing the more physiologically relevant divalent cation Mg2+ (21). The studies of Ryoo et al. were also supported by measurements of the damaging effects of SAMHD1 mutations on the in vitro RNA exonuclease activity, which were extended to cell based studies that indicated the RNase activity was essential for restricting viral infection (21). In contrast to the above reports, other groups have not detected any exonuclease activity, but were in agreement that SAMHD1 binds to single-stranded nucleic acid (18,20).
We were perplexed by the above reports and sought to perform more extensive measurements to characterize the exonuclease and nucleic acid binding properties of SAMHD1. Our findings show that (i) a contaminating 3′-5′ DNA exonuclease activity readily co-purifies with SAMHD1 and is largely removed after three chromatography steps, (ii) a low level 3′-5′ RNA exonuclease activity co-purifies with SAMHD1 after three chromatography steps, (iii) neither of the above exonuclease activities are affected by active site mutations that completely abolish the dNTPase activity of SAMHD1 and (iv) SAMHD1 shows preferential binding to ssRNA over ssDNA and that ssRNA binding selectively induces oligomerization of SAMHD1 on RNA. Our findings suggest that the single-stranded nucleic acid binding activity of SAMHD1, in addition to its dNTPase action, are likely determinants involved in HIV restriction and immune activation.
MATERIALS AND METHODS
General reagents
2′-Deoxyguanosine-5′-triphosphate (dGTP) was obtained from Promega, 2′-deoxyuridine-5′-triphosphate (dUTP) was obtained from Roche, tritiated 2′-deoxyuridine-5′-triphosphate tetraammonium salt ([5–3H] dUTP) and tritiated 2′-deoxyguanosine-5′-triphosphate tetraammonium salt ([8-3H] dGTP) were from Moravek Biochemicals, 2′-deoxyguanosine-5′-[α-thio] triphosphate lithium salt (dGTPαS) was from ChemCyte, guanosine-5′-triphosphate was from Sigma-Aldrich, and C18-reversed phase thin layer chromatography (TLC) plates were purchased from Macherey-Nagel. DEPC-treated water was obtained from Quality Biological, Inc. T7 RNA polymerase, T4 polynucleotide kinase, T7 DNA ligase and restriction enzymes were obtained from New England Biolabs. Pfu Ultra DNA polymerase was obtained from Agilent Technologies.
Oligonucleotides
All DNA oligonucleotides and the 10, 20 and 40 nt RNA oligonucleotides were obtained from Integrated DNA Technologies (IDT). The 90 nt RNA oligonucleotide was produced via in vitro transcription with T7 RNA Polymerase and a polymerase chain reaction (PCR)-amplified runoff transcript. The RNA was isolated by Qiagen RNeasy Mini Kit. The 5′-FAM labeled 147 nt DNA oligonucleotide was produced by splint mediated ligation with T4 DNA ligase, followed by polyacrylamide gel electrophoresis (PAGE) purification. The sequences of all oligonucleotides used in this study are listed in Supplementary Table S1.
SAMHD1 mutagenesis, expression and purification
SAMHD1 mutants were produced via QuikChange mutagenesis of the previously reported bacterial expression plasmid, and were verified by Sanger sequencing. The wt or mutant SAMHD1 plasmids were transformed into BL21(DE3) Escherichia coli, grown and isolated, lysed, and purified by Ni-NTA and Mono S CE chromatography as described previously (14). At each step during the purification (after the Ni-NTA column, after treatment with PreScission Protease, and after the Mono S column) protein samples were exchanged into storage buffer (50 mM Tris–HCl (pH 7.5), 150 mM KCl, 5 mM MgCl2, 1 mM DTT and 10% glycerol) by PD-10 column (GE Healthcare) and kept on ice for the remainder of the purification. For the hydrophobic interaction chromatography, the protein solution obtained from the CE purification was adjusted to 1 M (NH4)2SO4 and loaded on equilibrated phenyl sepharose resin (GE Healthcare). The column was washed with HIC buffer A (50 mM potassium phosphate pH 7.0, 1 M (NH4)2SO4, 5 mM MgCl2, 1 mM DTT, 10% glycerol) and eluted with a gradient of HIC buffer B (50 mM potassium phosphate pH 7.0, 5 mM MgCl2, 1 mM DTT, 10% glycerol). The eluted protein was exchanged into storage buffer by PD-10 column as described above. Each protein fraction was then aliquoted into small single-use volumes, flash frozen, and kept at −80°C until analysis. Protein concentrations were determined by absorbance measurements at 280 nm using the calculated (Protparam tool, ExPASy) molar extinction coefficients for full-length human SAMHD1 monomer (ϵ = 76 500 M−1 cm−1) and the ΔSAM (Δ112) monomer (ϵ = 62 000 M−1 cm−1).
dNTPase activity measurements
The dNTPase activity measurements were performed as described previously (14). Briefly, 3H-labeled dGTP or dUTP substrates were separated from the deoxynucleoside products by reverse-phase C18 TLC chromatography and quantified by phosphorimaging. The cpm ratio for the substrate and product spots was used to determine percent reaction as a function of time. For each condition tested initial rates were determined from linear fits of this data to no >25% reaction.
Nuclease activity measurements
Nuclease assays were carried out in identical reaction conditions to the dNTPase activity measurements described above (50 mM Tris–HCl [pH 7.5], 50 mM KCl, 5 mM MgCl2, 0.5 mM TCEP). Reactions of SAMHD1 with 5′ 32P-labeled ssDNA and ssRNA substrates were quenched at particular time points by the addition of one reaction volume to two volumes of formamide loading buffer. The substrate and cleavage products were resolved by 14% denaturing TBE polyacrylamide sequencing gels and imaged by phosphorimaging screen overnight. The screen was scanned on either a Storm or Typhoon Phosphorimager (GE Healthcare) and quantified with Quantity One Software (BioRad). Quantification of the individual bands in each lane was done from the trace areas. Each band trace area was normalized as a fraction of total trace area in that lane (to account for any minor differences in load volumes). From these normalized fractions, the number of nucleotides released (for the DNase activity) or the fraction of substrate remaining (for the RNase activity) were determined at each time point. The initial rates for each condition tested were obtained as linear fits to these plots. Because background hydrolysis of the RNA substrate was observed in buffer alone, the background rate was subtracted from the activity measurements in the presence of SAMHD1. The DNase activity measurements did not require any buffer background correction.
Nucleic acid binding measurements using fluorescence anisotropy
The fluorescence anisotropy measurements were carried out in quartz microcuvettes in a FluoroMax 3 Spectrofluorometer (Horiba Scientific) maintained at 20°C. Binding reactions were prepared in binding buffer (50 mM Tris–HCl [pH 7.5], 50 mM KCl, 1 mM EDTA) with either 50 nM (for ssDNA) or 10 nM (for ssRNA) of 5′ FAM-labeled oligonucleotides, unless otherwise indicated. Anisotropy of the FAM fluorophore was measured (excitation and emission wavelengths of 495 and 520 nm; 1 s integration time) as increasing concentrations of SAMHD1 were added. The total volume added was kept to <20%, and control experiments confirmed that the addition of SAMHD1 did not significantly decrease the FAM fluorescence signal. The resulting anisotropy vs. total [SAMHD1] curves were fit to a quadratic binding equation:
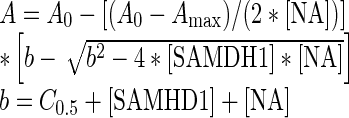 |
(1) |
where A is the observed anisotropy, A0 is the initial anisotropy value, Amax is the maximal anisotropy value at saturation, [NA] is the total nucleic acid concentration, [SAMHD1] is the total SAMHD1 concentration, and C0.5 is the SAMHD1 concentration at half-maximal saturation.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays were performed under the same binding conditions used for the fluorescence anisotropy measurements, except that a final concentration of 10% glycerol was added. Binding reactions were prepared, allowed to react for 1 min, then loaded in the wells of a 5% acrylamide (75:1 acrylamide:bisacrylamide) TAE gel. The gels were run in 1× TAE with a temperature probe to maintain the gel temperature no more than 37°C, then imaged directly (for FAM) or dried and imaged by phosphorimaging as described above (for 32P).
Glutaraldehyde crosslinking and silver staining
The analysis of the oligomeric state of SAMHD1 by glutaraldehyde crosslinking was performed as described previously (22). Briefly, reactions containing SAMHD1 and the indicated reaction components were prepared and mixed with 50 mM glutaraldehyde, then incubated for 15 min. The crosslinking reaction was quenched by addition of excess Tris–HCl, then resolved on Novex 4–12% Bis–Tris denaturing polyacrylamide gels. The proteins were visualized by silver staining using the previously-described protocol (22).
AFM substrate preparation
To prepare a duplex substrate containing a single-stranded DNA overhang, a region of the pMC454 plasmid (23) containing an Nt.BbvCI site and SalI site was amplified by PCR using oligonucleotide primers. Sequential digestion of the PCR product by SalI and Nt.BbvCI produced a 272 bp duplex with a 23 nt ssDNA 5′ overhang on one end. An oligonucleotide with a region complementary to this overhang and containing an additional 60 nt of poly(T) on the 3′ end was PAGE purified and 5′ phosphorylated with T4 polynucleotide kinase. A ligation reaction containing the digested PCR product and a 5-fold excess of the 5′ phosphorylated oligonucleotide was ligated with T7 DNA ligase and purified by agarose gel electrophoresis to isolate the product containing a 295 bp duplex region with a 60 nt 3′ overhang on one end.
To prepare a duplex substrate containing a single-stranded RNA overhang, a 302 nt ssRNA oligonucleotide was produced by in vitro transcription using T7 RNA polymerase with a PCR-amplified runoff transcript and was isolated by Qiagen RNeasy Mini Kit. The RNA oligonucleotide was annealed with a 1.6-fold excess of a PAGE-purified ssDNA 200mer, then the complex was purified by agarose gel electrophoresis.
AFM measurements
AFM studies were performed with the use of APS-mica methodology as described previously (24). SAMHD1 was mixed with DNA or RNA substrate in binding buffer (50 mM Tris–HCl [pH 7.5], 50 mM KCl, 1 mM EDTA) to make a 4:1 or 10:1 protein-to-DNA/RNA molar ratio and incubated in the test tube for 2 min at room temperature. Prior to the sample deposition, APS mica was placed on ice and 10 μl of the protein-nucleic acid mixture was deposited on the surface for 2 min followed by washing with deionized water (AquaMax™-ulta, CA). AFM images were acquired in air on a Multimode NanoScope VIII system operating in PeakForce mode (Bruker, Santa Barbara, CA, USA). MSNL probes (Bruker, Santa Barbara, CA, USA) with spring constant ∼0.5 N/m and resonance frequency ∼120 kHz were used.
AFM data analysis
Both the length of the duplex nucleic acid and protein volume (cross section option) were measured using the FemtoScan program (Advanced Technologies Center, Moscow, Russia) as described previously (25,26). The data were compiled into histograms for DNA length and protein volume using the Origin 8.5 program (Origin Lab, Northampton, MA, USA).
RESULTS AND DISCUSSION
Exonuclease and dNTPase activities of SAMHD1 and its active site mutants
The first goal of this study was to establish whether the ascribed exonuclease activities of SAMHD1 could be chromatographically separated from its dNTPase activity. Two mutant versions of SAMHD1 with substitutions in the essential HD-domain metal ligands His206 and Asp207 (D207A, and H206A/D207A) were purified and assayed in the same manner to test whether the dNTPase and exonuclease activities both depended on the conserved active site chemistry (7,12). This approach requires careful and simultaneous measurement of both activities using the same enzyme fractions obtained after each chromatographic step, which should ideally use distinct separation modalities (Figure 1A). To facilitate the nickel affinity chromatography step, we employed a SAMHD1 construct with a 10 residue amino terminal His-tag that could be cleaved away after affinity purification using a site-specific protease (PreScission Protease, PP). After gradient elution from the Ni-affinity column only a single major band corresponding to His10-SAMHD1 was observed with Coomassie blue staining when loading 6 μg of protein in the gel lane (lane 1, Figure 1B). Further purification steps involved proteolytic removal of the His-tag, followed by cation exchange (CE) and hydrophobic interaction (HIC) chromatography (lanes 2–4, Figure 1B). These additional chromatography steps removed the PreScission Protease and several trace impurities.
Figure 1.
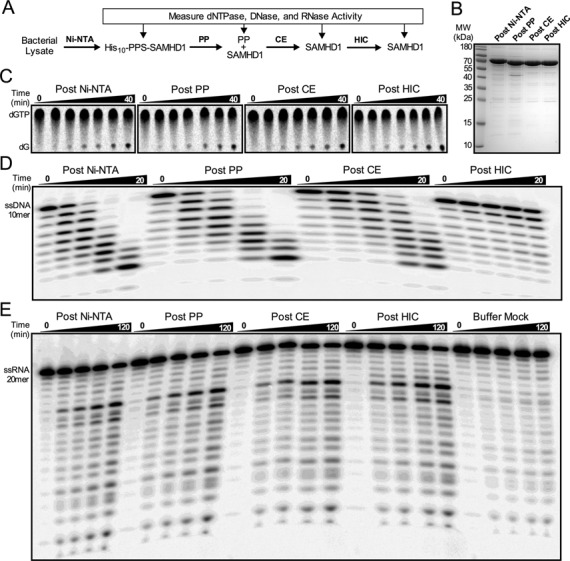
Copurification analysis of the dNTPase, DNase, and RNase activities. (A) SAMHD1 was purified by the indicated chromatography steps, with the three enzymatic activities tested at each point. (B) SDS-PAGE analysis of the purified SAMHD1 proteins (6 μg) with detection by Coomassie Brilliant Blue. Representative time course data is shown for the (C) RP-TLC dNTPase assay of SAMHD1 (0.5 μM) with dGTP (1 mM), (D) the PAGE ssDNase assay of SAMHD1 (0.5 μM) and ssDNA10 (1 μM), and (E) the PAGE ssRNase activity of SAMHD1 (0.5 μM) and ssRNA20 (1 μM).
For wild-type (wt)-SAMHD1, D207A, and H206A/D207A SAMHD1 the dNTPase, ssDNase, and ssRNase activities were measured on the same enzyme fractions obtained after each of the above chromatography steps with the expectation that the ratio of the activities would remain constant if they resided in the same protein (Figure 1C–E). The activity assays were not only performed on the same enzyme fraction, but also at the same time to negate any possible differences arising from a time-dependent loss in enzyme activity. For the dNTPase assay we used reversed-phase thin layer chromatography to separate the products resulting from time-dependent hydrolysis of 8-(3H)-dGTP, a preferred substrate and activator (Figure 1C) (14). For the DNase and RNase assays we used 5′-32P-labeled 10mer ssDNA and 20mer ssRNA, both of which are substrates for SAMHD1 exonuclease activities according to previous reports (19,21). The successive removal of nucleotides from the 3′-end of these oligonucleotides by SAMHD1 was quantitatively monitored by denaturing polyacrylamide gel electrophoresis (Figure 1D and E). (The corresponding dNTPase and single stranded exonuclease activity results for D207A SAMHD1 are shown in Supplementary Figure S1.) In all assays of SAMHD1, 5 mM Mg2+ was provided to satisfy the divalent cation requirement, which we confirmed was the preferred divalent metal for the dNTPase activity and has also been reported to be the preferred metal for the RNase and DNase activities (Supplementary Figure S2) (19,21). We also confirmed previous reports that SAMHD1 has no double-stranded DNA exonuclease activity using preparations that had undergone Ni-affinity and CE chromatography (Supplementary Figure S3) (18,19).
The relative dNTPase, DNase and RNase activities of wt-SAMHD1 and its D207A and H206A/D207A mutant forms are shown as bar graphs in Figure 2A–C. To facilitate comparisons, each enzyme activity was normalized to the corresponding activity of the wild-type enzyme measured after the Ni-NTA purification step. For wt-SAMHD1, a modest 50% enhancement of the dNTPase activity occurred over the three chromatography steps, but the DNase activity fell by 30-fold after the third HIC step, indicating that all, or the overwhelming majority of this activity, could not be attributed to SAMHD1 (black bars, Figure 2A and B). Importantly, our previously reported kcat of 2 s−1 for dGTP hydrolysis by SAMHD1 is equal to or greater than activities reported elsewhere, so the failure to observe DNase activity cannot be attributed to an overall low activity of our enzyme preparation (14,27,28). In contrast to the DNase activity, a weaker RNase activity persisted over the three purification steps, suggesting that this activity might be assignable to SAMHD1 (black bars, Figure 2C).
Figure 2.
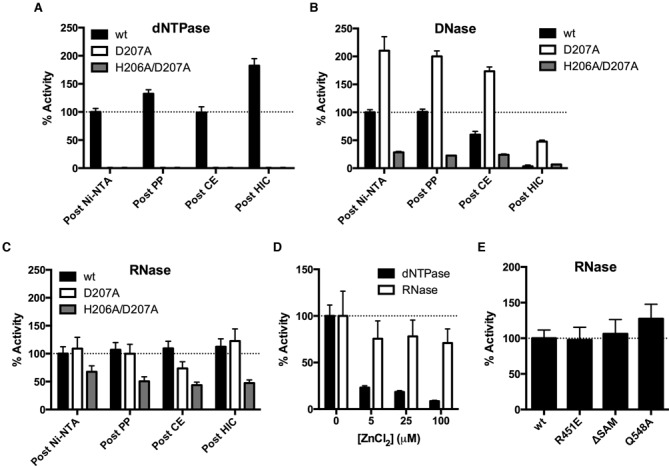
The exonuclease activities cannot be attributed to the SAMHD1 active site. (A) dNTPase activities of wild-type and mutant enzymes were measured after each purification step. The D207A and H206A/D207A enzymes had no detectable dNTPase activities. (B) ssDNA exonuclease activities and (C) ssRNA exonuclease activities after each purification step. Each wild-type and mutant enzyme activity was normalized to the corresponding activity of the wild-type enzyme measured after the Ni-NTA purification step. (D) Rates of dNTPase or ssRNA exonuclease activity were measured with the indicated concentration of ZnCl2 (in the presence of 5 mM MgCl2). (E) The normalized ssRNA exonuclease activities of the wild-type, R451E, ΔSAM or Q548A enzymes. All activities were measured after the CE chromatography step (Figure 1A).
We then investigated whether single or double mutagenesis of the catalytically essential active site metal chelating residues (D207A or H206A/D207A) had an effect on these activities. As expected, both the single and double mutations completely abolished the dNTPase activity (>100-fold reduction based on the limits of detection) at all purification steps (white and red bars, Figure 2A). In contrast, the D207A mutant showed a 2-fold greater DNase activity than wt-SAMHD1 after the first Ni-affinity column, while the H206A/D207A mutant showed about one-third the activity after this step. In both cases the DNase activity decreased by 5- to 10-fold after the third HIC step, which is similar to the results obtained with wt-SAMHD1 (white and grey bars, Figure 2B). These results confirm that all or most of the DNase activity cannot be attributed to SAMHD1, nor can it be assigned to the enzyme active site. Similarly, the D207A mutant showed nearly identical RNase activity as wt-SAMHD1 after the first Ni-affinity step, while the activity of the H206A/D207A mutant was only 2-fold less than wt-SAMHD1 (white and grey bars, Figure 2C). As observed with wt-SAMHD1, the RNase activities of the mutant enzymes were resistant to a pan-RNase inhibitor (RiboLock, Supplementary Figure S4), and persisted over the three chromatography steps (Figure 2C).
dNTPase activity is inhibited by Zn2+ and the RNase activity is not
As an orthogonal approach to determine whether the active site of SAMHD1 gave rise to the trace RNase activity we took advantage our recent finding that the catalytically inert metal Zn2+ binds very tightly to the SAMHD1 active site and strongly inhibits its dNTPase activity, even in the presence of a vast excess of Mg2+ (29). When 5–100 μM ZnCl2 was added to reactions with wt-SAMHD1, 90% of the dNTPase activity was inhibited in a dose dependent manner (black bars, Figure 2D). In contrast, the RNase rate was inhibited by <30% with no apparent concentration dependence over the same range. This disparity in Zn2+ inhibition further supports the mutagenesis results indicating that the RNase and dNTPase activities do not use the same HD domain active site chemistry.
Does the residual RNase activity arise from outside the HD-domain active site?
The above results do not exclude the possibility that SAMHD1 might possess another active site where the RNase activity resides. To explore this possibility we purified three mutant forms: (i) a deletion mutant lacking the N-terminal SAM domain (residues 1–112, ΔSAM), (ii) a charge reversal mutant in the activator 1 (A1) site that is known to prevent formation of dimers and tetramers (R451E) (30), and (iii) the Q548A mutant that is a second shell residue between the activator and active sites and has been previously reported to abolish the RNase activity (21). All of these enzymes were purified identically using nickel affinity and CE chromatography, and each retained nearly identical levels of RNase activity as measured with wt-SAMHD1 after the same purification steps (Figure 2E). If the RNase activity indeed arises from SAMHD1 and not a contaminant, we can now exclude several regions of SAMHD1 from consideration: (a) the HD-domain active site, (b) the SAM domain, and (c) the nucleotide activator sites. Further, the full RNase activity of the R451E mutant requires that any putative SAMHD1 RNase activity must be entirely independent of protein tetramerization, which contrasts completely with the dNTPase activity. Although we confirmed that the Q548A mutant retains dNTPase activity as previously reported (21), we note that our results diverge because we do not see the previously reported decrease in RNase activity with this mutant (Supplementary Figure S5) (21).
From the above results we make the following conclusions: (i) the residual RNase activity could belong to a trace enzyme that perfectly co-purifies with SAMHD1, (ii) if the RNase activity is assignable to SAMHD1, it does not utilize the same active site or tetrameric oligomeric state as the dNTPase activity, (iii) the RNase activity does not require an intact A1 or A2 activator site (the formation of the A2 site requires dimerization, which is prevented by the R451E mutation), and (iv) the activity does not require the SAM domain. These results do not support a previous conclusion that active site-localized RNase activity is required for the restriction of HIV by SAMHD1 (21). However, the above findings do not exclude a contribution of both nucleic acid binding and dNTPase activities to viral restriction and autoimmune disease (18).
What nucleic acid structures bind to SAMHD1?
There have been several previous reports that SAMHD1 binds preferentially to ssRNA and ssDNA (18–20), but a thorough investigation into this potentially important activity has not yet been reported. To explore this question, we investigated the relative binding affinities of SAMHD1 for different lengths of ssDNA, dsDNA, ssRNA, and RNA:DNA hybrid duplexes. With one exception that is noted below, all of the binding measurements were performed in the presence of excess EDTA to prevent residual nuclease activity. We used three general methods to assess binding: (i) direct binding was measured by the increase in fluorescence anisotropy of a 5′-fluorescein (FAM) label on the nucleic acid, (ii) competition displacement of a FAM-labeled oligonucleotide by an unlabeled oligonucleotide, and (iii) electrophoretic mobility shift assay (EMSA).
In the binding analysis below we characterize the relative affinities of various nucleic acid constructs using C0.5 values determined from fitting the data to Equation (1), which takes into account free ligand depletion. The C0.5 simply reflects the concentration of SAMHD1 monomer that is required to alter the observed anisotropy signal by 50% under the conditions of the given assay. This is the most appropriate way to make affinity comparisons because several aspects of this system complicate the determination of true thermodynamic parameters. These include: (i) the oligomeric state of SAMHD1 can change as a function of protein concentration during a titration with fixed nucleic acid, (ii) rigorous methods for measuring the relative affinities of SAMHD1 monomers, dimers and tetramers for nucleic acids are not available (31), (iii) the stoichiometry of protein binding is not obtainable because appropriate saturation conditions cannot be achieved (however, one exception is described below), (iv) the effect of nucleic acid binding on protein oligomerization is not known, and (v) the essential relationship between the measured fluorescence signals and fractional saturation is uncertain for such a complicated system (32,33).
None of the above complexities stood in the way of making the unambiguous conclusion that SAMHD1 binds to nucleic acids in the following order of binding affinity: ssRNA > ssDNA > RNA:DNA hybrid > dsDNA (Figure 3). Preferential binding to a 40mer ssRNA as compared to an RNA:DNA hybrid duplex of the same length was determined by fluorescence anisotropy measurements [C0.5 (ssRNA40) = 0.23 ± 0.07 μM and C0.5 (RNA:DNA40) = 2.0 ± 0.4 μM] (Figure 3A). A similar binding of SAMHD1 to a 5′-32P-labeled ssRNA 40mer was observed using EMSA, confirming that the presence of the 5′-FAM label does not significantly affect the binding interaction (Figure 3B). The fluorescence anisotropy measurements of SAMHD1 binding to ssDNA57 and dsDNA of the same length again also showed a large ∼34-fold preference for binding to the single-stranded nucleic acid [C0.5 (ssDNA57) = 0.45 ± 0.13 μM and (dsDNA57) = 18.0 ± 2 μM] (Figure 3C). [We were unable to obtain saturation in the case of dsDNA binding, however the extrapolated maximal anisotropy (0.25 ± 0.03) was similar to that obtained in the case of ssDNA.] Using EMSA, a similar concentration dependence of SAMHD1 binding to ssDNA57 was observed as compared to the fluorescence anisotropy measurements with 5′-FAM ssDNA57 (Figure 3D). We note that no cooperativity was observed in the fluorescence anisotropy binding curves for either single-stranded nucleic acid, and inclusion of a Hill coefficient did not improve curve- fitting statistics. This result should not be interpreted as evidence for the absence of cooperativity because it is possible, due to the nature of the anisotropy signal, that binding events subsequent to the initial protein binding event would not give rise to a significant further change in anisotropy. In this regard, the electrophoretic mobility assays display rather abrupt changes in band mobility, which does suggest cooperativity (Figure 3B and D). However, the moderate binding affinity of SAMHD1 for nucleic acids produces considerable smearing in these gels, which precludes any quantitative analysis.
Figure 3.
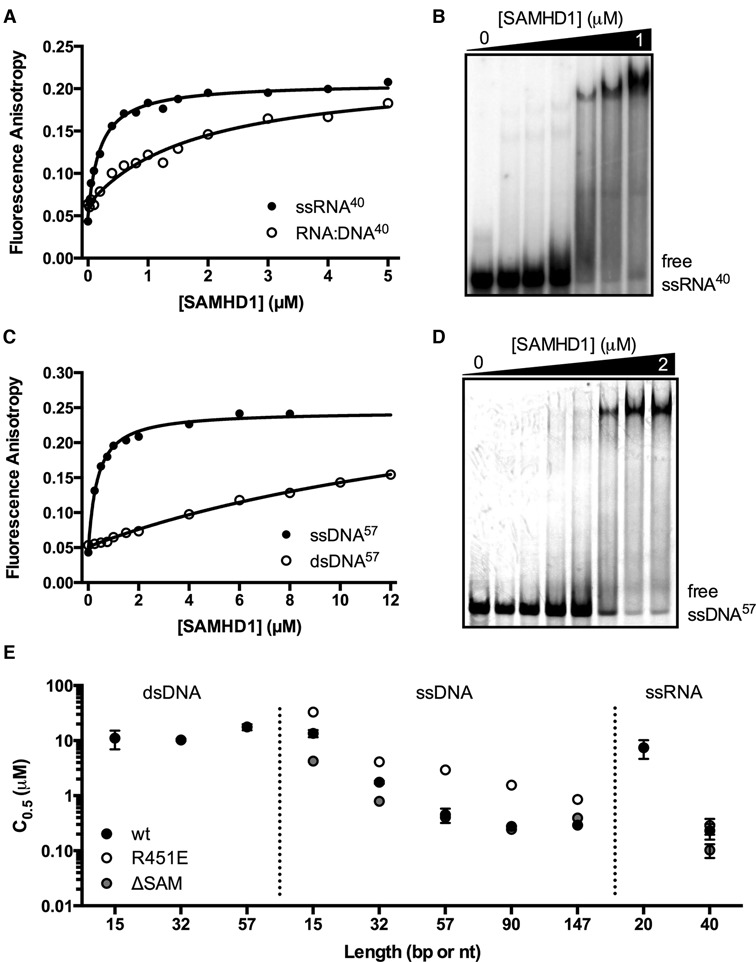
SAMHD1 is a single-stranded nucleic acid binding protein. (A) Binding of SAMHD1 to a 40mer 5′ FAM-labeled ssRNA (10 nM) and a 40 bp RNA:DNA hybrid duplex (10 nM) were measured by following the increase in fluorescence anisotropy of the FAM fluorophore upon nucleic acid binding. (B) Electrophoretic mobility shift (EMSA) measurements of SAMHD1 binding to ssRNA40 (10 nM). (C) The binding of SAMHD1 to a 57mer 5′ FAM-labeled ssDNA (50 nM) and a 57 bp DNA duplex (50 nM) were measured by following the increase in fluorescence anisotropy of the FAM fluorophore upon nucleic acid binding. (D) EMSA measurements of SAMHD1 binding to ssDNA57 (50 nM).
We investigated the length dependence for dsDNA, ssDNA, and ssRNA binding using FAM-labeled constructs with lengths in the range 10 to 147 nucleotides (nt) (Figure 3E, Supplementary Figure S6). Remarkably, dsDNA showed virtually no length dependence, while ssDNA and ssRNA showed increased binding affinities with length that began to reach a plateau when the length exceeded about 60 nt, suggesting an approximate binding footprint for ssDNA and ssRNA. EMSAs performed with varying lengths of ssRNA from 20 to 90 nt in length qualitatively mirrored the anisotropy results (Supplementary Figure S7).
Because of the similar structures and binding behaviors of these single-stranded nucleic acids, we reasoned that the ssDNA and ssRNA might bind to the same site on SAMHD1. To investigate this, we formed complexes of SAMHD1 with a 5′ FAM-labeled ssDNA57 or 5′ FAM-labeled ssRNA40 and monitored the decrease in anisotropy as increasing concentrations of unlabeled ssRNA or ssDNA competitors were added. As expected, the addition of unlabeled ssRNA to a complex of SAMHD1 and FAM-ssDNA 57mer decreased the anisotropy to baseline levels, indicating that ssRNA competes with ssDNA for binding (Supplementary Figure S8A). Similarly, the addition of unlabeled ssDNA to a complex of SAMHD1 and 5′-FAM ssRNA40 also decreased the anisotropy to baseline levels, indicating that ssDNA also competes with ssRNA for binding (Supplementary Figure S8B). The finding that ssDNA and ssRNA are mutually competitive is consistent with a shared binding site.
What oligomeric states and domains of SAMHD1 bind nucleic acids?
To test the domain and oligomeric state requirements for nucleic acid binding to SAMHD1 we employed the ΔSAM construct, which lacks the first 112 residues, and an R451E mutant that exists only in the monomeric form (see cross linking studies below). In fluorescence anisotropy measurements, the ΔSAM construct bound to ssRNA40 (Figure 4A) and ssDNA57 (Supplementary Figure S9) with similar affinities as observed for wt-SAMHD1. These findings establish that the SAM domain plays no discernable role in either single stranded nucleic acid binding or dNTP hydrolysis. The monomeric R451E construct also bound to ssRNA40 with an affinity identical to wt-SAMHD1 and ssDNA57 with only a 9-fold larger C0.5 than wt-SAMHD1 (Figure 4A, Supplementary Figure S9). Thus, from these mutational studies we conclude that the minimal unit that supports binding of SAMHD1 to single-stranded nucleic acids is the HD domain monomer.
Figure 4.
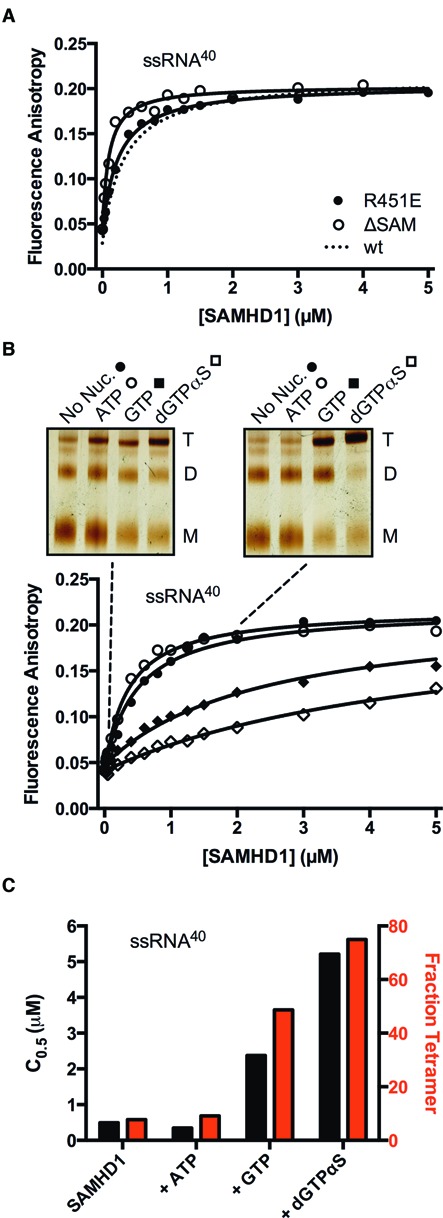
Binding of SAMHD1 to ssRNA40 does not require the SAM domain and is inhibited by guanine nucleotide-induced protein tetramerization. (A) Fluorescence anisotropy changes accompanying binding of the ΔSAM deletion mutant and the monomeric R451E mutant to ssRNA40 (10 nM). The binding curve for wt-SAMHD1 was very similar under the same conditions and is shown for comparison (dashed line). (B) Binding of wt-SAMHD1 to ssRNA40 was measured in the absence of nucleotide, and in the presence of ATP (non-activator), GTP, or dGTPαS (all at 1 mM concentration). For each nucleotide condition, the oligomeric state of SAMHD1 at two concentrations is indicated with the dashed lines (0.1 and 2 μM), which was determined by glutaraldehyde crosslinking and SDS-PAGE (insets). (C) The C0.5 values (black bars) and fraction of total SAMHD1 in the tetrameric form (red bars) for each nucleotide condition are correlated.
Since monomers were the minimal binding unit, we then asked whether the binding affinity for single-stranded nucleic acid might be altered by the guanine nucleotide-induced oligomerization of SAMHD1 to its dimeric and tetrameric forms. In this study, variable amounts of SAMHD1 were added to the FAM-ssRNA40 in the presence of 1 mM Mg2+ and various nucleotides provided at 1 mM concentration (inclusion of Mg2+ facilitates nucleotide binding). In the presence of Mg2+ alone, or upon addition of the negative control nucleotide ATP (which cannot bind to the activator sites and is not a substrate), the affinity of SAMHD1 for the FAM-ssRNA40 remained identical to that observed in the presence of EDTA (Figure 4B). In contrast, when the ribonucleotide GTP was added (which is not a substrate but binds to the A1 site and predominantly produces dimers) (14), the FAM-ssRNA40 binding curve showed 5-fold weaker affinity. When the non-hydrolyzable guanine analogue dGTPαS was added (which binds to the A1, A2 and catalytic sites and shifts the equilibrium to tetramer) (12,14), FAM-ssRNA40 binding was 10-fold weaker than in the absence of nucleotides (Figure 4B). We note that the above experiments performed in the presence of Mg2+ were executed in a time frame (<8 min) where less than 10% of the ssRNA was degraded by the residual RNase activity.
The oligomeric state of SAMHD1 that was present in each of the above nucleotide addition experiments was determined using glutaraldehyde crosslinking (22). We found that the inclusion of ATP did not change the oligomeric state over the absence of nucleotide, but the addition of GTP or dGTPαS pushed the enzyme towards its dimer and tetramer forms (Figure 4B, inset). These data establish that the C0.5 values for FAM-ssRNA40 binding increase as the amount of tetramer increases (i.e the nucleotide bound tetramer binds ssRNA more weakly than monomeric SAMHD1)(Figure 4C).
The above ssRNA binding data, where nucleotides are used to shift the oligomeric state of SAMHD1 from monomer to dimer to tetramer, should be interpreted in the context of the monomeric R451E mutant (Figure 4A): this mutant shows similar ssRNA binding activity as wt-SAMHD1 even though wild-type enzyme is predominantly in the dimeric form for a large portion of the binding reaction shown in Figure 4B (insets). Together, these observations suggest two equivalent mechanisms for ssRNA binding (i) free SAMHD1 dimers may directly bind to ssRNA or (ii) free monomers may bind in a sequential manner to form dimers on the RNA (see below). These two mechanisms are supported by the stoichiometry of binding to ssDNA57 and atomic force microscopy images (see below).
There have been previous reports that the Q548A mutant and phosphorylation of SAMHD1 at Thr592 negatively impact HIV restriction (21,34). To investigate whether these previous results might be attributed to disruption of ssRNA binding by these alterations, we measured ssRNA binding of the restriction-incompetent phosphomimetic mutant T592E as well as Q548A (21,34). By fluorescence anisotropy, the binding of the T592E and Q548A mutants to ssRNA40 and ssDNA57 were identical to wt-SAMHD1 (Supplementary Figure S10). Thus, both of these mutants maintain dNTPase and nucleic acid binding activity despite their apparent inability to restrict HIV infection when expressed in cells (21,34).
Single-stranded RNA binding induces distinct oligomeric states of SAMHD1
It is well-known that guanine nucleotide and dNTP binding drives formation of a dNTPase-active tetramer of SAMHD1 (14). We were curious whether single-stranded nucleic acids might induce oligomerization of SAMHD1 monomers on the nucleic acid scaffold, as has been observed with other enzymes involved in HIV restriction (35), or the interferon stimulatory DNA response (36). To compare the effects of nucleotide and single-stranded nucleic acid binding on the oligomeric state of wild-type and R451E SAMHD1, we used a previously described glutaraldehyde crosslinking method (22). The following results were obtained for wt-SAMHD1 (Figure 5A): (i) wt-SAMHD1 existed mostly as a mixture of monomers and dimers in the absence of any ligands as previously reported (14), (ii) addition of GTP largely served to shift the monomer-dimer equilibrium towards dimer (14), (iii) addition of GTP and dUTP drove the enzyme completely to tetramer (14), (iv) addition of ssDNA90 by itself had no effect on the oligomeric state, nor did it change the oligomeric states induced by dNTP binding and (v) addition of ssRNA oligomers as a sole agent (>40 nt) induced higher oligomeric forms of SAMHD1. The distinct oligomeric forms generated in the presence of ssRNA were not identical in mobility with the dimer and tetramers generated by the addition of dNTPs, nor do they prevent the formation of the dimer and tetramer forms upon addition of GTP and dUTP (Figure 5A). For R451E SAMHD1, addition of either nucleotides or nucleic acids did not shift this mutant from its monomeric form, indicating that a functional dimer interface is required for nucleotide and ssRNA induced oligomerization (Figure 5B).
Figure 5.
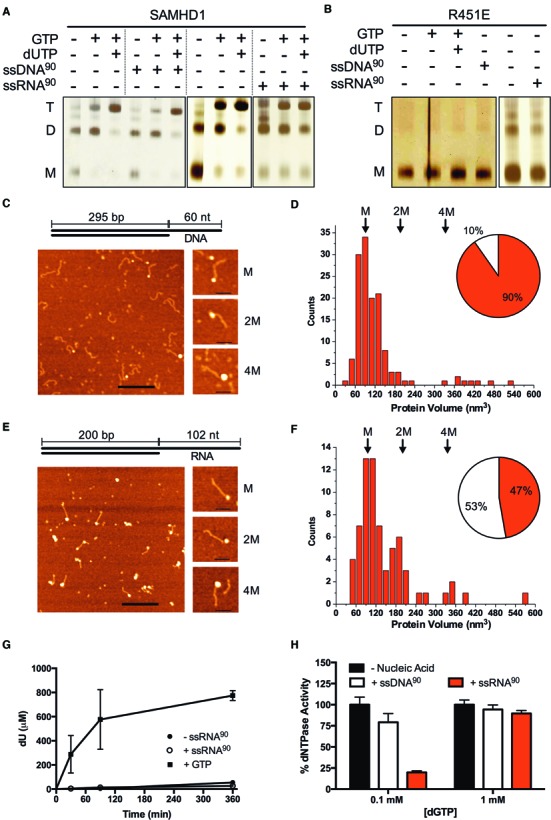
Longer ssRNA, but not ssDNA, induces higher oligomeric forms of SAMHD1 that are distinct from the tetramers induced by dNTP activation. (A) wt-SAMHD1 (1 μM) was crosslinked with glutaraldehyde in the presence of the indicated combinations of GTP (5 mM), dUTP (1 mM), ssDNA90 (2.5 μM), or ssRNA90 (2.5 μM) and the oligomeric forms were resolved by SDS-PAGE. Under conditions of this assay <10% of the dUTP was hydrolyzed by SAMHD1 and <5% of the ssRNA or ssDNA was hydrolyzed by the trace exonuclease activities that were present. (B) Glutaraldehyde crosslinking of the R451E mutant under identical conditions as wt-SAMHD1 showed that ssDNA, ssRNA and nucleotides were incapable of inducing higher order oligomeric forms. (C) AFM images of the wt-SAMHD1 complexes formed on the 60 nt ssDNA overhang of the 295 bp duplex AFM DNA construct (depicted) at 10:1 ratio. Bar size 200 nm. Shown to the right are representative images of complexes with protein volumes corresponding to SAMHD1 monomer (M), two monomers (2M) and four monomers (4M). Bar size 50 nm. (D) A histogram of the measured volumes of the globular ssDNA-protein complexes shows a distribution that is dominated by monomer complexes. The relative proportion of globular (red) and extended (white) protein complexes are shown in the inset pie chart. (E) AFM images of the wt-SAMHD1 complexes formed on the 102 nt ssRNA overhang of the 200 bp hybrid RNA:DNA duplex AFM construct (depicted) at 10:1 ratio. Bar size is 200 nm. On the right are representative images of complexes corresponding to SAMHD1 monomer (M), two monomers (2M) and four monomers (4M). Bar size is 50 nm. Complexes with protein volumes corresponding to monomer, dimer, and tetramer were observed in significant numbers. (F) A histogram of the measured volumes of the globular ssRNA-protein complexes showed a multimodal distribution with a shift towards higher order oligomeric species as compared to ssDNA. The relative proportion of globular (red) and extended (white) protein complexes are shown in the inset pie chart. (G) Despite inducing higher oligomeric forms of SAMHD1, ssRNA90 (2.5 μM) does not activate SAMHD1 (0.5 μM) for hydrolysis of the non-activating substrate dUTP. In contrast, addition of the activator GTP (0.1 mM) strongly induces dUTP hydrolysis. (H) SAMHD1 (0.5 μM) hydrolysis of low concentrations of the self-activating substrate dGTP (0.1 mM) is potently inhibited by ssRNA90 (2.5 μM) and to a lesser extent ssDNA90 (2.5 μM). Inhibition is not observed using high concentrations of dGTP (1 mM).
Further analysis of ssRNA-induced SAMHD1 oligomerization found that it was highly length-dependent: ssRNA10 and ssRNA20 were incapable of inducing a crosslinkable oligomeric state, ssRNA40 induced intermediate amounts, and ssRNA90 produced the largest levels (Figure 5A, Supplementary Figure S11). It is important to point out that the mobility shifts are unlikely to arise from crosslinking of SAMHD1 to RNA because (i) the R451E monomeric mutant has similar affinity for ssRNA as wt-SAMHD1 but induces no oligomerization (Figure 4A) and (ii) ssDNA binding to wt-SAMHD1 failed to induce oligomerization (Figure 5A). We conclude that oligomerization requires a minimal length of ssRNA to serve as a template and that crosslinking requires functional monomer-monomer contacts that are not stable with the R451E mutant.
Atomic force microscopy (AFM) imaging of SAMHD1-nucleic acid complexes
To complement the above measurements of ssDNA and ssRNA binding, we performed single-molecule AFM imaging of SAMHD1-nucleic acid complexes. Because single-stranded nucleic acid is not directly visible by AFM, we utilized a previous approach where a 295 bp dsDNA duplex with 60 nt ssDNA overhang on one end was used as imaging tag (26). These AFM experiments must also be performed using low concentrations of enzyme (8–20 nM) and DNA (2 nM) to prevent crowding on the matrix. Thus, comparisons between the solution and AFM measurements must take into account the different thermodynamic conditions of the experiments.
As a control for integrity of the single-strand overhang DNA duplex construct, we formed a complex with E. coli single-stranded DNA binding protein (SSB)(37). Complexes were deposited on APS-functionalized mica surface (38), and SSB proteins were observed bound to one end of each DNA molecule, confirming that the construct had the expected structural properties (Supplementary Figure S12A). Quantification of the protein volumes and the contour length of the duplex region in these images were consistent with the expected size of a 300 bp dsDNA B-form duplex with a single SSB tetramer bound to the single-strand end (Supplementary Figure S12B). When this same DNA construct was complexed with a 10-fold excess of SAMHD1, AFM images also showed the enzyme bound to only one end (Figure 5C), confirming the solution findings indicating that ssDNA is the preferred binding form (Figure 3C). The majority of the bound proteins were of a uniform globular size consistent with a SAMHD1 monomer, but a small number of larger protein complexes were also observed under these conditions (Figure 5C). For the globular complexes (90% of the total observed) the measured protein volumes showed a slightly skewed distribution with a maximum at the expected size of the 72 kDa SAMHD1 monomer (M, ∼100 nm3), and a tail stretched into to the expected size of two bound monomers (2M, Figure 5D). The observation that the major bound species was monomer is expected because SAMHD1 is almost entirely monomeric under the dilute conditions of the AFM experiments (14). A nearly identical distribution was obtained for the complexes at a lower 4:1 ratio of SAMHD1 to DNA, but fewer complexes were observed as expected using such dilute binding conditions (Supplementary Figure S13A and B). These AFM images confirm the solution studies where the SAMHD1 monomer was defined as the minimal unit for single-stranded DNA binding (Figure 4A).
The observation of a small population of 2M complexes on this 60mer ssDNA tail suggests that two monomers of SAMHD1 can reside on this length of ssDNA, perhaps using the known monomer-monomer interface (7). A stoichiometry of two monomers on an ssDNA of this length is supported by fluorescence anisotropy measurements using 5′-FAM ssDNA57 under stoichiometric binding conditions (Supplementary Figure S14). Although the SAMHD1 monomer is the dominant nucleic acid binding species due to its prevalent concentration and intrinsic binding affinity, both AFM images and fluorescence anisotropy measurements agree that two wt-SAMHD1 monomers can be bound in close proximity on ssDNA occupying about 60 nt of DNA.
To observe the complexes of SAMHD1 that form on ssRNA, we prepared a similar hybrid substrate containing 200 bp of an RNA:DNA duplex as an imaging tag with a 102 nt ssRNA overhang. This ssRNA length was selected because it is sufficient to form the higher-order oligomeric species observed in the crosslinking experiments, which were never observed for any length of ssDNA (Figure 5A). AFM images of this hybrid RNA:DNA construct showed the expected contour length of an A form duplex and E. coli SSB bound to a single end with volumes consistent with a single SSB tetramer (Supplementary Figure S12C and D). When the ssRNA overhang construct (2 nM) was complexed with SAMHD1 (20 nM), more protein–RNA complexes were formed as compared to the experiment with DNA (84% ssRNA ends bound compared to only 26% for ssDNA), indicating a higher binding affinity for RNA. A small number of complexes with SAMHD1 bound to both ends were observed, which is consistent with the weak affinity of the enzyme for RNA:DNA duplexes (Figure 3A). These results confirm the solution binding results where binding to ssRNA was favored (Figure 3C).
Considerably more size heterogeneity was observed in the ssRNA end-bound complexes as compared to ssDNA and this was more pronounced at a 10:1 ratio of enzyme to RNA as compared to 4:1 (Figure 5E and Supplementary Figure S13C and D). Quantification of the volumes of the most abundant globular complexes was consistent with the size of a SAMHD1 monomer as observed with ssDNA (see histogram in Figure 5F). However, a significant number of larger species were also apparent, some of which appeared elongated on the nucleic acid. The relative numbers of elongated and globular complexes observed with the ssDNA and ssRNA samples are shown as pie chart insets in Figure 5D and E, which indicates that ssRNA facilitates formation of the observed large species. The more abundant larger species observed in the AFM images with the ssRNA construct is consistent with the crosslinking experiments showing that oligomeric SAMHD1 complexes are formed preferentially on ssRNA.
Single-stranded RNA binding induces a dNTPase-inactive oligomeric state
To assess if the higher oligomeric forms induced by ssRNA90 binding are dNTPase competent, the non-activating substrate dUTP was incubated with SAMHD1 in the presence and absence of ssRNA, or in the presence of 0.1 mM GTP activator as a positive control (Figure 5G). The complete lack of dUTP hydrolysis in the presence of ssRNA90 alone indicated that the oligomeric forms observed by crosslinking are not equivalent to dNTPase-active tetramers. To further analyze the interplay between single-stranded nucleic acid binding and dNTPase activity, reactions with the self-activating substrate dGTP were carried out in the presence and absence of a saturating concentration of ssDNA or ssRNA 90mer (2.5 μM). Consistent with the observation that ssDNA90 was not capable of inducing oligomerization (Figure 5A), ssDNA did not inhibit the dNTPase reaction at either low or high concentrations of dGTP relative to its Km for hydrolysis (Figure 5H) (14). In contrast, ssRNA90 showed strong inhibition of the dGTPase activity at low dGTP concentrations, but not at higher concentrations (Figure 5H). These results indicate that the RNA-induced oligomeric forms are dNTPase-deficient, and that neither ssDNA nor ssRNA can prevent the formation of catalytically-competent tetramers when nucleotides are added at high concentrations. These results suggest that in resting immune cell targets of HIV that contain low dNTP pools SAMHD1 would primarily exist in dNTPase-inactive monomer/dimer forms and would be capable of binding ssRNA. When dNTP pool levels increase, SAMHD1 would shift to its dNTPase-competent tetrameric form.
Implications for SAMHD1 function
Much progress has been made in elucidating the mechanism of retroviral restriction by SAMHD1 in the relatively few years since its discovery. However, a determinate understanding of how its combined biochemical properties lead to restriction has not yet been achieved (18,19,21,39). One current model for SAMHD1 action invokes its RNase activity as the sole determinant of HIV-1 restriction (21). This conclusion was based on the observation that HIV-1 was able to efficiently infect U937 cells (a model cell line that resembles resting macrophages) when the Q548A mutant enzyme was ectopically expressed, but not when wt-SAMHD1 was expressed. This reasoning relies on the previous determination that the dNTPase activity is intact with Q548A, but the RNase activity is absent. Although we confirm the previous finding that Q548A has robust dNTPase activity, we also found no difference between the RNase activities of wt-SAMHD1 and this mutant. Our general findings are that (i) the RNase activity is weak and variable in different protein preparations, (ii) it not sensitive to Zn2+ inhibition like the dNTPase activity, (iii) it cannot be reasonably assigned to the active site or activator sites by mutational analysis and (iii) does not reside in the SAM domain. Although we cannot exclude the presence of another as of yet unknown exonuclease site on the HD domain, this seems unlikely based on structural and chemical evidence.
Our data suggest a bifunctional mechanism for HIV-1 restriction by SAMHD1 that utilizes its ssRNA binding and dNTPase activities (Figure 6). We propose that the dNTPase activity serves to deplete dNTP pools before the virus enters the cell and reduce SAMHD1 to its dNTPase-inactive monomer and dimer forms. When virus enters the cell, the low dNTP concentration would serve to slow viral first strand DNA synthesis. During this time window provided by the scarce dNTP pool, SAMHD1 monomers (M) can assemble on the HIV ssRNA template (∼2 M per 60 nt), and either slow or prevent further reverse transcription. Further, the detection of high-molecular weight SAMHD1 species with the size of four chemically crosslinked monomers (Figure 5A), combined with the observation of large globular and elongated RNA-protein complexes in AFM images (Figure 5E), suggests that the bound M and 2M forms of SAMHD1 can form further protein–protein contacts (Figure 6). Similar SAMHD1 structures would also be expected to form on the RNA genomes of endogenous retrotransposons. It is plausible that SAMHD1 RNA binding might target another RNase to the complex, but it now seems improbable that SAMHD1 performs this function itself.
Figure 6.
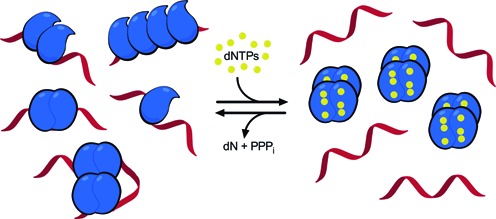
Possible binding modes of SAMHD1 to ssRNA and a model for the regulation of SAMHD1 dNTPase and RNA binding activities. In the low dNTP environment of resting immune cells, SAMHD1 exists in monomeric and dimeric forms that have high affinity for single-stranded RNA but possess no dNTPase activity. The figure depicts possible structures for RNA complexes with one, two and four monomers of SAMHD1 that are consistent with the biochemical, mutagenesis and AFM findings. The binding footprint for two adjacent monomers is ∼60 nt (Supplementary Figure S14), and two hypothetical binding modes for four monomers are depicted, consistent with chemical crosslinking (Figure 5A) and the elongated or globular complexes observed in AFM images (Figure 5E). Complexes of these types could form on HIV or LINE-1 ssRNA genomes. When dNTP levels increase, SAMHD1 can shift to its tetrameric form with high dNTPase activity but little affinity for single-stranded nucleic acids. Upon hydrolysis of available dNTP substrates by the activated SAMHD1 tetramers, the system will revert to the monomeric and dimeric states that are competent for ssRNA binding.
Our suggested function of SAMHD1 in RNA binding is consistent with previous in vivo studies of HIV-1 restriction and LINE-1 element replication. Although it remains unclear how the nonspecific binding of SAMHD1 is targeted to viral RNA, immunoprecipitation of epitope-tagged SAMHD1 from HIV-infected U937 cells demonstrated that SAMHD1 binds to the HIV-1 RNA genome during the early stages of reverse transcription, consistent with our proposed model (21). It is possible that protein-protein interactions between SAMHD1 and viral proteins target its binding to viral RNA genomes, but no evidence yet exists for such interactions. Furthermore, in cycling cells both dNTPase-deficient (D311A) and wt-SAMHD1 have been shown to strongly inhibit the replication of LINE-1 retroelements (10). Because the only activity shared by D311A and wt-SAMHD1 is nucleic acid binding, it can be inferred that the binding of SAMHD1 to LINE-1 ssRNA might also contribute to retroelement restriction. Future experiments will rigorously test various aspects of the proposed RNA binding model for SAMHD1 in cell-based systems of HIV infection and LINE-1 element replication.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Acknowledgments
National Institute of Health [T32 GM008763, RO1 GM056834 to J.T.S., R01 GM096039, P01 GM091743 to Y.L.L.]; K.J.S. is a recipient of an American Heart Association Predoctoral Fellowship [14PRE 20380664]. Funding for open access charge: National Institutes of Health [RO1 GM056834 to J.T.S.].
Conflict of interest statement. None declared.
REFERENCES
- 1.Hult K., Berglund P. Enzyme promiscuity: mechanism and applications. Trends Biotechnol. 2007;25:231–238. doi: 10.1016/j.tibtech.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 2.Pandya C., Farelli J.D., Dunaway-Mariano D., Allen K.N. Enzyme promiscuity: engine of evolutionary innovation. J. Biol. Chem. 2014;289:30229–30236. doi: 10.1074/jbc.R114.572990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richardson C.C., Kornberg A. A deoxyribonucleic acid phosphatase-exonuclease from escherichia coli. I. Purification of the enzyme and characterization of the phosphatase activity. J. Biol. Chem. 1964;239:242–250. [PubMed] [Google Scholar]
- 4.Suck D. DNA recognition by DNase I. J. Mol. Recognit. 1994;7:65–70. doi: 10.1002/jmr.300070203. [DOI] [PubMed] [Google Scholar]
- 5.Stivers J.T., Nagarajan R. Probing enzyme phosphoester interactions by combining mutagenesis and chemical modification of phosphate ester oxygens. Chem. Rev. 2006;106:3443–3467. doi: 10.1021/cr050317n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lassila J.K., Zalatan J.G., Herschlag D. Biological phosphoryl-transfer reactions: understanding mechanism and catalysis. Annu. Rev. Biochem. 2011;80:669–702. doi: 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldstone D.C., Ennis-Adeniran V., Hedden J.J., Groom H.C.T., Rice G.I., Christodoulou E., Walker P.A., Kelly G., Haire L.F., Yap M.W., et al. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature. 2011;480:379–382. doi: 10.1038/nature10623. [DOI] [PubMed] [Google Scholar]
- 8.Powell R.D., Holland P.J., Hollis T., Perrino F.W. Aicardi-Goutieres syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem. 2011;286:43596–43600. doi: 10.1074/jbc.C111.317628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature. 2011;474:654–657. doi: 10.1038/nature10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhao K., Du J., Han X., Goodier J.L., Li P., Zhou X., Wei W., Evans S.L., Li L., Zhang W., et al. Modulation of LINE-1 and Alu/SVA retrotransposition by Aicardi-Goutières syndrome-related SAMHD1. Cell Rep. 2013;4:1108–1115. doi: 10.1016/j.celrep.2013.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rice G.I., Bond J., Asipu A., Brunette R.L., Manfield I.W., Carr I.M., Fuller J.C., Jackson R.M., Lamb T., Briggs T.A., et al. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet. 2009;41:829–832. doi: 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ji X., Wu Y., Yan J., Mehrens J., Yang H., DeLucia M., Hao C., Gronenborn A.M., Skowronski J., Ahn J., et al. Mechanism of allosteric activation of SAMHD1 by dGTP. Nat. Struct. Mol. Biol. 2013;20:1304–1309. doi: 10.1038/nsmb.2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yan J., Kaur S., DeLucia M., Hao C., Mehrens J., Wang C., Golczak M., Palczewski K., Gronenborn A.M., Ahn J., et al. Tetramerization of SAMHD1 is required for biological activity and inhibition of HIV infection. J. Biol. Chem. 2013;288:10406–10417. doi: 10.1074/jbc.M112.443796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hansen E.C., Seamon K.J., Cravens S.L., Stivers J.T. GTP activator and dNTP substrates of HIV-1 restriction factor SAMHD1 generate a long-lived activated state. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E1843–E1851. doi: 10.1073/pnas.1401706111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Aravind L., Koonin E.V. The HD domain defines a new superfamily of metal-dependent phosphohydrolases. Trends Biochem. Sci. 1998;23:469–472. doi: 10.1016/s0968-0004(98)01293-6. [DOI] [PubMed] [Google Scholar]
- 16.Brown P.M., Caradoc-Davies T.T., Dickson J.M.J., Cooper G.J.S., Loomes K.M., Baker E.N. Crystal structure of a substrate complex of myo-inositol oxygenase, a di-iron oxygenase with a key role in inositol metabolism. Proc. Natl. Acad. Sci .U.S.A. 2006;103:15032–15037. doi: 10.1073/pnas.0605143103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wörsdörfer B., Lingaraju M., Yennawar N.H., Boal A.K., Krebs C., Bollinger J.M., Pandelia M.-E. Organophosphonate-degrading PhnZ reveals an emerging family of HD domain mixed-valent diiron oxygenases. Proc. Natl. Acad. Sci. U.S.A. 2013;110:18874–18879. doi: 10.1073/pnas.1315927110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goncalves A., Karayel E., Rice G.I., Bennett K.L., Crow Y.J., Superti-Furga G., Bürckstümmer T. SAMHD1 is a nucleic-acid binding protein that is mislocalized due to aicardi-goutières syndrome-associated mutations. Hum. Mutat. 2012;33:1116–1122. doi: 10.1002/humu.22087. [DOI] [PubMed] [Google Scholar]
- 19.Beloglazova N., Flick R., Tchigvintsev A., Brown G., Popovic A., Nocek B., Yakunin A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013;288:8101–8110. doi: 10.1074/jbc.M112.431148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tüngler V., Staroske W., Kind B., Dobrick M., Kretschmer S., Schmidt F., Krug C., Lorenz M., Chara O., Schwille P., et al. Single-stranded nucleic acids promote SAMHD1 complex formation. J. Mol. Med. 2013;91:759–770. doi: 10.1007/s00109-013-0995-3. [DOI] [PubMed] [Google Scholar]
- 21.Ryoo J., Choi J., Oh C., Kim S., Seo M., Kim S.-Y., Seo D., Kim J., White T.E., Brandariz-Nuñez A., et al. The ribonuclease activity of SAMHD1 is required for HIV-1 restriction. Nat. Med. 2014;20:936–941. doi: 10.1038/nm.3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seamon K.J., Hansen E.C., Kadina A.P., Kashemirov B.A., McKenna C.E., Bumpus N.N., Stivers J.T. Small molecule inhibition of SAMHD1 dNTPase by tetramer destabilization. J. Am. Chem. Soc. 2014;136:9822–9825. doi: 10.1021/ja5035717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson B.G., Stivers J.T. Variola type IB DNA topoisomerase: DNA binding and supercoil unwinding using engineered DNA minicircles. Biochemistry. 2014;53:4302–4315. doi: 10.1021/bi500571q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shlyakhtenko L.S., Gall A.A., Lyubchenko Y.L. Mica functionalization for imaging of DNA and protein-DNA complexes with atomic force microscopy. Methods Mol. Biol. 2013;931:295–312. doi: 10.1007/978-1-62703-056-4_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shlyakhtenko L.S., Gilmore J., Portillo A., Tamulaitis G., Siksnys V., Lyubchenko Y.L. Direct visualization of the EcoRII-DNA triple synaptic complex by atomic force microscopy. Biochemistry. 2007;46:11128–11136. doi: 10.1021/bi701123u. [DOI] [PubMed] [Google Scholar]
- 26.Shlyakhtenko L.S., Lushnikov A.Y., Li M., Lackey L., Harris R.S., Lyubchenko Y.L. Atomic force microscopy studies provide direct evidence for dimerization of the HIV restriction factor APOBEC3G. J. Biol. Chem. 2011;286:3387–3395. doi: 10.1074/jbc.M110.195685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miazzi C., Ferraro P., Pontarin G., Rampazzo C., Reichard P., Bianchi V. Allosteric regulation of the human and mouse deoxyribonucleotide triphosphohydrolase sterile α-motif/histidine-aspartate domain-containing protein 1 (SAMHD1) J. Biol. Chem. 2014;289:18339–18346. doi: 10.1074/jbc.M114.571091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ji X., Tang C., Zhao Q., Wang W., Xiong Y. Structural basis of cellular dNTP regulation by SAMHD1. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E4305–E4314. doi: 10.1073/pnas.1412289111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seamon K.J., Stivers J.T. A high-throughput enzyme-coupled assay for SAMHD1 dNTPase. J. Biomol. Screen. 2015 doi: 10.1177/1087057115575150. doi:10.1177/1087057115575150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu C., Gao W., Zhao K., Qin X., Zhang Y., Peng X., Zhang L., Dong Y., Zhang W., Li P., et al. Structural insight into dGTP-dependent activation of tetrameric SAMHD1 deoxynucleoside triphosphate triphosphohydrolase. Nat. Commun. 2013;4:2722. doi: 10.1038/ncomms3722. [DOI] [PubMed] [Google Scholar]
- 31.Lohman T.M., Mascotti D.P. Nonspecific ligand-DNA equilibrium binding parameters determined by fluorescence methods. Methods Enzymol. 1992;212:424–458. doi: 10.1016/0076-6879(92)12027-n. [DOI] [PubMed] [Google Scholar]
- 32.Kozlov A.G., Galletto R., Lohman T.M. SSB-DNA binding monitored by fluorescence intensity and anisotropy. Methods Mol. Biol. 2012;922:55–83. doi: 10.1007/978-1-62703-032-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bujalowski W., Jezewska M.J. Thermodynamic analysis of the structure-function relationship in the total DNA-binding site of enzyme-DNA complexes. Methods Enzymol. 2009;466:293–324. doi: 10.1016/S0076-6879(09)66013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.White T.E., Brandariz-Nuñez A., Valle-Casuso J.C., Amie S., Nguyen L.A., Kim B., Tuzova M., Diaz-Griffero F. The retroviral restriction ability of SAMHD1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe. 2013;13:441–451. doi: 10.1016/j.chom.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chaurasiya K.R., McCauley M.J., Wang W., Qualley D.F., Wu T., Kitamura S., Geertsema H., Chan D.S.B., Hertz A., Iwatani Y., et al. Oligomerization transforms human APOBEC3G from an efficient enzyme to a slowly dissociating nucleic acid-binding protein. Nat. Chem. 2014;6:28–33. doi: 10.1038/nchem.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Morrone S.R., Wang T., Constantoulakis L.M., Hooy R.M., Delannoy M.J., Sohn J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proc. Natl. Acad. Sci. U.S.A. 2014;111:E62–E71. doi: 10.1073/pnas.1313577111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raghunathan S., Kozlov A.G., Lohman T.M., Waksman G. Structure of the DNA binding domain of E. coli SSB bound to ssDNA. Nat. Struct. Biol. 2000;7:648–652. doi: 10.1038/77943. [DOI] [PubMed] [Google Scholar]
- 38.Lyubchenko Y.L., Shlyakhtenko L.S., Gall A.A. Atomic force microscopy imaging and probing of DNA, proteins, and protein DNA complexes: silatrane surface chemistry. Methods Mol. Biol. 2009;543:337–351. doi: 10.1007/978-1-60327-015-1_21. [DOI] [PubMed] [Google Scholar]
- 39.Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E.C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012;13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.