

Abstract
Glycogen storage diseases (GSD), a unique category of inherited metabolic disorders, were first described early in the 20th century. Since then, the biochemical and genetic bases of these disorders have been determined, and an increasing number of animal models for GSD have become available. At least 7 large mammalian models have been developed for laboratory research on GSDs. These models have facilitated the development of new therapies, including gene therapy, which are undergoing clinical translation. For example, gene therapy prolonged survival and prevented hypoglycemia during fasting for greater than one year in dogs with GSD type Ia, and the need for periodic re-administration to maintain efficacy was demonstrated in that dog model. The further development of gene therapy could provide curative therapy for patients with GSD and other inherited metabolic disorders.
Keywords: gene therapy, glycogen storage disease, animal model
Animal models for GSD
Glycogen storage disease (GSD) affecting companion animals have been well characterized, and these animal models of genetic disease have been highly valuable to medical research with regard to the elucidation of pathobiology and development of therapy. A discussion of specific examples of inherited disorders of metabolism in companion animals involving different metabolic pathways can be useful (Table 1). Similar to the case in human populations, consanguinity increases the likelihood of encountering rare autosomal recessive disorders like GSD. Thus, the consanguineous mating of dogs and cats has led to the appearance and recognition of inherited disorders of metabolism in several breeds. While the goal of animal breeding has been to eliminate genetic disease through selective breeding, colonies of these animals have been established for biomedical research. Indeed, such colonies have contributed greatly to the evaluation of safety and efficacy of new therapies for GSD. Specific examples of the research done in each model are described below.
Table 1.
GSD in Cats and Dogs
| Disorder | Enzyme Defect | Breed | Tissue Involvement |
|---|---|---|---|
| Carbohydrates | |||
| Type I; von Gierke | Glucose-6-phosphatase | Maltese terrier | Liver, kidney |
| Type II; Pompe | Acid α-glucosidase | Lapphund dog | Heart, skeletal muscle |
| Type IIIa; Cori | Glycogen debranching | Curly-coated retriever | Liver, skeletal muscle |
| Type IV; Andersen | Glycogen branching | Norwegian forest cat | Skeletal muscle, neurons |
| Type VII; Tarui | Phosphofructokinase | English Springer spaniel | Erythrocytes, skeletal muscle |
GSD Ia (von Gierke disease; OMIA 000418) was reported in Maltese dogs, when littermates developed growth failure and early demise in a pattern consistent with autosomal recessive inheritance (Brix et al. 1995). The presence of hypoglycemia and hepatomegaly suggested the diagnosis of GSD I, further supported by the presence of glycogen accumulations in the liver and kidney. The genetic basis was revealed to be a M121I missense mutation in the gene encoding the catalytic α-subunit of glucose-6-phosphatase (Kishnani et al. 1997).
The initial characterization of dogs with GSD Ia revealed that their clinical abnormalities most closely resemble severe, neonatal onset GSD Ia in humans (Brix et al. 1995, Kishnani et al. 2001). The original three Maltese puppies with GSD Ia died between 5 and 8 weeks of age with poor growth and feeding, and were found to have hepatomegaly and nephromegaly (Kishnani et al. 1997). Histology of liver and kidney revealed vacuolation consistent with glycogen storage, and liver glycogen was increased to 9.4% (normal 0 to 2.7%). Liver and kidney G6Pase were reduced to nearly undetectable levels, confirming a diagnosis of GSD Ia. Affected offspring of carrier dogs have hypoglycemia, hypercholesterolemia, and lactic acidosis during fasting, which is prevented by frequent feeding.
GSD II (Pompe disease; OMIA 000419) was demonstrated in Lapphund dogs, through fibroblast complementation studies in which human Pompe disease cells failed to complement acid α-glucosidase activity in heterokaryon cells (Walvoort et al. 1984). Clinical presentation included megaesophagus, exercise intolerance, and recurrent emesis (Walvoort 1985). Glycogen accumulations consisting of membrane bound vacuoles were present in the heart, skeletal, and smooth muscle. The genetic basis has been delineated as a c.2237G>A change corresponding to the nonsense mutation p.W746* in the acid α-glucosidase gene (Seppala et al. 2013).
GSD III (Cori disease; OMIA 001577) was characterized in curly coated retrievers that presented with exercise intolerance and lethargy at >12 months of age (Gregory et al. 2007). Liver transaminases, alkaline phosphatase and creatine kinase were elevated in serum by 6 months of age. Accumulations of non-membrane bound glycogen in liver and skeletal muscle, lacking short outer chains of α1,4-linked glucose, were accompanied by absence of glycogen debranching enzyme. A deletion of an adenine in exon 32 of the canine AGL gene predicted a truncation of the debranching enzyme by 126 amino acid residues (Gregory et al. 2007). Furthermore, progressive age-related liver fibrosis leading to cirrhosis was described in curly coated retrievers up to 16 months of age (Yi et al. 2012), which mimicked liver involvement in the human disorder (Markowitz et al. 1993).
GSD IV (Andersen disease; OMIA 420) in Norwegian forest cats presented with early demise, although surviving cats appeared normal until the onset of progressive neurological decline at 5 months of age (Fyfe et al. 2007). Affected kittens developed hypoglycemia, and glucose administration in the neonatal period promoted the survival of affected kittens to adulthood. Glycogen accumulations in skeletal muscle and neurons prompted the analysis of glycogen branching activity in skeletal muscle, which was severely deficient. An underlying mutation in the GBE1 gene was delineated, consisting of a 6.2 kbp deletion and 332 bp insertion that altered splicing of the mRNA and decreased glycogen branching enzyme in liver and muscle (Fyfe et al. 2007).
GSDV (McArdle disease; OMIA 001139) was reported in Charlois cattle with signs of rhabdomyolysis including exercise intolerance and myoglobinuria (Angelos et al. 1995). This was discovered to be caused by a mutation in the myophosphorylase gene caused by a C to T substitution in codon 489 leading to an arginine to trytophan substitution (Tsujino et al. 1996). An ovine model for McArdle disease was also described having a splice site mutation at the 3’ end of intron 19 of the myophosphorylase gene leading to disruption in the reading frame and premature truncation of the myophosphorylase protein. This mutation led to an increase in muscle glycogen and a lack of muscle glycogen phosphorylase discovered in a flock of Merino sheep exhibiting exercise intolerance (Tan et al. 1997).
GSD VII (Tarui disease; OMIA 000421) was reported in English springer spaniels in association with hemolytic crises and a metabolic myopathy that severely limited exercise (Giger et al. 1988). Phosphofructokinase deficiency in muscle caused ATP and phosphocreatine depletion during exercise, which caused rhabdomyolysis related to the block in glycolysis. The mutation in the M-PFK gene was demonstrated a nonsense mutation that caused premature termination with and protein instability (Smith et al. 1996).
Development of gene therapy for GSD Ia in the canine model
Proof-of-concept experiments demonstrated the efficacy of administering an AAV vector encoding G6Pase to newborn puppies with GSD Ia (Koeberl et al. 2008, Weinstein et al. 2010). Untreated puppies with GSD Ia develop hypoglycemia without fasting (blood glucose 25 +/− 12 mg/dl), and typically do not survive for >2 months when treated with dietary therapy only (Koeberl, Pinto et al. 2008). One of the limitations encountered with AAV vector-mediated gene therapy is a waning effect of transgene expression, which has been encountered with an AAV serotype vector pseudotyped as serotype 8 (AAV2/8) in canine GSD Ia (Weinstein et al. 2010). Prior experiments with AAV2/8 vectors encoding G6Pase revealed stable, efficacious G6Pase expression in the liver that prevented mortality from hypoglycemia in mice and dogs with GSD Ia (Koeberl et al. 2006, Koeberl et al. 2008). In contrast, Dr. David Weinstein and colleagues administered an AAV2/8 vector containing the chicken beta-actin promoter linked to a cytomegalovirus enhancer (CBA regulatory cassette) that constuitively expressed G6Pase expression, but failed to prevent hypoglycemia for >two months of age in one dog with GSD Ia (Weinstein et al. 2010). This contrasted with the experience following administration of an AAV2/8 vector containing the human G6Pase minimal promoter to drive expression of the human G6Pase cDNA, AAV-G6Pase prolonged survival for >11 months in 3 consecutive dogs with GSD Ia (Koeberl et al. 2008). Weinstein et al.’s AAV2/8 vector contained the CBA regulatory cassette, and subsequently it was associated with cytotoxic T cell responses and rapid clearance in the liver, when administered to young G6pase (−/−) mice (Yiu et al. 2010). However, it is likely that any traditional AAV vector will gradually lose efficacy due to loss of episomal vector genomes and diminishing liver transduction (Koeberl et al. 2006, Cunningham et al. 2008, Luo et al. 2011).
The necessity of readministering an AAV vector to maintain efficacy was confirmed by the report of a series of GSD Ia dogs that were re-treated over the first years of life (Demaster et al. 2012). Following repeat AAV vector of a new serotype administration the symptoms from GSD Ia resolved and duration of normal blood glucose during fasting increased (Demaster et al. 2012). Furthermore, one GSD Ia dog presented at 15 months of age with life-threatening pancreatitis (emesis, highly elevated amylase) as reported in humans with GSD Ia (Kikuchi et al. 1991). That dog’s symptoms and hypoglycemia resolved following administration of a second AAV vector (Figure), and she continues to thrive without hypoglycemia >3 years later. An additional complication of GSD Ia in dogs is growth hormone resistance, which persisted despite reversal of biochemical abnormalities of GSD Ia with gene therapy (Brooks et al. 2013). Thus, current research efforts include the development of integrating AAV vectors to achieve life-long correction of G6Pase deficiency in the GSD Ia liver (Landau et al. 2014).
Figure.
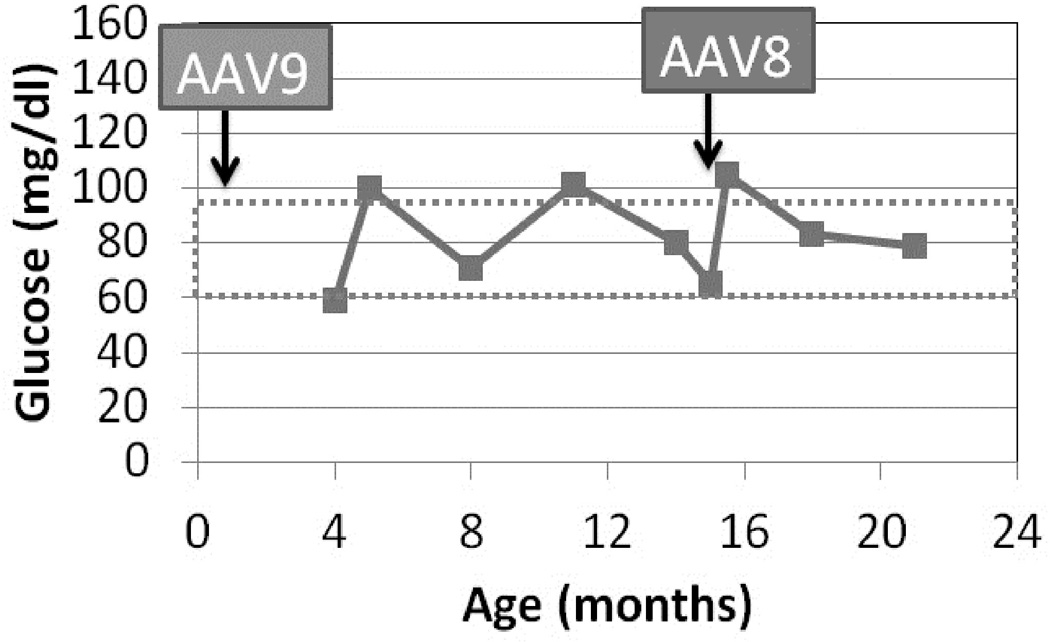
Plasma glucose following fasting for 6 hours. Dog W was treated by readministration of AAV-G6Pase, pseudotyped as AAV8, in response to life-threatening hypoglycemia, anorexia, and pancreatitis at 15 months of age. Initial treatment with AAV-G6Pase was as a neonate with AAV9 (Demaster, Luo et al. 2012).
An alternative approach to gene therapy for GSD Ia utilized a helper-dependent adenoviral (HDAd) vector to express canine G6Pase in the liver of newborn puppies with GSD Ia (Crane et al. 2012). The HDAd vector prolonged survival and prevented hypoglycemia in dogs with GSD Ia for >22 months. The initial HDAd vector was serotype 5, and the dog was successfully retreated with the serotype 2 HDAd vector for an additional 12 months before developing recurrent hypoglycemia. Another GSD Ia dog treated with both serotype 5 and 2 HDAd vectors survived until 12 months of age and then was euthanized as a result of renal complications associated with GSD Ia, which were not reversed by HDAd-mediated gene therapy (Crane et al. 2012).
Development of small molecule therapy for GSD III in the canine model
A recent study described potential therapeutic effects by inhibiting the mammalian target of rapamycin (mTOR), a known player in cell growth and proliferation, as well as protein synthesis pathways. Rapamycin, a specific inhibitor of mTOR, decreased muscle glycogen content and liver fibrosis in GSD IIIa-affected curly coated retrievers using high dose therapy at both early and late time points. Further, rapamycin inhibited glucose uptake by suppressing glycogen synthase and glucose transporter 1 expression in muscle cells cultured from GSD IIIa patients (Yi et al. 2014). These data suggest that small molecule drugs, such as rapamycin, may be useful in the treatment of GSD III.
Development of therapies in the ovine model of GSD V
The ovine model for GSD V has recently been used to develop in two ways: first, by using gene therapy to express muscle glycogen phosphorylase and second, by using pharmaceuticals to cause regeneration of muscles leading to re-expression of myophosphorylase in muscles. Intramuscular injection of an adenovirus 5 and/or an AAV2 vector expressing myophosphorylase caused expression of functional myophosphorylase in sheep affected with GSD V (Howell et al. 2008). Interestingly, in the same study, it was noted that damage to muscle fibers caused by injection with positive control vectors expressing LacZ caused re-expression of non-muscle isoforms of glycogen phosphorylase. Other studies in the GSD V ovine model have shown similar re-expression of non-muscle isoforms of myophosphorylase using valproate and notexin (Howell et al. 2008, Howell et al. 2014).
Conclusion
GSD in animals commonly presents with chronic growth failure and recurrent episodes of decompensation. Enzyme analyses implicate a specific enzyme deficiency, which leads to the identification of a causative mutation in related, affected animals. Gene therapy can be efficacious, if the gene defect is complemented through gene replacement in the involved tissues. The development of new therapies will benefit affected human patients, if preclinical experiments and clinical trials follow these proof-of-principle experiments.
Acknowledgements
GSD Ia research at Duke University has been supported by the Children's Fund for GSD Research, Children’s Miracle Network, and the Association for Glycogen Storage Disease. We wish to acknowledge technical support from Mr. Andrew Bird, Mr. Colby Bird, Ms. Bayley Crane, Mr. Danny Kozink, and Ms. Songtao Li. We wish to acknowledge inspiration and support from Dr. Emory and Mrs. Mary Chapman and their son Christopher, and from Dr. and Mrs. John Kelly. We deeply appreciate the dedication shown by the staff of the Duke Department of Laboratory Animal Resources, as well as undergraduate students at Duke University, who provided daily care for puppies with GSD Ia.
Funding:
Funding for preparation of this manuscript was provided by the Alice and YT Chen Pediatrics Genetics and Genomics Research Center.
Dwight Koeberl has received funding for research and honoraria for lectures from Genzyme and Amicus. He has received honoraria for lectures and consultancy from Amicus and Shire HGT. He has received funding for consultancy from Brawner Consulting, Clarion Healthcare LLC, Fitzpatrick Cella Harper & Scinto, Patterson Belknap Webb & Tyler LLP, ClearView Healthcare Partners, and AskBio.
Footnotes
Competing interest:
Elizabeth Brooks declares that she has no conflicts of interest.
Compliance with Ethics Guidelines
Animal Rights:
All institutional and national guidelines for the care and use of laboratory animals were followed.
Details of the contributions of individual authors:
Elizabeth Brooks prepared sections related to GSD III and V and proofread the entire article. Dwight Koeberl wrote and edited all other sections of the article.
References
- Angelos S, Valberg SJ, Smith BP, et al. Myophosphorylase deficiency associated with rhabdomyolysis and exercise intolerance in 6 related Charolais cattle. Musc Nerv. 1995;18(7):736–740. doi: 10.1002/mus.880180710. [DOI] [PubMed] [Google Scholar]
- Brix AE, Howerth EW, McConkie-Rosell A, et al. Glycogen storage disease type Ia in two littermate Maltese puppies. Vet Pathol. 1995;32(5):460–465. doi: 10.1177/030098589503200502. [DOI] [PubMed] [Google Scholar]
- Brooks ED, Little D, Arumugam R, et al. Pathogenesis of growth failure and partial reversal with gene therapy in murine and canine Glycogen Storage Disease type Ia. Mol Genet Metab. 2013;109(2):161–170. doi: 10.1016/j.ymgme.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crane B, Luo X, Demaster A, et al. Rescue administration of a helper-dependent adenovirus vector with long-term efficacy in dogs with glycogen storage disease type Ia. Gene Ther. 2012;19(4):443–452. doi: 10.1038/gt.2011.86. [DOI] [PubMed] [Google Scholar]
- Cunningham SC, Dane AP, Spinoulas A, Logan GJ, Alexander IE. Gene delivery to the juvenile mouse liver using AAV2/8 vectors. Mol Ther. 2008;16(6):1081–1088. doi: 10.1038/mt.2008.72. [DOI] [PubMed] [Google Scholar]
- Demaster A, Luo X, Curtis S, et al. Long-term efficacy following readministration of an adeno-associated virus vector in dogs with glycogen storage disease type Ia. Hum Gene Ther. 2012;23(4):407–418. doi: 10.1089/hum.2011.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyfe JC, Kurzhals RL, Hawkins MG, et al. A complex rearrangement in GBE1 causes both perinatal hypoglycemic collapse and late-juvenile-onset neuromuscular degeneration in glycogen storage disease type IV of Norwegian forest cats. Mol Genet Metab. 2007;90(4):383–392. doi: 10.1016/j.ymgme.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giger U, Argov Z, Schnall M, Bank WJ, Chance B. Metabolic myopathy in canine muscle-type phosphofructokinase deficiency. Musc Nerv. 1988;11(12):1260–1265. doi: 10.1002/mus.880111210. [DOI] [PubMed] [Google Scholar]
- Gregory BL, Shelton GD, Bali DS, Chen YT, Fyfe JC. Glycogen storage disease type IIIa in curly-coated retrievers. J Vet Intern Med. 2007;21(1):40–46. doi: 10.1892/0891-6640(2007)21[40:gsdtii]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Howell JM, Quinlivan R, Sewry C. Investigation of possible treatment regimes for McArdle's disease using the sheep model of the disease. Neuromuscul Disord. 2008;18(9–10):828–828. [Google Scholar]
- Howell JM, Walker KR, Creed KE, et al. Phosphorylase re-expression, increase in the force of contraction and decreased fatigue following notexin-induced muscle damage and regeneration in the ovine model of McArdle disease. Neuromuscul Disord. 2014;24(2):167–177. doi: 10.1016/j.nmd.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Howell JM, Walker KR, Davies L, et al. Adenovirus and adeno-associated virus-mediated delivery of human myophosphorylase cDNA and LacZ cDNA to muscle in the ovine model of McArdle's disease: expression and re-expression of glycogen phosphorylase. Neuromuscul Disord. 2008;18(3):248–258. doi: 10.1016/j.nmd.2007.10.006. [DOI] [PubMed] [Google Scholar]
- Kikuchi M, Hasegawa K, Handa I, Watabe M, Narisawa K, Tada K. Chronic pancreatitis in a child with glycogen storage disease type 1. Eur J Pediatr. 1991;150(12):852–853. doi: 10.1007/BF01955007. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Bao Y, Wu JY, Brix AE, Lin JL, Chen YT. Isolation and nucleotide sequence of canine glucose-6-phosphatase mRNA: identification of mutation in puppies with glycogen storage disease type Ia. Biochemi Mol Med. 1997;61(2):168–177. doi: 10.1006/bmme.1997.2600. [DOI] [PubMed] [Google Scholar]
- Kishnani PS, Faulkner E, VanCamp S, et al. Canine model and genomic structural organization of glycogen storage disease type Ia (GSD Ia) Vet Pathol. 2001;38(1):83–91. doi: 10.1354/vp.38-1-83. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Pinto C, Sun B, et al. AAV vector-mediated reversal of hypoglycemia in canine and murine glycogen storage disease type Ia. Mol Ther. 2008;16(4):665–672. doi: 10.1038/mt.2008.15. [DOI] [PubMed] [Google Scholar]
- Koeberl DD, Sun BD, Damodaran TV, et al. Early, sustained efficacy of adeno-associated virus vector-mediated gene therapy in glycogen storage disease type Ia. Gene Ther. 2006;13(17):1281–1289. doi: 10.1038/sj.gt.3302774. [DOI] [PubMed] [Google Scholar]
- Landau D, Brooks ED, Perez-Pinera P, et al. Zinc Finger Nuclease-Mediated Cleavage of the Rosa26 Locus to Allow Targeted Integration of a Glucose-6-Phosphatase Gene Promotes Survival in Mice With Glycogen Storage Disease Type-IA. Mol Ther. 2014;22(Suppl 1):57. doi: 10.1038/mt.2016.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Hall G, Li S, et al. Hepatorenal correction in murine glycogen storage disease type I with a double-stranded adeno-associated virus vector. Mol Ther. 2011;19(11):1961–1970. doi: 10.1038/mt.2011.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz AJ, Chen YT, Muenzer J, Delbuono EA, Lucey MR. A man with type III glycogenosis associated with cirrhosis and portal hypertension. Gastroenteroly. 1993;105(6):1882–1885. doi: 10.1016/0016-5085(93)91088-y. [DOI] [PubMed] [Google Scholar]
- Seppala EH, Reuser AJ, Lohi H. A nonsense mutation in the acid alpha-glucosidase gene causes Pompe disease in Finnish and Swedish Lapphunds. PLoS One. 2013;8(2):e56825. doi: 10.1371/journal.pone.0056825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith BF, Stedman H, Rajpurohit Y, et al. Molecular basis of canine muscle type phosphofructokinase deficiency. J Biol Chem. 1996;271(33):20070–20074. doi: 10.1074/jbc.271.33.20070. [DOI] [PubMed] [Google Scholar]
- Tan P, Allen JG, Wilton SD, Akkari PA, Huxtable CR, Laing NG. A splice-site mutation causing ovine McArdle's disease. Neuromuscul Disord. 1997;7(5):336–342. doi: 10.1016/s0960-8966(97)00062-x. [DOI] [PubMed] [Google Scholar]
- Tsujino S, Shanske S, Valberg SJ, Cardinet GH, 3rd, Smith BP, DiMauro S. Cloning of bovine muscle glycogen phosphorylase cDNA and identification of a mutation in cattle with myophosphorylase deficiency, an animal model for McArdle's disease. Neuromuscul Disord. 1996;6(1):19–26. doi: 10.1016/0960-8966(95)00014-3. [DOI] [PubMed] [Google Scholar]
- Walvoort HC. Glycogen storage disease type II in the Lapland dog. Vet Q. 1985;7(3):187–190. doi: 10.1080/01652176.1985.9693981. [DOI] [PubMed] [Google Scholar]
- Walvoort HC, Slee RG, Sluis KJ, Koster JF, Reuser AJ. Biochemical genetics of the Lapland dog model of glycogen storage disease type II (acid alpha-glucosidase deficiency) Am J Med Genet. 1984;19(3):589–598. doi: 10.1002/ajmg.1320190323. [DOI] [PubMed] [Google Scholar]
- Weinstein DA, Correia CE, Conlon T, et al. Adeno-associated virus-mediated correction of a canine model of glycogen storage disease type ia. Hum Gene Ther. 2010;21(7):903–910. doi: 10.1089/hum.2009.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Brooks ED, Thurberg BL, Fyfe JC, Kishnani PS, Sun B. Correction of glycogen storage disease type III with rapamycin in a canine model. J Mol Med (Berl) 2014;92(6):641–650. doi: 10.1007/s00109-014-1127-4. [DOI] [PubMed] [Google Scholar]
- Yi H, Thurberg BL, Curtis S, et al. Characterization of a canine model of glycogen storage disease type IIIa. Dis Model Mech. 2012;5(6):804–811. doi: 10.1242/dmm.009712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu WH, Lee YM, Peng WT, et al. Complete normalization of hepatic G6PC deficiency in murine glycogen storage disease type Ia using gene therapy. Mol Ther. 2010;18(6):1076–1084. doi: 10.1038/mt.2010.64. [DOI] [PMC free article] [PubMed] [Google Scholar]