

Abstract
Site-1 protease (S1P) cleaves membrane-bound lipogenic sterol regulatory element-binding proteins (SREBPs) and the α/β-subunit precursor protein of the N-acetylglucosamine-1-phosphotransferase forming mannose 6-phosphate (M6P) targeting markers on lysosomal enzymes. The translocation of SREBPs from the endoplasmic reticulum (ER) to the Golgi-resident S1P depends on the intracellular sterol content, but it is unknown whether the ER exit of the α/β-subunit precursor is regulated. Here, we investigated the effect of cholesterol depletion (atorvastatin treatment) and elevation (LDL overload) on ER-Golgi transport, S1P-mediated cleavage of the α/β-subunit precursor, and the subsequent targeting of lysosomal enzymes along the biosynthetic and endocytic pathway to lysosomes. The data showed that the proteolytic cleavage of the α/β-subunit precursor into mature and enzymatically active subunits does not depend on the cholesterol content. In either treatment, lysosomal enzymes are normally decorated with M6P residues, allowing the proper sorting to lysosomes. In addition, we found that, in fibroblasts of mucolipidosis type II mice and Niemann-Pick type C patients characterized by aberrant cholesterol accumulation, the proteolytic cleavage of the α/β-subunit precursor was not impaired. We conclude that S1P substrate-dependent regulatory mechanisms for lipid synthesis and biogenesis of lysosomes are different.
Keywords: mannose 6-phosphate, LDL receptor, cholesterol, statins, Niemann-Pick type C1 disease, Golgi apparatus, endocytosis, mucolipidosis type II, lysosome
Lysosomes function in the degradation of macromolecules, such as proteins, lipids, glycosaminoglycans or nucleic acids, intracellular organelles, and pathogens obtained by endocytosis, autophagy, and phagocytosis through the concerted action of more than 50 acid hydrolases (1). To maintain their function, lysosomes require a continuous replenishment of newly synthesized components transported from the endoplasmic reticulum (ER) via the Golgi apparatus to the endosomal/lysosomal compartment. The efficient transport of soluble lysosomal enzymes to lysosomes requires mannose 6-phosphate (M6P) residues on their N-linked glycans, which serve as recognition markers for M6P-specific receptors (MPRs) (2). The formation of M6P residues is catalyzed by two enzymes, the N-acetylglucosamine-1-phosphotransferase (termed “GlcNAc-1-phosphotransferase”) and the GlcNAc-1-phosphodiester α-N-acetylglucosaminidase (termed “uncovering enzyme”) localized in the cis-Golgi apparatus and trans-Golgi network, respectively (3).
The GlcNAc-1-phosphotransferase is a heterohexameric complex of three subunits (α2β2γ2), which are encoded by two genes (4–6). The α- and β-subunits of the GlcNAc-1-phosphotransferase are synthesized in the ER as a single, highly N-glycosylated, 190 kDa, type III membrane protein (6). For the transport to the Golgi apparatus, combinatorial dileucine and dibasic sorting motifs in the N- and C-terminal cytosolic domains, respectively, are required (7). Upon arrival in the cis-Golgi compartment, the α/β-subunit precursor is proteolytically cleaved by the site-1 protease (S1P) into mature α- and β-subunits (8), which are prerequisite for the enzymatic activity of the GlcNAc-1-phosphotransferase complex (9).
S1P [also known as subtilisin kexin isoenzyme-1 (SKI-1)] is a type I membrane serine protease and plays a crucial role in the proteolytic activation of the sterol regulatory element-binding proteins (SREBP)-1 and -2, which are necessary for synthesis of fatty acids, triglycerides, and cholesterol, respectively (10, 11). SREBPs are type III membrane-bound transcription factors with both cytosolic N-terminal transcription factor domains and C-terminal regulatory domains (12). At conditions of cholesterol abundance, SREBPs form ER-anchored complexes with the polytopic sterol-sensing SREBP cleavage-activating protein (SCAP) and INSIG, an insulin-induced gene product. Upon cholesterol depletion, the complex dissociates, and SCAP escorts SREBPs from the ER to the Golgi apparatus, followed by S1P-initiated release of the active transcription factor domain entering the nucleus (13, 14).
In cells deficient for S1P, the α/β-subunit precursor of GlcNAc-1-phosphotransferase cannot be cleaved and activated, which is subsequently associated with the lack of M6P residues, missorting of newly synthesized lysosomal enzymes, and accumulation of nondegraded storage material in lysosomes (8), which are hallmarks of mucolipidosis type II (MLII). MLII (also called I-cell disease) is a severe multisystemic inherited disorder of childhood caused by mutations in the GNPTAB gene encoding the α/β-subunit precursor protein of the GlcNAc-1-phosphotransferase (6, 15). Although the nature of storage material in MLII cells and tissues has not been fully characterized, analysis of fibroblasts of MLII patients and MLII mice revealed, in addition to increased levels of phospholipids, fucosylated oligosaccharides, and sialic acid-containing glycosphingolipids, an elevation and lysosomal accumulation of cholesterol (16–18).
In the present study, we investigated whether alterations in the cholesterol content of murine and human fibroblasts affect the ER-Golgi transport, the proteolytic activation of the α/β-subunit precursor of GlcNAc-1-phosphotransferase, and the subsequent lysosomal targeting of lysosomal enzymes. The results were compared with data from fibroblasts of Niemann-Pick type C1 (NPC1) patients and MLII mice that were characterized by abnormal cholesterol metabolism (18, 19). These analyses demonstrate that the export and the S1P-mediated cleavage of the α/β-subunit precursor protein of GlcNAc-1-phosphotransferase is independent of the cellular cholesterol content. Furthermore, missorting of lysosomal enzymes and lysosomal dysfunction in MLII fibroblasts lead to up-regulation of endocytic receptors, such as LDL receptor (LDLR) and the 300 kDa MPR (MPR300), and accumulation of their degradation intermediates.
MATERIALS AND METHODS
Reagents
Sodium 125I (74 TBq/mmol) and [35S]methionine (37 Bq/mmol) were purchased from Hartmann Analytik. Atorvastatin, newborn calf serum (NBCS), saponine, paraformaldehyde, mannose 6-phosphate sodium salt, penicillin/streptomycin, cycloheximide, TRI® Reagent, protease inhibitor cocktail (P2714), and BSA were obtained from Sigma-Aldrich. Maxima™ Probe qPCR Master Mix, Iodination reagent, and prestained protein molecular mass marker Pageruler™ were from Thermo Fisher Scientific. Prestained molecular mass marker Full-Range Rainbow™, FCS, and methionine-free DMEM were from GE Healthcare. TaqMan® Gene Expression Assays, High Capacity cDNA Reverse Transcription Kit, DMEM, and Opti-MEM® were purchased from Life Technologies. Transfection reagent JetPEI® was purchased from Peqlab. Roti® Quant Protein Assay was from Roth. Lipoprotein-deficient serum (LPDS) and human LDL were prepared from human plasma as previously described (20). Recombinant human arylsulfatase B (ASB) was kindly provided by Dr. Vellard (BioMarin/Genzyme LLC, Novato, CA).
Antibodies
A myc-tagged, single-chain antibody fragment against M6P residues (scFv M6P-1) and the monoclonal rat antibody against the α-subunit of the GlcNAc-1-phosphotransferase have been described recently (21, 22). The monoclonal anti-myc antibody was purchased from Cell Signaling. The polyclonal antibody against cathepsin Z was obtained from R&D Systems. Monoclonal antibodies against glyceraldehyde 3-phosphate dehydrogenase (GAPDH), LDLR and the Golgi marker protein (GM130) were obtained from Santa Cruz Biotechnology, Epitomics-Abcam, and BD Biosciences, respectively. The monoclonal antibody against β-tubulin was obtained from the Developmental Studies Hybridoma Bank (University of Iowa). Anti-rat Mpr300 antibodies were a kind gift from Dr. von Figura (Göttingen, Germany). Secondary antibodies conjugated to HRP or Alexa Fluor® were purchased from Dianova and Life Technologies, respectively.
Cell culture and cDNA transfection
Mouse embryonic fibroblasts (MEFs) of WT and MLII “knock-in” mice were prepared as previously described (18). Human fibroblasts were obtained from skin biopsies with informed consent of two controls and three NPC1 patients carrying the following mutations encoded by the NPC1 gene: Patient 1 (P1) was heterozygous for p.G910S and p.G1034R; patient 2 (P2) and patient 3 (P3) were homozygous for p.S940L and p.I1095del, respectively. MEFs and fibroblasts from healthy controls and NPC1-defective patients were cultured in DMEM containing 10% FCS and penicillin/streptomycin. To change the cholesterol content, MEFs were grown on 6- or 3.5-cm plates or glass coverslips, rinsed with PBS (pH 7.4), and further incubated in DMEM containing 10% NBCS or LPDS. To deplete or overload the cholesterol content in cells, 10 µM atorvastatin or 100 µg/ml human LDL was added to the DMEM/LPDS medium for 48 h, respectively. Twenty-four hours after addition of atorvastatin (ATV) or LDL, MEFs and human fibroblasts were transfected with cDNA of the α*/β-subunit construct #3 (8, 22) using JetPEI® according to the manufacturer´s instructions. The α*/β-subunit miniconstruct lacks amino acids 431–819 and results in a shortened α*-subunit exhibiting identical topology, N- and C-terminal ER exit structures, and structural requirements for efficient proteolytic cleavage by S1P (8, 22). The medium was replaced 6 h after transfection, and cells were further incubated with cholesterol depletion medium or LDL loading medium for 18 h.
mRNA analysis
Total RNA isolation, cDNA synthesis, and real-time PCR were performed as previously described (8). TaqMan™ Gene Expression Assays, including predesigned probes and primer sets for mouse Ldlr (Mm00440169_m1), Gnptab (Mm01773334_m1), Actb (Mm00607939_s1) and human LDLR (Hs00181192_m1) and ACTB (Hs9999903_m1), were used. The relative expression of human and murine LDLR and Gnptab mRNA was normalized to the level of ACTB mRNA in the same cDNA using the comparative CT method (2 –ΔΔCT).
Internalization assay of [125I]ASB
Recombinant human ASB was iodinated with Iodination Reagent and sodium 125I to a specific activity of 6 µCi/µg as previously described (23). Cells grown on 3.5-cm plates were washed with PBS, preincubated with DMEM containing 0.1% BSA (DMEM/BSA) for 30 min at 37°C, and incubated with [125I]ASB (625,000 cpm/ml DMEM/BSA) in the presence or absence of 10 mM M6P for 20 min at 37°C. After four washes with PBS to remove nonbound [125I]ASB. cells were either harvested or chased in DMEM/BSA for 2 to 16 h at 37°C. Cells were lysed in 0.1 ml PBS containing 0.2% Triton X-100 and protease inhibitors for 30 min at 4°C and centrifuged at 15,000 g at 4°C for 15 min. The supernatants were analyzed by SDS-PAGE and autoradiography.
Western blotting
Cells were lysed for 30 min at 4°C in PBS containing 1% Triton X-100 and protease inhibitor cocktail. After centrifugation at 10,000 g, supernatants were used for measurement of the protein content by the Roti® Quant Protein Assay. Aliquots of cell extracts (75 µg protein) were solubilized, separated by SDS-PAGE, and analyzed by Western blotting either with monoclonal antibody against the α-subunit of the GlcNAc-1-phosphotransferase (dilution 1:25) (22), cathepsin Z (1:500), LDLR (1:1,000), or Mpr300 (1:500). β-Tubulin or GAPDH were used as loading controls. After incubation with secondary HRP-conjugated antibodies, the immunoreactive bands were visualized by enhanced chemoluminescence (Molecular Imager ChemiDoc XRS, Bio-Rad). The content of M6P-containing proteins in cell extracts was analyzed by scFv M6P-1 Western blotting (21).
Other methods
Confocal immunofluorescence microscopy of transfected cells, activity determination of the enzyme β-hexosaminidase, and metabolic [35S]methionine labeling of MEF followed by immunoprecipitation of cathepsin Z were described recently (18, 22). Densitometric analyses were performed with the Image J software (The National Institutes of Health) to quantify band intensities in Western blots and X-ray films.
Statistical analysis
For statistical analysis, Student’s t-test was performed. P values <0.05 were considered significant.
RESULTS
High LDL receptor expression in MLII mouse embryonic fibroblasts
To investigate whether the α/β-subunit precursor protein of GlcNAc-1-phosphotransferase is transported from the ER to the Golgi apparatus independently of cholesterol/LDL, the effects of the cholesterol-depleting drug ATV and the LDL-mediated cholesterol overload were examined in MEFs. The efficiency of both treatments has been demonstrated by determination of LDL receptor gene (Ldlr) expression, which is known to be regulated by cellular cholesterol level (24). Treatment of WT MEFs with 10 µM ATV for 48 h increased the mouse Ldlr mRNA expression 6-fold (Fig. 1). When cholesterol levels are high after LDL loading, the Ldlr mRNA expression dropped to 20% of controls. For comparison, the transcript concentration of Gnptab was not affected by changes in the cholesterol level in WT cells. These data indicate that both treatments to increase or decrease the cellular cholesterol concentration in MEFs were effective and reproducible. In MEFs of MLII mice harboring the mutant Gnptabc.3082insC (18), the Gnptab transcript level was reduced, whereas the Ldlr mRNA concentration was 3-fold higher under basal conditions (DMEM supplemented by 10% NBCS) than in WT MEFs, which could be further increased by 50% after treatment with ATV (Fig. 1), suggesting low amounts of cholesterol in ER membranes under basal conditions of MLII fibroblasts. Incubation of MLII MEFs with LDL reduced the Ldlr mRNA level to 20% of nontreated MLII cells.
Fig. 1.
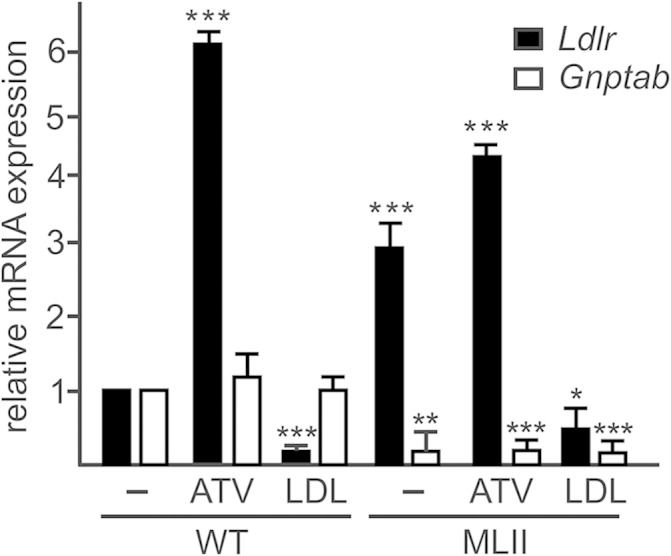
Effect of ATV and LDL on Ldlr mRNA expression in fibroblasts. MEFs from WT and MLII mice were cultured in DMEM containing 10% NBCS or 10% LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 48 h. The relative mRNA level of Ldlr and for comparison of Gnptab were determined by real-time PCR and normalized to β-actin mRNA expression. The relative mRNA expression in nontreated WT cells, incubated in DMEM containing 10% NBCS, was set at1. The data are the mean of triplicate PCRs obtained from three independent experiments and expressed as the fold change ± SD. * P ≤ 0.05, ** P ≤ 0.01, and *** P ≤ 0.005.
The cholesterol-dependent alterations in the Ldlr mRNA expression in WT MEFs could be confirmed on the LDLR protein level (Fig. 2). ATV treatment resulted in a 12-fold increase, whereas LDL overload suppressed the LDLR concentration to 30% of control cells (Fig. 2A). No or marginal effects on the LDLR expression have been observed when the cells were incubated in the presence of 10% NBCS or LPDS alone (supplementary Fig. 1). In MLII MEFs, the basal LDLR protein level was 3-fold higher than in wild-type MEFs (Fig. 2A, B). Incubation of MLII MEFs with ATV or LDL led to a 7-fold increase and a 6-fold reduction of LDLR expression, respectively.
Fig. 2.

Effect of ATV and LDL on LDLR protein expression in fibroblasts. MEFs from WT (A) and MLII (B) were cultured in DMEM containing 10% NBCS or 10% LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 48 h. The protein expression of LDLR was analyzed by Western blotting and evaluated by densitometry. The LDLR expression in cells incubated with DMEM containing NBCS was set at 1. The positions of the molecular mass marker proteins (in kDa) are indicated. The content of GAPDH in the samples served as loading control. Representative Western blots of four experiments (n = 4) are shown. ** P ≤ 0.01 and *** P ≤ 0.005.
Cholesterol-independent transport and proteolytic cleavage of the GlcNAc-1-phosphotransferase
The site-1 protease substrates SREBPs are translocated from the ER to the Golgi apparatus in dependency of the membrane cholesterol content (12). To examine whether the α/β-subunit precursor is transported to the Golgi apparatus in a cholesterol-dependent manner, MEFs of WT mice (not shown) and human fibroblasts were treated with and without ATV and LDL and were transfected with the human α*/β-subunit precursor construct (8, 22) of the GlcNAc-1-phosphotransferase. Immunofluorescence microscopy revealed complete costaining of α*-subunit immunopositive material with the cis-Golgi marker protein GM130 independent of high (LDL) or low (ATV) levels of cholesterol (Fig. 3A). When fibroblasts of a NPC1 patient characterized by a high level of nonesterified cholesterol in lysosomes (19) and 2- to 3.5-fold increased Ldlr mRNA level (supplementary Fig. 2) were analyzed, α*-subunit immunoreactive material was found in GM130-positive Golgi membranes (Fig. 3B). These data were complemented by Western blot analysis of cell extracts of ATV- or LDL-treated WT MEFs transiently transfected with the α*/β-subunit precursor construct. At steady state, small amounts of immunoreactive 120 kDa α*/β-subunit precursor polypeptides were observed in WT MEFs (Fig. 3C) and human control fibroblasts (Fig. 3D). In contrast, strong signals of mature 90 kDa α*-subunits were detected both in MEFs (Fig. 3C) and human fibroblasts (Fig. 3D). The proteolytic pattern of α*/β-subunit precursor and mature α*-subunit was not affected by high or low cholesterol content (Fig. 3C). Similarly, α*/β-subunit precursor expressed in NPC1 fibroblasts were proteolytically cleaved to mature α*-subunits (Fig. 3D). Identical cleavage patterns have been observed in two other NPC1 fibroblast cell lines (Fig. 3E). Of note, the endogenous α/β-subunit precursor or mature α-subunit cannot be visualized by the anti-α-subunit monoclonal antibody due to their low expression. Finally, we tested whether MLII MEFs exhibiting high lysosomal cholesterol content (18) and characterized by low cholesterol sensing (Fig. 2A, B) were impaired in their capability to cleave the α*/β-subunit precursor protein. As shown in Fig. 3C, the majority of immunoreactive material is presented by the mature α*-subunit. These data demonstrate that neither increase nor reduction of cellular cholesterol level affected the ER-Golgi transport and the proteolytic activation of the α*/β-subunit precursor protein of GlcNAc-1-phosphotransferase.
Fig. 3.

ATV or LDL overload did not affect the localization or proteolytic cleavage of the GlcNAc-1-phosphotransferase α*/β-subunit precursor protein. A–E: Human fibroblasts of healthy controls and NPC1 patients, WT and MLII MEFs were cultured in DMEM containing 10% NBCS or 10% LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 24 h as indicated. Cells were then transfected with cDNA of α*/β-subunit precursor construct and analyzed after 24 h. A and B: The localization of α*/β-subunit precursor of the GlcNAc-1-phosphotransferase (α*/β, green) was determined in control patient (A) and in NPC1 patient 1 (P1) (B) fibroblasts by costaining with the cis-Golgi marker protein GM130 (red) using immunofluorescence microscopy. Colocalization in merged images appears yellow. Scale bars, 5 µm. C and D: The proteolytic cleavage of the α*/β-subunit precursor as indicator for correct transport to the Golgi apparatus was analyzed by α-subunit Western blot of transfected WT and MLII MEFs (C), human control and NPC1 (P1) fibroblasts (D), and three different NPC1 cell lines (P1, P2 and P3) (E). GAPDH/GAPDH Western blot analysis was used as a loading control. Extracts of nontransfected cells were used as a negative control. The positions of molecular mass marker proteins (in kDa), α*/β-subunit precursor, and mature α*-subunit are indicated.
Biogenesis of lysosomes is independent of cellular cholesterol content
The enzymatic activity of the GlcNAc-1-phosphotransferase was determined indirectly by mannose 6-phosphate (M6P) Western blotting, showing that neither the reduction of cholesterol content by ATV nor the LDL treatment affected the intensity or pattern of M6P-containing proteins in WT fibroblasts (Fig. 4A). MLII MEFs were used as negative controls lacking M6P-containing proteins (18). To analyze the M6P-dependent targeting of lysosomal enzymes, we performed [35S]methionine pulse-chase experiments followed by immunoprecipitation of the lysosomal protease cathepsin Z (CtsZ) from cell extracts and media. In WT MEFs, the 38 kDa CtsZ precursor (p) was transported to lysosomes, proteolytically processed to the 36 kDa mature (m) form, and partially secreted during the 4 h chase period (Fig. 4B). The synthesis rate of CtsZ, the proteolytic processing to the mature form, and the amount of secreted precursor forms were not affected by ATV or LDL treatment of WT MEFs (Fig. 4B). In contrast, in MLII MEFs used as control, the newly synthesized CtsZ was completely missorted into the medium during the 4 h chase period, and no CtsZ immunoreactive polypeptides were retained intracellularly (Fig. 4B). This was confirmed by the steady-state expression level of CtsZ. The amount of intracellular detectable 36 kDa mature CtsZ in ATV- and LDL-treated and nontreated WT MEFs was similar (Fig. 4C), whereas no CtsZ immunoreactive material was observed in MLII MEFs. Furthermore, the activity of another lysosomal enzyme, β-hexosaminidase, in ATV- or LDL-treated MEFs and in their respective media was comparable with nontreated control cells (Fig. 4D). In contrast, the β-hexosaminidase activity in MLII MEFs was markedly reduced by approximately 80% and missorted into the medium. These data indicate that neither ATV nor LDL treatment affected the GlcNAc-1-phosphotransferase activity in the Golgi apparatus and the transport and processing of lysosomal enzymes along the biosynthetic pathway to lysosomes.
Fig. 4.

ATV or LDL overload did not affect GlcNAc-1-phosphotransferase activity and transport of lysosomal enzymes. A–D: WT and MLII MEFs were cultured in DMEM containing 10% NBCS or 10% LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 48 h. A: The GlcNAc-1-phosphotransferase activity in cell extracts was indirectly determined by M6P Western blotting. The positions of the molecular mass markers (in kDa) are indicated. B: The biosynthesis and sorting of the lysosomal protease CtsZ was analyzed by labeling of MEFs with [35S]methionine for 1 h followed by 4 h incubation in nonradioactive medium and immunoprecipitation of CtsZ from cell extracts and media, SDS-PAGE, and fluorography. The amounts of secreted CtsZ precursors were estimated by densitometry and expressed as percentage of totally synthesized CtsZ. The positions of the molecular mass marker proteins (in kDa), precursor (p), and mature (m) form of CtsZ are indicated. The identity of the 45 kDa 35S-labeled polypeptide immunoprecipitated from cell extracts (closed circle) is unknown. C: The total intracellular CtsZ expression at steady state was analyzed by Western blotting. Anti β-tubulin Western blot was used as a loading control. The positions of the molecular mass markers (in kDa) are indicated. D: The relative enzyme activity of the lysosomal enzyme β-hexosaminidase was measured in cell extracts and media conditioned for 24 h. The activities in control WT MEFs incubated with DMEM containing 10% NBCS were set at 1. The specific activity of β-hexosaminidase in these cells and media were 57.7 mU/h/mg protein ± 14.0 and 4.1 mU/24 h/mg protein ± 1.3, respectively. *** P ≤ 0.005.
To examine whether the transport of lysosomal enzymes along the endocytic pathway is altered in dependency of the cholesterol membrane content, WT MEFs were incubated for 20 min with 125I-labeled M6P-containing ASB followed by variable chase periods. ASB is internalized in an M6P-dependent manner, as shown by the complete inhibition of ASB uptake in the presence of 10 mM M6P in the medium (Fig. 5). Subsequently, the internalized ASB is proteolytically activated via 47 kDa intermediate to mature 15 kDa forms (Fig. 5A) (25). The amount of internalized [125I]ASB was up to 2-fold higher in LDL-treated cells compared with nontreated and ATV-treated cells, indicating that an elevated cellular cholesterol content resulted in increased [125I]ASB endocytosis whereas the subsequent proteolytic processing of [125I]ASB was not impaired by ATV or LDL treatment. In MLII MEFs, however, the amount of internalized [125I]ASB was 8-fold higher than in WT MEFs and the proteolytic maturation was almost completely inhibited even 22 h after internalization of the ASB precursor (Fig. 5B). These data suggest that sufficient amounts of proteases involved in the maturation of the ASB precursor are present in MLII lysosomes, whereas proteases mediating the degradation of ASB polypeptides are missing. Of note, in NPC1 cells (P1), both the amount of internalized [125I]ASB and the stability of proteolytically generated ASB forms were comparable with that in human control fibroblasts (Fig. 5C). The addition of the vATPase inhibitor bafilomycin A1 during the chase periods completely inhibited the maturation and degradation of ASB polypeptides by acid proteases. These data indicate that the deficiency of various lysosomal enzymes, in particular lysosomal proteases in MLII cells rather than the lysosomal accumulation of nonesterified cholesterol, affects the proteolytic maturation of the internalized indicator protein ASB.
Fig. 5.

Endocytosis and proteolytic processing of arylsulfatase B. A: WT MEFs were cultured in DMEM containing 10% NBCS or 10% LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 48 h. A, B: WT and MLII MEFs were incubated with [125I]ASB (625,000 cpm/ml) in the presence or absence of 10 mM M6P for 20 min, washed, and either harvested (-) or chased for the indicated time points (n = 3). C: Human healthy control and NPC1 (P1) fibroblasts were incubated with [125I]ASB (625,000 cpm/ml), washed, and either harvested (-) or chased in the presence or absence of 100 nM bafilomycin A1 (Baf A1). Cell extracts containing equal amounts of β-hexosaminidase activity (A: 21.0 mU/h/mg protein ± 3.1; B: 22 mU/mg protein ± 1.9 for WT and 4.5 mU/mg protein ± 0.6 for MLII; C: 19.4 mU/mg protein ± 3.3) were separated by SDS-PAGE, and the internalized ASB was visualized by autoradiography. The positions of the molecular mass markers (in kDa) and the migration of precursor (p) and mature (arrow) forms of ASB are indicated.
The increased M6P-dependent endocytosis of [125I]ASB correlated with a 3-fold increase of Mpr300 expression in MLII MEFs (Fig. 6A). The Mpr300 expression was not affected by ATV-induced reduction of cholesterol content or by LDL overload. In addition to the major 300 kDa Mpr immunoreactive protein, ∼240 and 160 kDa Mpr300 immunoreactive polypeptide bands were observed in MLII MEFs (Fig. 6A). Furthermore, the cholesterol-dependent variations in the 160 kDa LDLR expression (Fig. 2) correlated with the appearance of high amounts of 17 kDa and minor 40 kDa LDLR fragments and intermediates, respectively, in MLII MEFs (Fig. 6B) (26). Small percentages of the 17 kDa LDLR fragment were detectable in ATV-treated WT MEFs. These data support previous observations that both Mpr300 and LDLR undergo ectodomain shedding at the plasma membrane or endosomes (26–28) followed by lysosomal degradation of the C-terminal fragments accumulating in high amounts in MLII MEFs.
Fig. 6.
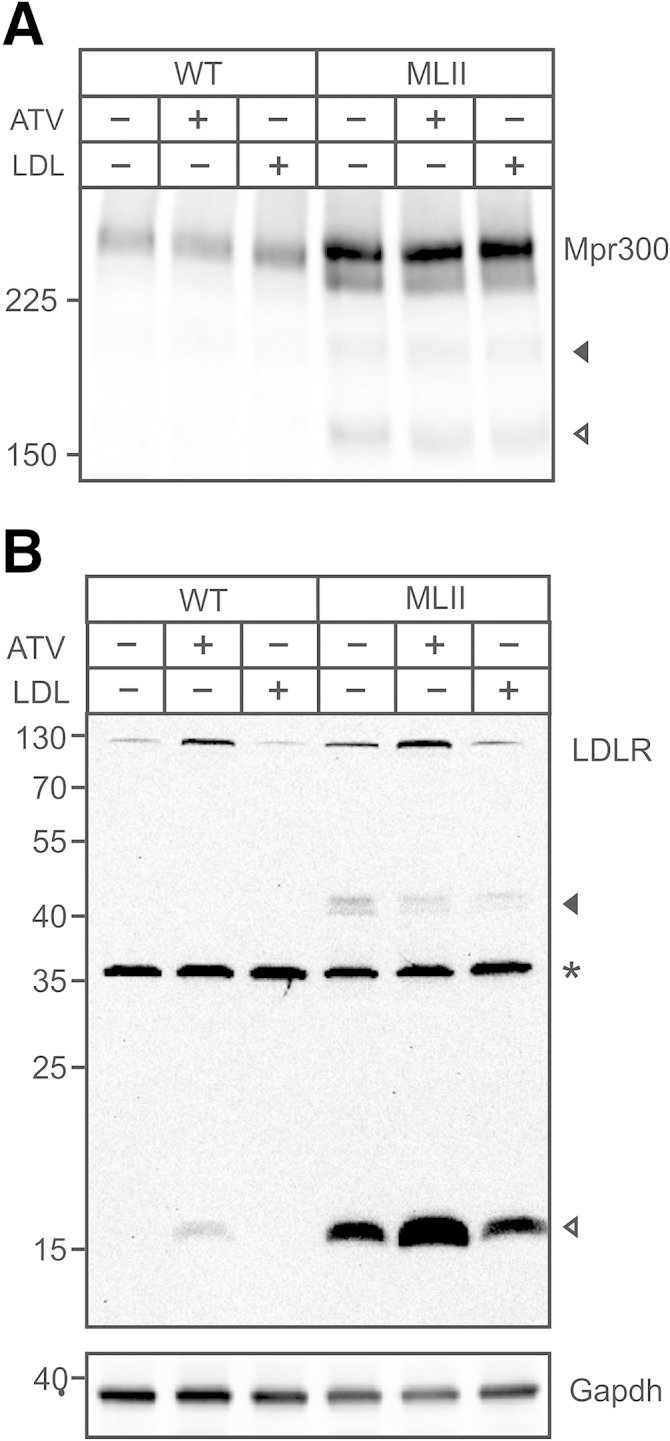
Accumulation of LDLR in MLII embryonic fibroblasts. WT and MLII MEFs were cultured in DMEM containing NBCS or LPDS supplemented with 10 µM ATV or 100 µg/ml human LDL for 48 h. A: Cell extracts were separated by SDS-PAGE (5% acrylamide) under nonreducing conditions and analyzed by Mpr300 Western blotting. B: Cell extracts were separated by SDS-PAGE (15% acrylamide) followed by LDLR Western blotting. Then the membrane was stripped and reused for GAPDH Western blotting as loading control. Western blot analyses were repeated twice with similar results. The positions of the molecular mass markers (in kDa) and the Ldlr fragments are indicated. * Unspecific polypeptide band.
DISCUSSION
It is well known how the prototypical precursor membrane proteins of lipogenic transcription factors, SREBP-1 and SREBP-2, are released from the ER and proteolytically activated by S1P localized in the Golgi apparatus. In the presence of cholesterol, the formation of a complex between SREBPs, the sterol-sensing protein SCAP, and INSIG results in retention in the ER. When cells are depleted of sterols, the complex dissociates and exposes a Golgi sorting signal, which allows the ER exit of SREBP/SCAP complexes, and subsequent first cleavage of SREBPs by S1P (29, 30). Here we examined whether the transport of the GlcNAc-1-phosphotransferase α/β-subunit precursor from the ER to the Golgi apparatus is also regulated by the intracellular sterol content because the loss of GlcNAc-1-phosphotransferase activity is associated with accumulation of cholesterol (16–18). We demonstrated, however, that neither the induced reduction of the cholesterol content by the HMG-CoA reductase inhibitor atorvastatin nor the LDL-mediated overload of cholesterol affected the ER-Golgi transport of the GlcNAc-1-phosphotransferase α/β-subunit precursor protein. Immunofluorescence microscopy and Western blots showed colocalization of immunoreactive α-subunits with the cis Golgi marker protein GM130 and the appearance of mature α-subunits, respectively (Fig. 3). Subsequently, neither the pattern of M6P modifications on lysosomal proteins, the transport kinetics of the newly synthesized lysosomal protease cathepsin Z, and its lysosomal steady state concentration nor the activity of the lysosomal marker enzyme β-hexosaminidase were altered in cells of low or high cholesterol content in comparison with control cells (Fig. 4). In a second control approach, we used fibroblasts from GlcNAc-1-phosphotransferase knock-in mice, which mimic the biochemical and clinical symptoms of human MLII disease (18, 31, 32). Fibroblasts of the MLII mouse express truncated, inactive α-subunits that are localized in the ER due to the loss of the combinatorial ER exit motif (7, 18). The missorting and lysosomal depletion of the soluble Npc2 protein in these cells (33) result in lysosomal accumulation of nonesterified cholesterol (18). In consequence, the impaired egress from lysosomal compartments causes a lower cholesterol content in ER membranes, which stimulates SREBP processing (13). In MLII cells, activated SREBP signaling leads to a 3-fold up-regulated LDLR expression that is further increased by atorvastatin treatment (Figs. 1, 2), indicating an intact cholesterol-sensing mechanism in these mutant cells.
The overexpression of the human α/β-subunit precursor protein of GlcNAc-1-phosphotransferase in MLII MEFs, however, did not affect the transport to the Golgi apparatus and the S1P-mediated formation of mature α-subunits (Fig. 3C). As a second cell model accumulating massive amounts of cholesterol and other lipids, we used fibroblasts of patients with mutations in the polytopic membrane protein NPC1, which mediates the egress of nonesterified cholesterol from lysosomes to the ER and plasma membranes (19). Similar to MLII MEFs, the expression of α/β-subunit precursor protein in NPC1 patient fibroblasts resulted in the cleavage into mature subunits (Fig. 3D), confirming the independency of the α/β-subunit precursor transport from the ER to the Golgi apparatus of the cholesterol content.
Of interest, with the exception of LDL overload of WT MEFs, both the induced alterations in the cellular cholesterol content and the lysosomal accumulation of cholesterol in NPC1 fibroblasts failed to impair the receptor-mediated internalization and the transport of an M6P-containing lysosomal enzyme, arylsulfatase B (ASB), along the endocytic pathway or the proteolytic processing into mature enzyme forms (Fig. 5A–C). It is likely that the increased cellular cholesterol content in LDL overloaded WT MEFs results in a moderate redistribution of Mpr300 toward the endosomal compartment with a subsequent higher number of receptors recycling via the cell surface. In MLII MEFs, however, the internalization rate of ASB is increased due to the elevated MPR300 level (Fig. 6A) caused by the higher Mpr300 mRNA level (33), and the intermediate and mature forms of ASB generated by lysosomal protease(s) accumulate. This observation is unexpected because the majority of lysosomal proteases (e.g., cathepsins D, L, B, S, and C) reach lysosomes in MLII cells in an M6P/sortilin-independent manner, with the exception of cathepsin Z (Fig. 4B, C), carboxypeptidase Q, and dipeptidylpeptidase 7 (33). It remains to be examined whether these missorted peptidases/proteases are involved in the degradation of ASB polypeptides or if the 17-kDa LDLR fragments (Fig. 6B) formed in the process of proprotein convertase subtilisin/kexin type 9 (PCSK9) initiated and mediated internalization and degradation of LDLR (26, 34).
Similarly to SREBPs, which are retained in the ER by SCAP, the ER exit of another S1P substrate, ATF6, is prevented by binding of the ER chaperone BiP, which dissociates in response to ER stress and allows the translocation of ATF6 to the Golgi apparatus (35). Our data showed that the transport of the α/β-subunit precursor of GlcNAc-1-phosphotransferase is independent of cholesterol and ER stress (unpublished data). However, mutational analysis suggests that the transport of the α/β-subunit precursor is not constitutive but depends on the binding of a yet unknown protein in the lumen of the ER that is required for the ER transport of the precursor protein to the Golgi apparatus (36).
Supplementary Material
Acknowledgments
The authors thank Sandra Ehret and Johannes Brand for expert technical assistance and the Microscopic Imaging Facility of the University Medical Center Hamburg-Eppendorf for support.
Footnotes
Abbreviations:
- ASB
- arylsulfatase B
- ATV
- atorvastatin
- CtsZ
- cathepsin Z
- ER
- endoplasmic reticulum
- GlcNAc
- N-acetylglucosamine
- GM130
- Golgi marker protein 130
- LPDS
- lipoprotein-deficient serum
- LDLR
- LDL receptor
- M6P
- mannose 6-phosphate
- MEF
- mouse embryonic fibroblast
- MLII
- mucolipidosis type II
- MPR
- M6P receptor
- NBCS
- newborn calf serum
- NPC1
- Niemann-Pick disease type C1
- S1P
- site-1 protease
- S2P
- site-2 protease
- SCAP
- SREBP cleavage-activating protein
- SREBP
- sterol regulatory element-binding protein
This work was supported by Deutsche Forschungsgemeinschaft grants SFB877-B3 (S.K., T.B., and S.P.) and GRK1459 (S.M., J.H., T.B., and S.P.).
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of two figures.
REFERENCES
- 1.Saftig P., Klumperman J. 2009. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function. Nat. Rev. Mol. Cell Biol. 10: 623–635. [DOI] [PubMed] [Google Scholar]
- 2.Braulke T., Bonifacino J. S. 2009. Sorting of lysosomal proteins. Biochim. Biophys. Acta. 1793: 605–614. [DOI] [PubMed] [Google Scholar]
- 3.Pohl S., Marschner K., Storch S., Braulke T. 2009. Glycosylation- and phosphorylation-dependent intracellular transport of lysosomal hydrolases. Biol. Chem. 390: 521–527. [DOI] [PubMed] [Google Scholar]
- 4.Raas-Rothschild A., Cormier-Daire V., Bao M., Genin E., Salomon R., Brewer K., Zeigler M., Mandel H., Toth S., Roe B., et al. 2000. Molecular basis of variant pseudo-hurler polydystrophy (mucolipidosis IIIC). J. Clin. Invest. 105: 673–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kudo M., Bao M., D’souza A., Ying F., Pan H., Roe B. A., Canfield W. M. 2005. The alpha- and beta-subunits of the human UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase [corrected] are encoded by a single cDNA. J. Biol. Chem. 280: 36141–36149. [DOI] [PubMed] [Google Scholar]
- 6.Tiede S., Storch S., Lübke T., Henrissat B., Bargal R., Raas-Rothschild A., Braulke T. 2005. Mucolipidosis II is caused by mutations in GNPTA encoding the alpha/beta GlcNAc-1-phosphotransferase. Nat. Med. 11: 1109–1112. [DOI] [PubMed] [Google Scholar]
- 7.Franke M., Braulke T., Storch S. 2013. Transport of the GlcNAc-1-phosphotransferase alpha/beta-subunit precursor protein to the Golgi apparatus requires a combinatorial sorting motif. J. Biol. Chem. 288: 1238–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marschner K., Kollmann K., Schweizer M., Braulke T., Pohl S. 2011. A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism. Science. 333: 87–90. [DOI] [PubMed] [Google Scholar]
- 9.Kudo M., Canfield W. M. 2006. Structural requirements for efficient processing and activation of recombinant human UDP-N-acetylglucosamine:lysosomal-enzyme-N-acetylglucosamine-1-phosphotransferase. J. Biol. Chem. 281: 11761–11768. [DOI] [PubMed] [Google Scholar]
- 10.Sakai J., Nohturfft A., Goldstein J. L., Brown M. S. 1998. Cleavage of sterol regulatory element-binding proteins (SREBPs) at site-1 requires interaction with SREBP cleavage-activating protein. Evidence from in vivo competition studies. J. Biol. Chem. 273: 5785–5793. [DOI] [PubMed] [Google Scholar]
- 11.Seidah N. G., Mowla S. J., Hamelin J., Mamarbachi A. M., Benjannet S., Toure B. B., Basak A., Munzer J. S., Marcinkiewicz J., Zhong M., et al. 1999. Mammalian subtilisin/kexin isozyme SKI-1: a widely expressed proprotein convertase with a unique cleavage specificity and cellular localization. Proc. Natl. Acad. Sci. USA. 96: 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown M. S., Goldstein J. L. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 89: 331–340. [DOI] [PubMed] [Google Scholar]
- 13.Brown M. S., Goldstein J. L. 1999. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. USA. 96: 11041–11048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., Brown M. S. 2002. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 110: 489–500. [DOI] [PubMed] [Google Scholar]
- 15.Braulke T., Raas-Rothschild A., Kornfeld S. 2013. I-cell disease and pseudo-Hurler polydystrophy: Disorders of lysosomal enzyme phosphorylation and localization. In D. Valle, B. Vogelstein, K. W. Kinzler, S. E. Antonarakis, A. Ballabio, C. R. Scriver, W. S. Sly, F. Bunz, K. M. Gibson, and G. Mitchell, editors. The online metabolic and molecular basis of inherited diseases. McGraw-Hill, NY. Chapter 138. Accessed February 2013 at www.ommbid.com.
- 16.Inui K., Nishimoto J., Okada S., Yabuuchi H. 1989. Impaired cholesterol esterification in cultured skin fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy. Biochem. Int. 18: 1129–1135. [PubMed] [Google Scholar]
- 17.Otomo T., Higaki K., Nanba E., Ozono K., Sakai N. 2011. Lysosomal storage causes cellular dysfunction in mucolipidosis II skin fibroblasts. J. Biol. Chem. 286: 35283–35290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kollmann K., Damme M., Markmann S., Morelle W., Schweizer M., Hermans-Borgmeyer I., Röchert A. K., Pohl S., Lübke T., Michalski J. C., et al. 2012. Lysosomal dysfunction causes neurodegeneration in mucolipidosis II ‘knock-in’ mice. Brain. 135: 2661–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanier M. T. 2015. Complex lipid trafficking in Niemann-Pick disease type C. J. Inherit. Metab. Dis. 38: 187–199. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein J. L., Basu S. K., Brown M. S. 1983. Receptor-mediated endocytosis of low-density lipoprotein in cultured cells. Methods Enzymol. 98: 241–260. [DOI] [PubMed] [Google Scholar]
- 21.Müller-Loennies S., Galliciotti G., Kollmann K., Glatzel M., Braulke T. 2010. A novel single chain antibody fragment for detection of mannose 6-phosphate-containing proteins: application in mucolipidosis type II patients and mice. Am. J. Pathol. 177: 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Pace R., Coutinho M. F., Koch-Nolte F., Haag F., Prata M. J., Alves S., Braulke T., Pohl S. 2014. Mucolipidosis II-related mutations inhibit the exit from the endoplasmic reticulum and proteolytic cleavage of GlcNAc-1-phosphotransferase precursor protein GNPTAB. Hum. Mutat. 35: 368–376. [DOI] [PubMed] [Google Scholar]
- 23.Braulke T., Gartung C., Hasilik A., von Figura K. 1987. Is movement of mannose 6-phosphate-specific receptor triggered by binding of lysosomal enzymes? J. Cell Biol. 104: 1735–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown M. S., Goldstein J. L. 1975. Regulation of the activity of the low density lipoprotein receptor in human fibroblasts. Cell. 6: 307–316. [DOI] [PubMed] [Google Scholar]
- 25.Steckel F., Hasilik A., von Figura K. 1983. Biosynthesis and maturation of arylsulfatase B in normal and mutant cultured human fibroblasts. J. Biol. Chem. 258: 14322–14326. [PubMed] [Google Scholar]
- 26.Tveten K., Strøm T. B., Berge K. E., Leren T. P. 2013. PCSK9-mediated degradation of the LDL receptor generates a 17 kDa C-terminal LDL receptor fragment. J. Lipid Res. 54: 1560–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scott C. D., Baxter R. C. 1996. Regulation of soluble insulin-like growth factor-II/mannose 6-phosphate receptor in hepatocytes from intact and regenerating rat liver. Endocrinology. 137: 3864–3870. [DOI] [PubMed] [Google Scholar]
- 28.Clairmont K. B., Czech M. P. 1991. Extracellular release as the major degradative pathway of the insulin-like growth factor II/mannose 6-phosphate receptor. J. Biol. Chem. 266: 12131–12134. [PubMed] [Google Scholar]
- 29.Rawson R. B. 2003. The SREBP pathway-insights from Insigs and insects. Nat. Rev. Mol. Cell Biol. 4: 631–640. [DOI] [PubMed] [Google Scholar]
- 30.Motamed M., Zhang Y., Wang M. L., Seemann J., Kwon H. J., Goldstein J. L., Brown M. S. 2011. Identification of luminal Loop 1 of Scap protein as the sterol sensor that maintains cholesterol homeostasis. J. Biol. Chem. 286: 18002–18012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kollmann K., Pestka J. M., Kühn S. C., Schöne E., Schweizer M., Karkmann K., Otomo T., Catala-Lehnen P., Failla A. V., Marshall R. P., et al. 2013. Decreased bone formation and increased osteoclastogenesis cause bone loss in mucolipidosis II. EMBO Mol. Med. 5: 1871–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Otomo T., Schweizer M., Kollmann K., Schumacher V., Muschol N., Tolosa E., Mittrücker H. W., Braulke T. 2015. Mannose 6 phosphorylation of lysosomal enzymes controls B cell functions. J. Cell Biol. 208: 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Markmann S., Thelen M., Cornils K., Schweizer M., Brocke-Ahmadinejad N., Willnow T., Heeren J., Gieselmann V., Braulke T., Kollmann K. 2015. Lrp1/LDL receptor play critical roles in mannose 6-phosphate-independent lysosomal enzyme targeting. Traffic. 16: 743–759. [DOI] [PubMed] [Google Scholar]
- 34.Canuel M., Sun X., Asselin M. C., Paramithiotis E., Prat A., Seidah N. G. 2013. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. 8: e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen J., Chen X., Hendershot L., Prywes R. 2002. ER stress regulation of ATF6 localization by dissociation of BiP/GRP78 binding and unmasking of Golgi localization signals. Dev. Cell. 3: 99–111. [DOI] [PubMed] [Google Scholar]
- 36.Velho R. V., De Pace R., Klünder S., Sperb-Ludwig F., Lourenço C. M., Schwartz I. V., Braulke T., Pohl S. 2015. Analyses of disease-related GNPTAB mutations define a novel GlcNAc-1-phosphotransferase interaction domain and an alternative site-1 protease cleavage site. Hum. Mol. Genet. 24: 3497–3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.