

Abstract
Neuronal cell death plays a role in many chronic neurodegenerative diseases with the loss of particular subsets of neurons. The loss of the neurons occurs during a period of many years, which can make the mode(s) of cell death and the initiating factors difficult to determine. In vitro and in vivo models have proved invaluable in this regard, yielding insight into cell death pathways. This review describes the main mechanisms of neuronal cell death, particularly apoptosis, necrosis, excitotoxicity and autophagic cell death, and their role in neurodegenerative diseases such as ischaemia, Alzheimer's, Parkinson's and Huntington's diseases. Crosstalk between these death mechanisms is also discussed. The link between cell death and protein mishandling, including misfolded proteins, impairment of protein degradation, protein aggregation is described and finally, some pro-survival strategies are discussed.
Keywords: Alzheimer's disease (AD), apoptosis, autophagic cell death (ACD), autophagy, endoplasmic reticulum (ER) stress, excitotoxicity, heat shock proteins (Hsps), Huntington's disease (HD), ischaemia, Parkinson's disease (PD), protein aggregation, ubiquitin-proteasome system (UPS), unfolded protein response (UPR)
Introduction: neurodegeneration in disease
-
Mechanisms of cell death in neuronal cells
Necrosis
Apoptosis
Excitotoxicity
Autophagic cell death
Crosstalk between different cell death mechanisms
-
Mechanisms of neuronal cell death in neurodegenerative disease: the evidence
Alzheimer's disease
Parkinson's disease
Huntington's disease
Ischaemia
-
Recurring themes around protein mishandling in neurodegenerative diseases
Endoplasmic reticulum stress, the unfolded protein response and ER stress-induced cell death
Protein degradation: the ubiquitin-proteasome system and autophagy
Ubiquitin-proteasome system
Autophagy
Protein aggregation
Towards pro-survival therapies for neurodegenerative diseases
Introduction: neurodegeneration in disease
Neurodegenerative diseases, such as Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD) and ischaemia, are characterized by the loss or dysfunction of particular groups of neurons. Neuronal dysfunction may not necessarily be the result of neuronal cell death. It can be caused by synaptic loss, impairment of long-term potentiation (LTP) or disruption of neuronal signalling as a result of disease pathology. These events often precede neuronal cell loss in chronic neurodegenerative diseases [1]. Aging is a factor in these diseases, suggesting that accumulation of neuronal stresses overtime causes cell death. However, during normal aging there is relatively little neuronal loss, which is in contrast to a high degree of neuronal death in neurodegenerative diseases [2]. This review will focus on mechanisms of cell death in neurons and will attempt to bring together some common themes around protein handling that may help to shed light on the causes of neuronal cell death in neurodegenerative diseases. The underlying genetic basis of some neurodegenerative diseases such as familial AD, familial PD and HD has helped greatly in furthering our understanding of what causes neuronal cell death in these diseases and the type of cell death that is induced.
Mechanisms of cell death in neuronal cells
Although it is recognized that neurons die in neurodegenerative diseases, the mode of cell death is often unclear. There are a number of recognized ways in which neuronal cells can die, including apoptosis, necrosis, autophagic cell death (ACD) and excitotoxic-ity. Other forms of cell death such as oncosis and paraptosis have not been studied much in neurons, and a role for these modes of cell death in neurodegenerative diseases is not known, largely because of the lack of specific markers [1].
Necrosis
Necrosis is an acute form of cell death. It is characterized by loss of ATP, dissipation of ion gradients, cell swelling and ultimately cell lysis (Fig.1A). Necrosis thus causes inflammation in the surrounding region due to release of cell contents into the environment. Necrotic cell death is usually caused by severe cellular stress, e.g. by high levels of toxins. Necrosis is not a programmed form of cell death in that there is no identifiable biochemical pathway, and once initiated, this form of cell death does not offer much potential for therapeutic intervention since it tends to be acute in nature.
Figure 1.
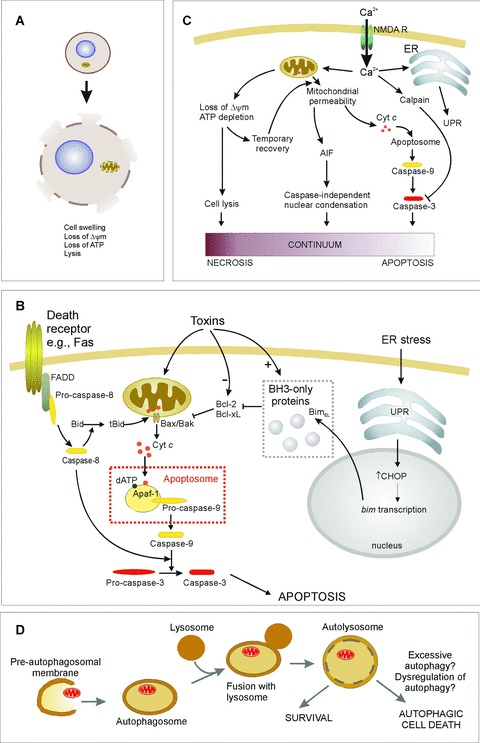
Mechanisms of cell death. See text for details. (A) Necrosis is characterized by loss of ATR which leads to cell lysis and release of cellular contents into the extracellular environment. (B) Apoptosis induction involves different pathways leading to caspase activation. In the extrinsic pathway ligand binding to death receptors {e.g. Fas) induces their trimerization and recruitment of caspase-8 through death domain-containing adaptor proteins (e.g. FADD). This leads to auto-activation of caspase-8, which can directly activate effector caspases. Alternatively in some cells Bid cleavage by caspase-8 is required for death. Truncated Bid translocates to the mitochondria and activates the intrinsic pathway. The intrinsic pathway is activated by toxins or conditions that cause stress to the mitochondria or endoplasmic reticulum (ER). Such stresses often alter the levels of, or cause post-translational modification of, Bcl-2 family proteins, initiating the release of cytochrome c from the mitochondria. For example, ER stress up-regulates the unfolded protein response (UPR), which increases expression of BimEL, a BH3-only protein that antagonizes Bcl-2 and Bcl-xL, permitting Bax-mediated cytochrome c release. Once in the cytosol, cytochrome c stimulates formation of the apoptosome complex, leading to caspase-9 activation. Initiator caspases (e.g. caspase-8 and -9) are shown in yellow, downstream effector caspases (e.g. caspase-3) are shown in red. (C) Excitotoxicity is caused by the influx of Ca2+ through the NMDA receptor. This can result in apoptosis, necrosis, or cell death that exhibits apoptotic nuclear morphology without caspase activation, suggesting a continuum of death patterns. (D) Autophagic cell death may be due to excessive autophagic digestion, as a result of dysregulation of the pathway by unknown mechanisms.
Apoptosis
Apoptosis was originally defined as ‘shrinkage necrosis’, exhibiting condensed and fragmented nuclei in shrunken cells. In contrast to necrosis, apoptosis is characterized by maintenance of cellular ATP and the initiation of a biochemical pathway leading to cell shrinkage, budding off of apoptotic bodies and phagocytosis of these cell particles. There is no associated cell lysis or inflammation. Biochemically, apoptosis is associated with the activation of caspase proteases that exist in a functional hierarchy of initiator and downstream effector caspases (Fig.1B). Effector caspases cleave a wide variety of protein substrates to cause the degradation and demise of the cell [3]. There are at least two well-characterized ways in which caspase activation is initiated in mammalian cells. These are the intrinsic (or mitochondrial) pathway and the extrinsic (or death receptor) pathway.
In the intrinsic pathway, caspase activation is triggered by the release of cytochrome c from the mitochondria into the cytosol, where it plays an essential role in the formation of a multimeric caspase activation complex called the apoptosome [4, 5]. Specifically, cytosolic cytochrome c interacts with Apaf-1 which, in the presence of dATP, leads to clustering and autoactivation of initiator caspase-9 [4, 6]. Active caspase-9 causes cleavage and activation of downstream effector caspases, such as caspase-3. This occurs in response to several apoptotic stimuli including neu-rotoxins such as colchicine, glutamate, 6-hydroxydopamine [7–9]. It should be noted that other pro-apoptotic factors such as Smac/Diablo, apoptosis-inducing factor (AIF) and Omi/Htra2 are also released from the mitochondria during apoptosis [10]. One of these, AIF can induce chromatin condensation without caspase activation (Fig.1C) and in fact, apoptotic nuclear morphology in neuronal cells is not always associated with caspase activity [11, 12]. In the extrinsic pathway, ligand binding of death receptors (e.g. Fas, TNF-receptor, TRAIL receptors) leads to trimerization of these receptors and the formation of an intracellular multiprotein complex. The initiator caspases-8 or -10 are part of this complex, and proximity-induced autoactivation leads to their cleavage and full activation (Fig.1B) [13].
Apoptosis can be regulated at various points in the pathway. Mitochondrial integrity, and thus the intrinsic pathway, is regulated by the Bcl-2 family, which includes both pro- (e.g. Bax, Bak) and anti-apoptotic (e.g. Bcl-2, BcI-xl) members, which either promote or inhibit release of pro-apoptotic factors into the cytosol (Fig.1B) [14–16]. Downstream of the release of cytochrome c, caspase activation and activity are regulated by the inhibitor of apoptosis protein (IAP) family of proteins [17].
Excitotoxicity
Particular to neuronal cells is death by excitotoxicity, which istox-icity caused by excessive stimulation of the NMDA subtype of glutamate receptors. This form of neuronal cell death occurs as a result of ATP depletion that occurs during ischaemia. The loss of ATP causes general depolarization of neurons, thus stimulating neurotransmitter release, and impairing ATP-dependent neuro-transmitter re-uptake systems. Since glutamate is the most abundant excitatory neurotransmitter in the brain, this leads to overex-citation of NMDA receptors (Fig.1C). These ligand-gated ion channels allow influx of Ca2+. Cytosolic Ca2+ is then sequestered by the mitochondria and by the endoplasmic reticulum (ER). If excessive, this can cause ER stress and/or mitochondrial permeability transition [18]. It is open to discussion, however, whether excitotoxicity can really be considered a separate form of cell death, as it is linked to activation of both apoptosis and necrosis (Fig.1C). For example, in vitro evidence shows that glutamate can cause both early necrosis and delayed apoptosis in neurons [19].
Autophagic cell death
The word autophagy comes from the Greek meaning ‘self-eating’. It is primarily a mechanism by which organelles, protein aggregates and some proteins are degraded by the cell (see section below on autophagy). It may also be a pathway to cell death termed ACD, which is thought to due to excessive autophagy (Fig.1D) [20]. ACD has a distinct morphology, including organelle swelling, vacuolation of the cytoplasm and membrane disruptions leading to local inflammation [21]. Although autophagy is generally associated with conditions of starvation, where it plays a pro-survival role in providing amino acids and other building blocks of cellular macromolecules, markers of autophagy have been observed during cell death. ACD has mainly been associated with developmental cell death, where massive autophagy causes irreversible cellular atrophy, leading to cell death of large numbers of cells [22]. Recently, induction of cell death that is dependent on autophagy (Atg) genes, such as Atg7 or Beclinl has been reported [23, 24].
Crosstalk between different cell death mechanisms
Many cell death stimuli can induce more than one mode of cell death depending on the conditions, such as the seventy of the stress, duration of the stress, the underlying ‘health’ of the cell, particularly with regard to redox levels and mitochondrial integrity. Some researchers have proposed that there is a continuum of cell death mechanisms in ischemic and excitotoxic death, with apoptosis and necrosis at opposite ends of a scale that incorporates cell death mechanisms featuring some but not all of the characteristics of apoptosis and necrosis [25–27]. Notably, certain neurotoxins, such as glutamate that induces excitotoxicity, and 6-hydroxy-dopamine that is a Parkinson's mimetic, can induce both apoptosis and necrosis in vitro[8, 19]. The mode of death induced by glutamate has been shown to depend on mitochondrial transmembrane potential (ΔΨm) [19, 28]. The ΔΨm controls three interrelated mitochondrial functions of relevance to neuronal survival: namely ATP synthesis, Ca2+ accumulation and superoxide generation [29]. During exposure of cerebellar granule cells to glutamate, both ΔΨm and ATP levels decline rapidly (Fig.1C) [19]. In those neurons with irreversibly dissipated ΔΨm, necrosis rapidly ensues, while the surviving population recover both ΔΨm and energy levels and subsequently undergo delayed apoptosis [19]. Since many of the cytoplasmic and nuclear changes observed during apoptosis require energy in the form of ATP, the ability of mitochondria to generate sufficient ATP may be one important factor that directs neurons towards necrosis or apoptosis [19, 30], such that if ATP levels are at least partially maintained, the cell is capable of undergoing apoptosis and if ATP levels fall profoundly, necrotic killing ensues [31]. This is supported by the observation that while treatment of cerebellar granule cells with glutamate induces delayed apoptosis, treatment with a combination of glutamate and the irreversible mitochondrial uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP) results in necrosis [19].
There is also mounting evidence of crosstalk between apoptosis, autophagy and ACD. Inhibition of caspases induces cell death that is dependent on the autophagy genes Atg7nnd Beclin 1 (also known as Atg6) [23]. Conversely, inhibition of autophagy can trigger apoptosis [32]. At a basic level autophagic digestion of injured mitochondria may also help to ensure that only healthy mitochondria endure within cells. The intrinsic pathway of apoptosis requires the presence of Bax/Bak, but the mode of cell death switches to ACD in the absence of Bax/Bak [33]. Similarly, in apoptosis-deficient cells that lack Bax and Bak, prolonged stress to the ER results in cell death that resembles necrosis and is associated with autophagy [34]. Inhibition of autophagy with 3-methy-ladenine or Atg5 knockdown in these cells promoted survival, which suggests that in apoptosis-competent cells autophagy has a pro-survival function, while in apoptosis-deficient cells autophagy is part of the cell death process, i.e. ACD [34].
During recent years, details of the molecular mechanisms that mediate crosstalk between different cell death mechanisms are beginning to be elucidated. One group of molecules that play a role at the intersection of apoptosis, autophagy and ACD is the Bcl-2 family. The best understood function of Bcl-2 proteins is in the regulation of apoptosis through control of the release of pro-apoptotic factors from mitochondria. Recently, Bcl-2 has been shown to also inhibit autophagy [32, 35] and ACD [36]. It has been suggested that the anti-autophagy function of Bcl-2 may help maintain autophagy at levels that are compatible with cell survival, rather than cell death [35]. However, the effect of Bcl-2 on autophagy is controversial as it has also been reported to sensitize cells to autophagy and permit autophagy to develop through inhibition of apoptosis [33, 37]. Bcl-2 inhibition of autophagy is mediated through its interaction with Beclin 1 [35]. Beclin 1 was the first identified mammalian autophagy gene product and plays an essential role in autophagosome formation [39]. However, it was originally identified in a screen for Bcl-2-interacting proteins [39]. Recently, Beclin 1 has been shown to be a novel BH3-only protein and to interact with a number of anti-apoptotic Bcl-2 family members including Bcl-2, Bcl-xL, Bcl-w and Mcl-1 [40–43]. Thus, one possible scenario is that a Bcl-2:Beclin 1 rheostat acts as a switch between autophagy and apoptosis.
The autophagy gene product, Atg5, is another protein having roles in both autophagy and apoptosis. Atg5 is involved in the early stages of autophagosome formation [44]. Atg5 has been reported to interact with FADD (Fas-associated protein with death domain), mediating interferon—/-induced cell death [45]. Atg5-mediated cell death, but not vacuole formation, was blocked in FADD-deficient cells [45]. In contrast, inhibition of autophagy with 3-methyladenine or expression of an Atg5 mutant blocks both cell death and vacuole formation [45]. A more recent study shows that calpain-mediated cleavage of Atg5 produces a truncated form of Atg5 that can translocate from the cytosol to mitochondria, associate with Bcl-xL and trigger cytochrome c release [46]. Thus, truncated Atg5 may promote apoptosis by acting in a manner reminiscent of some BH3-only proteins that inactivate anti-apoptotic Bcl-2 proteins and permit Bax/Bak-mediated cytochrome c release [47]. Since Ca2+ influx is a major factor in excitotoxicity it will be particularly interesting to observe the role, if any, of truncated Atg5 in excitotoxic neuronal cell death. One might speculate that calpain-mediated cleavage of Atg5 during excitotoxicity diverts cytoprotective autophagy and promotes cell death. However, cal-pains have also been shown to inactivate caspases and in this way, convert apoptotic death to necrotic death [48]. Therefore, it is difficult to predict whether apoptosis or necrosis would be the outcome of calpain-mediated cleavage of Atg5 and caspases within the same neuron. It is likely that mitochondrial outer membrane permeabilization (even perhaps involving truncated Atg5) permits the release of pro-apoptotic factors such as AIF, which induces nuclear condensation. This scenario would support the suggested continuum model of cell death patterns (Fig.1C) and the experimental observation of neurons that exhibit characteristics of both apoptosis (condensed nuclei) and necrosis (cell lysis, lack of cas-pase activity) [11, 26, 27].
Increasingly, we are learning that there are compensatory cell death pathways that are induced if one route to cell death is blocked. Research in this area is gathering momentum, particularly with regard to crosstalk between apoptosis and autophagy (Fig.2). In addition to the Bcl-2 family, Beclin 1 and Atg5 there continue to be new proteins identified that participate in both apoptosis and ACD, e.g. the novel transmembrane protein TMEM166 [49]. The current models suggest that apoptosis and autophagy may be mutually exclusive pathways, which would further suggest that apoptosis and ACD are also mutually exclusive. This idea is supported by the accumulating evidence such as the effect of Bcl-2:Beclin 1 interaction and calpain-mediated cleavage of Atg5. However, at least one study has reported the accumulation of autophagic vacuoles preceding apoptotic cell death [50]. Is this indicating that there is also a continuum of death patterns between apoptosis and ACD? It will be interesting during the coming years to learn the role, if any, of crosstalk between different cell death pathways in neurodegenerative diseases.
Figure 2.

Crosstalk between apoptosis, autophagy and autophagic cell death. The schematic depicts some points of crosstalk between these pathways (see text for details).
Mechanisms of neuronal cell death in neurodegenerative disease: the evidence
There is growing evidence from post-mortem patient tissue describing the modes of cell death observed in neurodegenerative diseases. Such evidence has been difficult to accumulate, partly due to the protracted nature of most neurodegenerative diseases, such that only a tiny proportion of the whole cell population is in the process of dying at any one time. Since cell death occurs over many years, the snap-shot representation obtained from such samples can only reveal cells that have recently died. Usually these tissues are representative of the end-stage of the disease, far-removed in time from when we would ideally like to be treating such patients to attempt to prevent neuronal cell death. Often the dying/dead cells display mixed signals and defy classification into distinct patterns of cell death. Another difficulty with patient samples is that markers used for different modes of cell death can be overlapping, non-specific, and therefore, prone to misinterpretation. For example, terminal transferase dUTP nick-end labelling (TUNEL staining) has been used as a marker of apoptosis in human brain tissue. However, TUNEL can also label necrotic cells [51]. Therefore, it is imperative that the correct criteria are used to monitor different modes of cell death. Usually more than one characteristic feature of the type of cell death should be considered. TUNEL staining needs to be combined with others, e.g. ultrastruc-tural analysis of nuclear morphology, or demonstration of activated or cleaved caspases, such as caspase-3 or -8, using specific antibodies for the identification of apoptosis [52]. Although criteria for the identification of autophagy have recently been published, and markers for autophagy have been identified in different neurodegenerative diseases [53], it is not yet possible to be certain that these are indicative of the occurrence of ACD.
Much evidence regarding mechanisms of cell death in neurodegenerative diseases comes from models, either in vitro cell culture systems or in vivo models of the disease. These models use acute toxin exposure and/or genetic mutations found in disease and have contributed much to promote the view that apoptosis and other forms of cell death play important roles in neuronal cell death in neurodegenerative diseases. However, models are limited since they tend to examine the effect of just one toxin or one gene on cell death, and they generally involve acute exposure to insults, which may not be relevant to the protracted degeneration observed in patients. Nevertheless, they have greatly increased our knowledge of the biochemical mechanisms of neuronal cell death in neurodegenerative diseases. In the following section, some of the major neurodegenerative diseases and the link to neuronal cell death mechanisms are described.
Alzheimer's disease
AD is the most common neurodegenerative disease. It is clinically characterized by progressive memory loss and cognitive dysfunction, resulting in severe dementia. The pathology of AD includes the loss of medium and large pyramidal neurons from hippocam-pal regions [54–56] and the presence of extracellular placques containing amyloid filaments and intracellular neurofibrillary tangles composed of hyperphosphorylated tau [57]. Familial AD is caused by mutations in amyloid precursor protein or presenilin-1 or -2. Presenilins play a crucial role in 7-secretase activity, which is responsible for proteolytic processing of amyloid precursor protein, producing the β-amyloid peptide [58]. AD mutations lead to altered processing that is accompanied by increased production of a slightly larger peptide, p-amyloid-42, which is prone to oligomerization, forming fibrils and placques [59]. It is uncertain what leads to neuronal cell death in AD.
Studies of post-mortem brain tissue indicate that neuronal apoptosis occurs in AD [60]. Active caspases have been detected in post-mortem tissue from AD brains [61–63]. However, this has been disputed by others who report no apoptotic morphology in AD cases [64]. In vitro models show that β-amyloid can induce apoptosis [65, 66]. Presenilin mutations sensitize neurons to induction of apoptosis and to excitotoxicity [67, 68]. In fact, it has been suggested that accumulation of β-amyloid leads to increased sensitivity to excitotoxicity in AD [69] and excessive activation of NMDA receptors has been specifically linked to β-amyloid-induced degeneration in a rat model [70]. It has also been suggested that excitotoxicity might have a more important role in later stages of AD [71]. Although autophagy has been linked to models of AD, there is no evidence that this is part of a cell death pathway [72].
Parkinson's disease
PD is the second most common neurodegenerative disease, mainly affecting people over 55 years. Pathologically it is characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta and the presence of eosinophilic intracytoplasmic inclusions (Lewy bodies) within the surviving neurons. This leads to dopamine depletion in the striatum, which is the direct cause of the clinical features of bradykinesia, resting tremor and rigidity. The clinical symptoms of PD become evident when there is a loss of about 50–60% of the dopaminergic neurons within the substantia nigra. Thus, PD is a good example of a neuro-degenerative disease where neuronal cell loss is directly respon-sible for the clinical features observed. The cause of PD is most probably multifactorial, with genetic predisposition, environmental toxins and aging being important factors in disease initiation and progression for both hereditary and sporadic PD. To date, mutations in at least 13 PARK genes have been linked to the pathogenesis of familial PD [73]. These include mutations in genes that encode the proteins α-synu-clein, parkin, PTEN-induced kinase 1 (PINK1), DJ-1, leucine-rich repeat kinase2 (LRRK2) and ubiquitin carboxy-terminal hydrolase L1 (UCHL1) [73]. Of these, parkin and UCHL1 are linked to the ubiquitin-proteasome system (UPS) that degrades damaged or misfolded proteins [74]. In addition, several of these genes, including parkin, PINK1, DJ-1 and Omi/Htra2 are linked to the mitochondria, and may have roles in mitochondrial function and resistance to oxidative stress [75]. The molecular mechanisms that initiate dopaminergic neuron loss in PD are not known. Evidence from various sources suggest a role for toxin-induced death, oxidative stress, defects in mitochondrial complex I, protein aggregation and impairment of the UPS [76–78].
The presence of apoptotic chromatin clumps along with TUNEL staining in neuromelanin-containing cells in the substantia nigra PD post-mortem tissue suggests that apoptotic cell death contributes to neuronal loss in PD [79–82]. However, others have been unable to confirm these findings [83, 84]. Activated caspase-3 has also been observed in these cells [81]. There is also evidence for a role for the extrinsic apoptotic pathway in postmortem studies of PD brain. These are related to the increased levels of TNFα, soluble Fas and the Fas adaptor protein FADD in the midbrain of PD patients [85–87]. Importantly, increased active caspase-8 has been detected in neuromelanin-containing substantia nigra neurons in PD brains [88, 89]. In vitro models of PD using the Parkinson mimetics MPP+ (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) and 6-hydroxydopamine have reported both apoptosis and necrosis [8, 90–92]. The pesticide rotenone induces caspase-3-mediated apoptosis in ventral mesencephalic dopaminergic neurons [93]. Inducible expression of mutant a-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis [94]. Targeted overexpres-sion of α-synuclein in the nigrostriatal system has provided a new animal model of PD [95]. In this model there is selective degeneration of nigral dopamine neurons with activation of caspase-9 [96]. Some early studies proposed a role for excitotoxicity in PD although subsequent research has not supported this hypothesis [97, 98]. However, there may be a role for ACD in PD. Ultrastructural examination revealed autophagosome-like structures in melanized neurons of the substantia nigra in PD patients [99–101]. It has been reported that the neuromelanin granules in catecholaminergic neurons of the substantia nigra are delimited by double membranes, and may in fact be autophagosomes sequestering dopamine and metabolites to protect the cell from their lethal accumulation in the cytosol [101]. In support of these findings, dopamine and MPP+ have been shown to induce ACD in human neuroblastoma cells [102, 103].
Huntington's disease
HD is one of a number of trinucleotide repeat disorders. It is caused by a dominantly inherited expansion of a translated CAG repeat in the N-terminus of the huntingtin protein. The length of the CAG expansion inversely correlates with age of onset of disease symptoms [104]. The main clinical symptom is the occurrence of generalized involuntary movements, which is a consequence of the selective loss of the medium spiny neurons in the neostriatum [104, 105]. The disease pathology is characterized by the presence of nuclear inclusions containing mutant huntingtin [106]. Neuronal cell death in HD may be linked to the accumulation of toxic mutated huntingtin fragments since expansion of the huntingtin CAG repeat above 35 results in neuronal cell death [107–109]. Although a number of interacting proteins have been identified, the physiological role of huntingtin is not known.
Caspases have been shown to be activated in HD brains [110, 111]. Interestingly, huntingtin itself is a substrate for caspases, generating truncated fragments containing the polyglutamine tract that may be toxic [112–114]. Excitotoxicity has been suggested to play a role in the pathogenesis of HD. The medium spiny neurons that are lost in HD are innervated by glutamate-releasing neurons from the cortex, and they selectively degenerate upon the administration of glutamate receptor agonists into the rat striatum [115]. Post-mortem analysis of the early stage HD patient brains indicates increased levels of the NMDA receptor agonist, quinolinic acid, and its precursor 3-hydroxy-kynurenine, suggesting that it may act as an endogenous excitotoxin in HD [116]. Moreover expression of mutant huntingtin is related to alterations in glutamate neurotransmission, including decreased glutamate uptake, altered trafficking and functionality of glutamate receptors [117]. Although cultured medium spiny from transgenic mice expressing different mutations in huntingtin are more sensitive to excitotoxic damage [118], in vivo experiments have produced conflicting results [119, 120]. However, a direct link between mutant huntingtin expression and excitotoxicity in HD patients has not been shown. Autophagy is stimulated by huntingtin, but it is not known if this is related to ACD [121]. Paradoxically, certain mutant forms of huntingtin can protect neurons from cell death [122].
Ischaemia
Ischaemia, or stroke, is an acute condition caused by an interruption in blood supply, leading to localized deprivation of oxygen, glucose and trophic support in the brain. Since neurons are reliant on a constant supply of oxygen and glucose to generate ATP, the loss of ATP during ischaemia causes rapid dissipation of ion gradients, leading to uptake of water and cell swelling, lysis and cell death.
Ischaemia is characterized by acute neuronal cell death and delayed death that occurs many days after the initial insult [123, 124]. The initial phase of cell death is necrotic and the delayed death is apoptotic. Neuronal cell death at the ischemic core tends to be necrotic, while in the penumbra region it is apoptotic, suggesting that the severity of the insult determines the mode of cell death [123, 124]. Both phases of cell death can be blocked by antagonism of NMDA receptors [125]. This cell death is excitotoxic and thus shares the same characteristics in terms of a continuum of necrosis and apoptosis being induced [25, 26]. Up-regulation of Beclin 1 is observed in the ischemic penumbra, along with and subcellular redistribution of the autophagy marker LC3 to vacuolic structures in ischemic neurons indicating that autophagy is induced during ischaemia [126–128]. However, ACD has not been conclusively shown.
Recurring themes around protein mishandling in neurodegenerative diseases
Key to the prevention and treatment of neurodegenerative diseases is an understanding of the mechanisms that trigger neuronal cell death. Notably, different neurodegenerative diseases, with different causes, genetic mutations, different patterns of neuronal death, often display a number of common features that include ER stress, oxidative stress, mitochondrial dysfunction, impairment of the proteasome and protein aggregation. These changes are often interrelated, causing disruption of normal neuronal function and eventually leading to cell death. The picture is further complicated by the fact that neurons mount pro-survival responses to protect themselves from these detrimental, disease-related changes. Therefore, neurons in the final stages of their demise often present a complex picture reflecting a struggle between pro-death factors and pro-survival responses, and teasing out the sequence of events can be a challenge. Here we will discuss neurodegenerative diseases with relation to problems in protein handling.
Protein homeostasis in cells is a balance between protein synthesis and protein degradation. Disruption of this equilibrium leads to problems of accumulation and aggregation of excess or mis-folded proteins. Many neurodegenerative diseases including AD, PD and HD are characterized by the accumulation and aggregation of misfolded proteins. Misfolding and/or aggregation of proteins can be the result of problems during synthesis due to impairment of the synthesis/folding machinery, or mutations in the sequence of the protein causing incorrect folding or a tendency to aggregate. Proteins can also become unfolded at some point during their life cycle, or undergo abnormal cleavage generating proteins that cannot fold properly or have a tendency to aggregate. Similarly, mutations in proteins such as proteases can cause the inappropriate accumulation of proteins that cannot be processed further. In addition, proteasome impairment can render a cell unable to degrade misfolded proteins efficiently, thus compounding the problem.
Endoplasmic reticulum stress, the unfolded protein response and endoplasmic reticulum stress-induced cell death
The ER is the site of synthesis and folding of secreted, membranebound and some organelle-targeted proteins. It also acts as a major store for intracellular Ca2+. The protein-folding capacity of the ER is highly sensitive to alterations in cellular ATP levels, Ca2+ levels, and the redox state of the cell, all of which result in the accumulation and aggregation of unfolded proteins, a condition referred to as ER stress [129]. In response cells mount the unfolded protein response (UPR) [129]. In mammalian cells, this is a three-pronged protective strategy resulting in halt of general protein synthesis to reduce the backlog of proteins, induction of Grp78 and Grp94 chaperone proteins to aid folding of proteins already in the system and ER-associated degradation to remove proteins that cannot be refolded by the ER. This concerted cellular response is mediated through three ER transmembrane receptors: pancreatic ER kinase (PERK), activating transcription factor-6 (ATF6) and inositol-requir-ing enzyme 1 (I re 1) [130]. In resting cells, all three ER stress receptors are maintained in an inactive state through their association with the ER chaperone, Grp78. On accumulation of unfolded proteins, Grp78 dissociates from the three receptors, which leads to their activation and triggers the UPR.
Although UPR activation is primarily a protective response by the cell, if the stress is unresolved, prolonged activation of the UPR may trigger cell death [129]. In fact, in recent years, a third apoptosis pathway has been proposed, whereby ER stress leads to direct activation of initiator caspase-12 that is located at the ER [131]. However, since full-length caspase-12 is not expressed in the majority of human beings, and since many reports demonstrate that mitochondrial release of cytochrome c, apoptosome formation and activation of caspase-9 are required for ER stress-induced apoptosis, it is likely that this death occurs Wathe intrinsic pathway [132–134]. For example, we and others have recently shown that ER stress-induced apoptosis requires up-regulation of the Bcl-2 family member Bim that further implicates a requirement for the mitochondrial pathway [133, 135]. The details of how UPR signalling is switched from pro-survival to pro-death is unclear and is a subject of much research, reviewed in [129]. Induction of the transcription factor, CHOP, has been shown to be required for ER stress-induced apoptosis [136]. Recently, the attenuation of IRE1 activity by persistent ER stress has been identified as a potential switch [137]. If this is involved in neuronal cell death in neurodegenerative diseases that exhibit ER stress, it suggests the exciting possibility of altering disease pathogenesis by manipulating the UPR.
ER stress is linked to the pathogenesis of several different chronic neurodegenerative disorders, where UPR markers are seen to be up-regulated. For example, markers of UPR activation (phos-phorylated PERK and Grp78) are increased in AD brains [138]. These changes were seen in normal-appearing neurons, suggesting a role for the UPR early in AD neurodegeneration. Furthermore, presenilin-1 and -2 reside in the ER, and AD mutations appear to impair the ER stress response and enhance vulnerability to stress-induced apoptosis [67, 68]. It has also been suggested that presenilin-1 mutations may deregulate ER Ca stores [139]. Immunoreactivity for phosphorlyated PERK and phosphorylated elF2a has also been detected in neuromelanin-containing dopaminergic neurons in the substantia nigra of PD cases [140]. The phospho-PERK immunoreactivity was colocalized with increased α-synuclein immunoreactivity in dopaminergic neurons [140]. In vitro models using two PD mimetic compounds, 6-hydroxydopamine and MPP+, trigger ER stress in dopaminergic neurons [141, 142]. Furthermore, neuronal cultures from PERK knockout mice, display an increased sensitivity to 6-hydroxy-dopamine [142], while a null mutation in CHOP results in a reduction in 6-hydroxydopamine-induced apoptosis in vivo[143]. However, protection was not observed in the chronic MPTP model, despite robust expression of CHOP [143]. In trinucleotide repeat disorders such as HD, the intracellular accumulation of polygluta-mine triggers ER stress by inhibiting protein degradation and has been linked to the induction of neuronal cell death by N-terminal mutant huntingtin [144–146]. ER stress is also a factor in neuronal cell death during ischaemia, with upregulation of CHOP [147, 148].
Protein degradation: the ubiquitin-proteasome system and autophagy
Misfolded or unwanted proteins are degraded within the cell via two pathways. The ubiquitin-proteasome system (UPS) is the main degradatory pathway for the majority of intracellular proteins. This system is linked to ER stress responses since ER-associated degradation feeds proteins into the UPS for degradation. Secondly, autophagy is used by the cell to eliminate unwanted or damaged organelles via lysosomal degradation and can also be used for degradation of proteins.
Ubiquitin-proteasome system
The UPS involves conjugation of ubiquitin to target substrates, followed by destruction of the tagged protein by the 26S proteasome system [149]. Conjugation of ubiquitin to proteins involves a series of enzyme reactions. Ubiquitin-activating enzyme E1 generates a high-energy intermediate, E1 -ubiquitin. Next, E2 enzymes transfer ubiquitin from E1 to E3 ligase that is bound to specific substrates, to which it ligates activated ubiquitin [149]. Cells usually contain just one E1, a few E2 and many E3s, the latter of which are specific for just a small number of substrates. Tagged proteins are then degraded into peptides by the proteasome, a barrel-shaped, multiprotein, proteolytic complex.
Alterations in the UPS have been connected to several neuro-degenerative diseases [150,151]. Proteasome inhibition leads to the intracellular accumulation of ubiquitinated proteins and causes cell death [152, 153]. The accumulation of ubiquitin conjugates is often found in protein aggregates, which include the neurofibril-lary tangles of AD, Lewy bodies in PD and nuclear inclusions in HD [154, 155]. It is likely that accumulation of ubiquitin conjugates reflects failed attempts by the UPS to remove the damaged/abnormal proteins that have been tagged for destruction (Fig.3). This could be caused by malfunction or overload of the UPS, or from structural changes in specific protein substrates, halting their degradation.
Figure 3.

Protein metabolism in neurons. Misfolded proteins can occur as a result of mutations, oxidative damage, erroneous proteolysis or other post-translational modifications. These can be refolded by chaperones such as Hsps, or targeted for proteasomal degradation by the addition of ubiqui-tin (Ub). Impairment of UPS-mediated degradation can lead to the formation of protein aggregates, which may be degraded via autophagy. The accumulation of misfolded proteins or protein aggregates leads to neuronal cell death by an unknown mechanism.
Proteasome activity is impaired in AD [156]. In the brains of some AD patients a mutated ubiquitin with an extra 20 amino acids at its C-terminus has been observed [157]. This mutated ubiquitin forms polyubiquitin chains that cannot be disassembled and potently inhibit proteasomal degradation of proteins to which they are attached [158]. UPS impairment may also play a role in PD. The PD-associated gene parkin is an E3 ligase, involved in the addition of ubiquitin chains to a select number of protein substrates [159, 160]. It is frequently mutated in early onset PD with loss of ligase function and a reduction in degradation of target proteins [159, 161, 162]. One target of parkin is Pael-R (Parkin-associated endothelin receptor-like receptor) and loss of parkin function results in accumulation of Pael-R, ER stress and neuronal cell death [163, 164]. Conversely, overexpression of parkin suppresses ER stress-induced cell death [164]. Parkin is abundantly expressed within mesencephalic dopaminergic neurons that may explain part of their selective vulnerability [159]. Another mutated-in-PD gene, UCH-L1, was originally characterized as a deubiquiti-nating enzyme [165]. However, it has also been found to act as a ubiquitin ligase [166] and a mono-ubiquitin stabilizer [167]. Discovery of these mutations in genes linked to UPS function has led to the hypothesis that some PD mutations cause aberrant accumulation of substrates that may be toxic [168]. In addition, reports that α-synuclein is degraded through the proteasome led to the idea that abnormalities in UPS-mediated degradation of a-synuclein underlie PD [169–171]. Systemic administration of proteasome inhibitors produces pathological features that are similar to those found in PD [172, 173]. However, some groups have been unable to reproduce these results [174]. The intracellular accumulation of polyglutamine in HD inhibits protein degradation and has been linked to the induction of neuronal cell death by N-terminal mutant huntingtin [144–146].
Autophagy
Autophagy mediates the intracellular degradation of organelles and protein complexes that are too big to pass through the narrow pore of the proteasome. It is normally associated with nutrient and growth factor deprivation where it promotes cell survival through provision of molecular building blocks [20]. However, it can also be induced by the presence of protein aggregates such as are found in neurodegenerative diseases (Fig.3). Three types of autophagy have been described in mammalian cells: macroau-tophagy, microautophagy and chaperone-mediated autophagy (CMA) [175]. They all share the lysosome as a common end-point, but differ in the substrates targeted, their regulation and the conditions under which they are preferentially activated. In macro-autophagy, intracellular components are sequestered by a limiting membrane to form an autophagic vacuole, the autophagosome, that then fuses with lysosomes to degrade the contents [176]. In microautophagy, substrates are directly internalized through invaginations of the lysosomal membrane. CMA is specialized for the degradation of selective substrate proteins that contain a KFERQ (or similar) motif that is recognized by heat shock cognate protein, HSC70, which translocates the proteins into the lysosomes [177].
Autophagy has been linked to different neurodegenerative diseases. However, it is unclear if this phenomenon is the result of increased autophagic activity, or decreased fusion of autophago-somes with lysosomes [178]. It has been reported that β-amyloid generation may occur in autophagic vacuoles [179]. Autophagy is also linked to PD [99, 101]. α-Synuclein is degraded by both CMA and the proteasome [180, 181]. Pathogenic α-synuclein mutations block CMA-mediated degradation of α-synuclein and of other substrates which may underlie the toxic gain-of-function of these mutants [181]. Increased autophagosomes are seen in HD [121], which may be linked to the processing of huntingtin fragments by autophagy [182]. Autophagic neuronal death may play a role in neonatal brain ischaemia [127, 128].
The presence of autophagosomes in neurons in neurodegenerative disorders has raised questions about whether they are contributing to neuronal cell death or protecting against it. Emerging evidence supports the view that induction of autophagy is a neuro-protective response and that inadequate or defective autophagy, rather than excessive autophagy, promotes neuronal cell death in most of these disorders. A cytoprotective role for autophagy by catabolizing intracellular substrates for energy, and by removing damaged mitochondria and other factors that trigger apoptosis, is clear [183]. Autophagy also prevents toxic effects of misfolded or abnormal proteins by sequestering them inside autophagosomes delivering them to the lysosome for degradation [184]. This suggests that increased autophagy observed in neurodegenerative diseases may be linked to an increase in the levels of misfolded and aggregated proteins, and/or to increased numbers of damaged mitochondria, both of which are associated with aging.
Although the UPS and autophagy were thought to work in parallel, recent investigations suggest a functional link between the two. For example, proteasome inhibition activates autophagy and the suppression of autophagy causes polyubiquitinated protein aggregates [185]. Toxicity due to proteasome inhibition can be rescued by autophagy induction [152, 186]. The obvious interpretation is that autophagy upregulation simply provides an alternative route for protein degradation when the UPS is impaired. However, it is also possible that by removing mitochondria from the cell autophagy induction reduces cellular susceptibility to subsequent apoptotic stimuli mediated by the mitochondrial pathway [32,187].
Protein aggregation
Proteins that are misfolded and escape degradation may be prone to aggregation. This occurs because misfolded proteins often expose hydrophobic surfaces that would normally be protected within the interior of the protein. This leads to abnormal protein-protein interactions and formation of intracellular inclusions [188]. The formation of intracellular inclusions may result from the build-up of high concentrations of proteins in the cell, and/or aberrant protein folding.
Many neurodegenerative diseases are characterized by the presence of aggregated protein deposits, such as the extracellular amyloid plaques and intracellular neurofibrillary tangles found in AD, Lewy bodies found in most forms of PD nuclear inclusions found in HD. Protein aggregates often contain high levels of ubiquitin, as well as of misfolded proteins. [154, 155]. The two most striking pathological features of AD are aggregated extracellular deposits of β-amyloid plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau proteins. Both of these are caused by the misfolding and conversion of highly soluble proteins into insoluble, filamentous polymers. Mutations in the amyloid precursor protein in certain forms of familial AD lead to its aberrant cleavage by the 7-secretase complex, resulting in the production of p-amyloid42 that is prone to aggregation and leads to formation of extracellular amyloid plaques. p-amyloid42 has been shown to induce apoptosis in neurons in culture, and to cause oxidative stress. In PD α-synuclein is recognized as a major component of Lewy bodies [189]. It has a tendency to form protein aggregates that are increased in mutant forms of the protein [190, 191].
It is likely that the accumulation of protein aggregates is linked in some way to neurodegeneration. However, a direct link between aggregate formation and cell death has not been shown (Fig.3). Protein aggregates may disrupt normal functioning of neurons, and this stress may lead to the initiation of cell death. For example, protein aggregation can cause UPS impairment [145]. It has also been suggested that the accumulation of mutant protein into cellular aggregates may be neuroprotective, probably depending on the nature and the function of the protein in question. In this scenario intracellular protein deposits would become deleterious only after reaching a certain level within the cells.
Towards pro-survival therapies for neurodegenerative diseases
There is a large body of evidence linking impairment of protein handling with neuronal loss in neurodegenerative diseases. Model systems clearly demonstrate that neuronal cell death can be induced acutely by excessive ER stress, inhibition of the UPS, inhibition of autophagy or excessive autophagy. The protracted nature of many neurodegenerative diseases indicates that neurons can protect themselves from a certain amount of damage before there is severe disruption to function and viability. This suggests that the life or death of an individual neuron is dependent upon the overall burden of accumulated misfolded or aggregated proteins, balanced against the overall capacity of the cell to deal with this effectively and safely.
Therapeutic approaches aimed at modulating the activity of protein degradation systems, the UPS and autophagy, are therefore of particular relevance to neurodegenerative disease. However, since the UPS has roles in synaptic activity and neuronal plasticity, pro-teasome-modulating drugs could have deleterious effects in the nervous system [150, 192]. Macroautophagy-activating drugs such as rapamycin have been shown to increase clearance of aggregate-prone polyglutamine mutant proteins and mutant α-synuclein [193, 194]. In fact, in a transgenic HD mouse model, autophagy-enhancing drugs decrease aggregate formation and improved HD-associated behavioural tasks [193]. Thus, this class of drugs may prove particularly useful in promoting degradation of protein aggregates, and thus allowing improved neuronal function.
Neurons themselves mount protective responses to various stresses. Increases in chaperone proteins such as heat shock proteins (Hsps) are observed in the presence of misfolded proteins to help with refolding. In several neurodegenerative diseases there is evidence for increased expression of Hsps [195]. For example, focal cerebral ischaemia increases the levels of several classes of Hsps and their corresponding mRNAs [196]. Notably, the inducibility of Hsps decreases with aging, which may contribute to the inability of aged neurons to fully protect themselves from stresses such as protein misfolding, aggregation and oxidative stress [197]. Hsps promote survival via several different mechanisms [198]. They aid in the refolding and/or degradation of misfolded proteins [199]. Hsp27 and Hsp70 can regulate apoptosis through their ability to inhibit release of pro-apoptotic factors from the mitochondria and to interact with key components of the apop-tosome [200–204]. The small Hsps, Hsp27 and αβ-crystallin can also modulate intracellular redox potential and help maintain cytoskeletal integrity [198]. Hsps can promote survival in models of various neurodegenerative diseases [195]. Hsps reduce neuronal cell death in models of PD. Heat shock (which leads to elevated expression of inducible Hsp27 and Hsp70) is protective in in vitro models of PD [8, 90] while Hsp27 itself is protective against 6-hydroxydopamine-induced apoptosis in vitro[8]. Hsp27 but not Hsp70 is protective against α-synuclein-induced cell death in neuronal cells [205]. Viral vector-delivered Hsp70 is protective against MPTP toxicity in an in vivo model of PD [206]. Hsp27 also protects against polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin [207], while Hsp70 can alter the endosome-lysosomal localization of huntingtin [208]. Hsp70 overexpression is neuroprotective in a model of cerebral ischaemia [209].
Currently, available therapies for neurodegenerative diseases treat only the symptoms, but not the cause of the disease. As we understand more clearly the cause of these diseases, and improve diagnostic ability at pre-symptomatic and early stages, we may be able to develop therapies that slow or halt the underlying neurodegeneration. From a therapeutic point of view, the use of apoptosis inhibitors such as caspase inhibitors has failed to prevent neurodegeneration. Likely, the damage is too far advanced for the inhibition of these late-stage participants to be effective and the cell will find another way to die, e.g. by necrosis. For example, recent clinical failures with apoptosis inhibitors in PD underscore the need for a shift in thinking with regard to drug discovery for neurodegenerative diseases [210]. Targeting the causes of cell death is the next major challenge in generating effective therapies, and in this regard, strategies incorporating chaperones and/or protein degradation-enhancing drugs may prove a worthwhile endeavour.
Acknowledgments
I would like to thank Afshin Samali for helpful advice. Space limitations have precluded the citation of many primary publications in favour of recent reviews. My research is supported by National University of Ireland, Galway Millenium Fund and the Irish Research Council for Science and Engineering Technology.
References
- 1.Bredesen DE, Rao RV, Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- 3.Stroh C, Schulze-Osthoff K. Death by a thousand cuts: an ever increasing list of caspase substrates. Cell Death Differ. 1998;5:997–1000. doi: 10.1038/sj.cdd.4400451. [DOI] [PubMed] [Google Scholar]
- 4.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-depend-ent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 5.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates pro-caspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 6.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of cas-pase-3. Cell. 1997;90:405–13. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]
- 7.Gorman AM, Bonfoco E, Zhivotovsky B, Orrenius S, Ceccatelli S. Cytochrome c release and caspase-3 activation during colchicine-induced apoptosis of cerebellar granule cells. Eur J Neurosci. 1999;11:1067–72. doi: 10.1046/j.1460-9568.1999.00512.x. [DOI] [PubMed] [Google Scholar]
- 8.Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxy-dopamine-induced cytochrome c release and apoptosis in PC12 cells. Blochem Biophys Res Commun. 2005;327:801–10. doi: 10.1016/j.bbrc.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 9.Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in exci-totoxic neuron death: cytochrome c release and a secondary increase in super-oxide production. J Neurosci. 2000;20:5715–23. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ekert PG, Vaux DL. The mitochondrial death squad: hardened killers or innocent bystanders? Curr Opin Cell Biol. 2005;17:626–30. doi: 10.1016/j.ceb.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 11.Dare E, Gorman AM, Ahlbom E, Gotz M, Momoi T, Ceccatelli S. Apoptotic morphology does not always require caspase activity in rat cerebellar granule neurons. NeurotoxRes. 2001;3:501–14. doi: 10.1007/BF03033206. [DOI] [PubMed] [Google Scholar]
- 12.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–6. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 13.Schulze-Osthoff K, Ferrari D, Los M, Wesselborg S, Peter ME. Apoptosis signalling by death receptors. EurJBiochem. 1998;254:439–59. doi: 10.1046/j.1432-1327.1998.2540439.x. [DOI] [PubMed] [Google Scholar]
- 14.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–25. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galonek HL, Hardwick JM. Upgrading the BCL-2 Network. Nat Cell Biol. 2006;8:1317–9. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 16.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 17.Listen P, Young SS, Mackenzie AE, Korneluk RG. Life and death decisions: the role of the lAPs in modulating programmed cell death. Apoptosis. 1997;2:423–41. doi: 10.1023/a:1026465926478. [DOI] [PubMed] [Google Scholar]
- 18.Nicholls DG, Budd SL. Neuronal excito-toxicity: the role of mitochondria. BioFactors. 1998;8:287–99. doi: 10.1002/biof.5520080317. [DOI] [PubMed] [Google Scholar]
- 19.Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–73. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- 20.Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–52. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- 21.Hornung JP, Koppel H, Clarke PG. Endocytosis and autophagy in dying neurons: an ultrastructural study in chick embryos. J Comp Neurol. 1989;283:425–37. doi: 10.1002/cne.902830310. [DOI] [PubMed] [Google Scholar]
- 22.Clarke PG. Developmental cell death: morphological diversity and multiple mechanisms. AnatEmbryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 23.Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–2. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 24.Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell. 2007;131:1137–48. doi: 10.1016/j.cell.2007.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Portera-Cailliau C, Price DL, Martin LJ. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evi-dence for an apoptosis-necrosis continuum. J Comp Neurol. 1997;378:88–104. [PubMed] [Google Scholar]
- 26.Portera-Cailliau C, Price DL, Martin LJ. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol. 1997;378:70–87. [PubMed] [Google Scholar]
- 27.Cheung NS, Pascoe CJ, Giardina SF, John CA, Beart PM. Micromolar L-gluta-mate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology. 1998;37:1419–29. doi: 10.1016/s0028-3908(98)00123-3. [DOI] [PubMed] [Google Scholar]
- 28.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Curr Mol Med. 2004;4:149–77. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 29.Nicholls DG, Budd SL, Castilho RF, Ward MW. Glutamate excitotoxicity and neuronal energy metabolism. Ann N Y Acad Sci. 1999;893:1–12. doi: 10.1111/j.1749-6632.1999.tb07813.x. [DOI] [PubMed] [Google Scholar]
- 30.Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6. doi: 10.1084/jem.185.8.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B. Mitochondrial dysfunction in the patho-genesis of necrotic and apoptotic cell death. J Bioenerget Biomembr. 1999;31:305–19. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]
- 32.Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Yoshimori T, Pierron G, Codogno P, Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–40. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6:1221–8. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 34.Ullman E, Fan Y, Stawowczyk M, Chen HM, Yue Z, Zong WX. Autophagy promotes necrosis in apoptosis-deficient cells in response to ER stress. Cell Death Differ. 2008;15:422–5. doi: 10.1038/sj.cdd.4402234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapop-totic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–39. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 36.Akar U, Chaves-Reyes A, Barria M, Tari A, Sanguino A, Kondo Y, Kondo S, Arun B, Lopez-Berestein G, Ozpolat B. Silencing of Bcl-2 expression by small interfering RNA induces autophagic cell death in MCF-7 breast cancer cells. Autophagy. 2008;4:669–79. doi: 10.4161/auto.6083. [DOI] [PubMed] [Google Scholar]
- 37.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gelinas C, Fan Y, Nelson DA, Jin S, White E. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2001;2:330–5. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol. 1998;72:8586–96. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erlich S, Mizrachy L, Segev O, Lindenboim L, Zmira O, Adi-Harel S, Hirsch JA, Stein R, Pinkas-Kramarski R. Differential interactions between Beclin 1 and Bcl-2 family members. Autophagy. 2007;3:561–8. doi: 10.4161/auto.4713. [DOI] [PubMed] [Google Scholar]
- 41.Maiuri MC, Criollo A, Tasdemir E, Vicencio JM, Tajeddine N, Hickman JA, Geneste O, Kroemer G. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-X(L) Autophagy. 2007;3:374–6. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 42.Maiuri MC, Le Toumelin G, Criollo A, Rain JC, Gautier F, Juin P, Tasdemir E, Pierron G, Troulinaki K, Tavernarakis N, Hickman JA, Geneste O, Kroemer G. Functional and physical interaction between Bcl-X(L) and a BH3-like domain in Beclin-1. EMBO J. 2007;26:2527–39. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-Beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chein. 2007;282:13123–32. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 44.Ohsumi Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol. 2001;2:211–6. doi: 10.1038/35056522. [DOI] [PubMed] [Google Scholar]
- 45.Pyo JO, Jang MH, Kwon YK, Lee HJ, Jun Jl, Woo HN, Cho DH, Choi B, Lee H, Kim JH, Mizushima N, Oshumi Y, Jung YK. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280:20722–9. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 46.Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8:1124–32. doi: 10.1038/ncb1482. [DOI] [PubMed] [Google Scholar]
- 47.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 48.Lankiewicz S, Marc Luetjens C, True Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a cas-pase-independent cell death. J Biol Chem. 2000;275:17064–71. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- 49.Wang L, Yu C, Lu Y, He P, Guo J, Zhang C, Song Q, Ma D, Shi T, Chen Y. TMEM166, a novel transmembrane protein, regulates cell autophagy and apoptosis. Apoptosis. 2007;12:1489–502. doi: 10.1007/s10495-007-0073-9. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez-Polo RA, Boya P, Pauleau AL, Jalil A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J Cell Sci. 2005;118:3091–102. doi: 10.1242/jcs.02447. [DOI] [PubMed] [Google Scholar]
- 51.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–8. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 52.Samali A, Zhivotovsky B, Jones D, Nagata S, Orrenius S. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6:495–6. doi: 10.1038/sj.cdd.4400520. [DOI] [PubMed] [Google Scholar]
- 53.Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, Bamber BA, Bassham DC, Bergamini E, Bi X, Biard-Piechaczyk M, Blum JS, Bredesen DE, Brodsky JL, Brumell JH, Brunk UT, Bursch W, Camougrand N, Cebollero E, Cecconi F, Chen Y, Chin LS, Choi A, Chu CT, Chung J, Clarke PG, Clark RS, Clarke SG, Clave C, Cleveland JL, Codogno P, Colombo MI, Coto-Montes A, Cregg JM, Cuervo AM, Debnath J, Demarchi F, Dennis PB, Dennis PA, Deretic V, Devenish RJ, Di Sano F, Dice JF, Difiglia M, Dinesh-Kumar S, Distelhorst CW, Djavaheri-Mergny M, Dorsey FC, Droge W, Dron M, Dunn WA, Jr, Duszenko M, Eissa NT, Elazar Z, Esclatine A, Eskelinen EL, Fesus L, Finley KD, Fuentes JM, Fueyo J, Fujisaki K, Galliot B, Gao FB, Gewirtz DA, Gibson SB, Gohla A, Goldberg AL, Gonzalez R, Gonzalez-Estevez C, Gorski S, Gottlieb RA, Haussinger D, He YW, Heidenreich K, Hill JA, Hoyer-Hansen M, Hu X, Huang WP, Iwasaki A, Jaattela M, Jackson WT, Jiang X, Jin S, Johansen T, Jung JU, Kadowaki M, Kang C, Kelekar A, Kessel DH, Kiel JA, Kim HP, Kimchi A, Kinsella TJ, Kiselyov K, Kitamoto K, Knecht E, Komatsu M, Kominami E, Kondo S, Kovacs AL, Kroemer G, Kuan CY, Kumar R, Kundu M, Landry J, Laporte M, Le W, Lei HY, Lenardo MJ, Levine B, Lieberman A, Lim KL, Lin FC, Liou W, Liu LF, Lopez-Berestein G, Lopez-Otin C, Lu B, Macleod KF, Malorni W, Martinet W, Matsuoka K, Mautner J, Meijer AJ, Melendez A, Michels P, Miotto G, Mistiaen WP, Mizushima N, Mograbi B, Monastyrska I, Moore MN, Moreira PI, Moriyasu Y, Motyl T, Munz C, Murphy LO, Naqvi NI, Neufeld TP, Nishino I, Nixon RA, Noda T, Nurnberg B, Ogawa M, Oleinick NL, Olsen LJ, Ozpolat B, Paglin S, Palmer GE, Papassideri I, Parkes M, Perlmutter DH, Perry G, Piacentini M, Pinkas-Kramarski R, Prescott M, Proikas-Cezanne T, Raben N, Rami A, Reggiori F, Rohrer B, Rubinsztein DC, Ryan KM, Sadoshima J, Sakagami H, Sakai Y, Sandri M, Sasakawa C, Sass M, Schneider C, Seglen PO, Seleverstov O, Settleman J, Shacka JJ, Shapiro IM, Sibirny A, Silva-Zacarin EC, Simon HU, Simone C, Simonsen A, Smith MA, Spanel-Borowski K, Srinivas V, Steeves M, Stenmark H, Stromhaug PE, Subauste CS, Sugimoto S, Sulzer D, Suzuki T, Swanson MS, Tabas I, Takeshita F, Talbot NJ, Talloczy Z, Tanaka K, Tanaka K, Tanida I, Taylor GS, Taylor JP, Terman A, Tettamanti G, Thompson CB, Thumm M, Tolkovsky AM, Tooze SA, Truant R, Tumanovska LV, Uchiyama Y, Ueno T, Uzcategui NL, Van Der Klei I, Vaquero EC, Vellai T, Vogel MW, Wang HG, Webster P, Wiley JW, Xi Z, Xiao G, Yahalom J, Yang JM, Yap G, Yin XM, Yoshimori T, Yu L, Yue Z, Yuzaki M, Zabirnyk O, Zheng X, Zhu X, Deter RL. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–75. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.West MJ, Coleman PD, Flood DG, Troncoso JC. Differences in the pattern of hippocampal neuronal loss in normal ageing and Alzheimer's disease. Lancet. 1994;344:769–72. doi: 10.1016/s0140-6736(94)92338-8. [DOI] [PubMed] [Google Scholar]
- 55.Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- 56.Gomez-lsla T, Price JL, McKeel DW, Jr, Morris JC, Growdon JH, Hyman BT. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci. 1996;16:4491–500. doi: 10.1523/JNEUROSCI.16-14-04491.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Selkoe DJ. Alzheimer's disease: genes: proteins, and therapy. Physiol Rev. 2001;81:741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 58.Blennow K, de Leon MJ, Zetterberg H. Alzheimer's disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 59.Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ. Aggregation of secreted amyloid beta-protein into sodium dodecyl sul-fate-stable oligomers in cell culture. J Biol Chein. 1995;270:9564–70. doi: 10.1074/jbc.270.16.9564. [DOI] [PubMed] [Google Scholar]
- 60.Colurso GJ, Nilson JE, Vervoort LG. Quantitative assessment of DNA fragmentation and beta-amyloid deposition in insular cortex and midfrontal gyrus from patients with Alzheimer's disease. Life Sci. 2003;73:1795–803. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- 61.Stadelmann C, Deckwerth TL, Srinivasan A, Bancher C, Bruck W, Jellinger K, Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. AmJPathol. 1999;155:1459–66. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rohn TT, Head E, Nesse WH, Cotman CW, Cribbs DH. Activation of caspase-8 in the Alzheimer's disease brain. Neurobiol D/s. 2001;8:1006–16. doi: 10.1006/nbdi.2001.0449. [DOI] [PubMed] [Google Scholar]
- 63.Rohn TT, Rissman RA, Davis MC, Kim YE, Cotman CW, Head E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol Dis. 2002;11:341–54. doi: 10.1006/nbdi.2002.0549. [DOI] [PubMed] [Google Scholar]
- 64.Woodhouse A, Dickson TC, West AK, McLean CA, Vickers JC. No difference in expression of apoptosis-related proteins and apoptotic morphology in control pathologically aged and Alzheimer's disease cases. Neurobiol Dis. 2006;22:323–33. doi: 10.1016/j.nbd.2005.11.010. [DOI] [PubMed] [Google Scholar]
- 65.Li YP, Bushnell AF, Lee CM, Perlmutter LS, Wong SK. Beta-amyloid induces apop-tosis in human-derived neurotypic SH-SY5Y cells. Brain Res. 1996;738:196–204. doi: 10.1016/s0006-8993(96)00733-0. [DOI] [PubMed] [Google Scholar]
- 66.Forloni G, Bugiani O, Tagliavini F, Salmona M. Apoptosis-mediated neuro-toxicity induced by beta-amyloid and PrP fragments. Molecular and chemical neuropathology. 1996;28:163–71. doi: 10.1007/BF02815218. [DOI] [PubMed] [Google Scholar]
- 67.Guo Q, Sopher BL, Furukawa K, Pham DG, Robinson N, Martin GM, Mattson MP. Alzheimer's presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–22. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, Mattson MP. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in prese-nilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6. doi: 10.1038/4789. [DOI] [PubMed] [Google Scholar]
- 69.Olney JW, Wozniak DF, Farber NB. Glumate receptor dysfunction and Alzheimer's disease. Restorat Neurol Neurosci. 1998;13:75–83. [PubMed] [Google Scholar]
- 70.Miguel-Hidalgo JJ, Alvarez XA, Cacabelos R, Quack G. Neuroprotection by memantine against neurodegeneration induced by beta-amyloid(1–40) Brain Res. 2002;958:210–21. doi: 10.1016/s0006-8993(02)03731-9. [DOI] [PubMed] [Google Scholar]
- 71.Ikonomovic MD, Mizukami K, Warde D, Sheffield R, Hamilton R, Wenthold RJ, Armstrong DM. Distribution of glutamate receptor subunit NMDAR1 in the hippocampus of normal elderly and patients with Alzheimer's disease. Exp Neurol. 1999;160:194–204. doi: 10.1006/exnr.1999.7196. [DOI] [PubMed] [Google Scholar]
- 72.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Naslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy-a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mochizuki H. Gene therapy for Parkinson's disease. Expert Rev Neurotherap. 2007;7:957–60. doi: 10.1586/14737175.7.8.957. [DOI] [PubMed] [Google Scholar]
- 74.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nature. 2006;7:306–18. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]
- 75.Dodson MW, Guo M. Pinkl, Parkin, DJ-1 and mitochondrial dysfunction in Parkinson's disease. Curr Opin Neurobiol. 2007;17:331–7. doi: 10.1016/j.conb.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 76.Gandhi S, Wood NW. Molecular pathogen-esis of Parkinson's disease. Hum Mol Genet. 2005;14:2749–55. doi: 10.1093/hmg/ddi308. [DOI] [PubMed] [Google Scholar]
- 77.Przedborski S. Pathogenesis of nigral cell death in Parkinson's disease. Parkinsonism Relat Disord. 2005;11(Suppl 1):S3–7. doi: 10.1016/j.parkreldis.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 78.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–50. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 79.Tompkins MM, Basgall EJ, Zamrini E, Hill WD. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigral neurons. Am J Pathol. 1997;150:119–31. [PMC free article] [PubMed] [Google Scholar]
- 80.Tatton NA, Maclean-Fraser A, Tatton WG, Perl DP, Olanow CW. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson's disease. Ann Neurol. 1998;44:S142–8. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 81.Tatton NA. Increased caspase 3 and Bax immunoreactivity accompany nuclear GAPDH translocation and neuronal apoptosis in Parkinson's disease. Exp Neurol. 2000;166:29–43. doi: 10.1006/exnr.2000.7489. [DOI] [PubMed] [Google Scholar]
- 82.Lev N, Melamed E, Often D. Apoptosis and Parkinson's disease. Prog Neuro-psychopharmacol Biol Psychiatry. 2003;27:245–50. doi: 10.1016/S0278-5846(03)00019-8. [DOI] [PubMed] [Google Scholar]
- 83.Kosel S, Egensperger R, von Eitzen U, Mehraein P, Graeber MB. On the question of apoptosis in the Parkinsonian substantia nigra. Ada Neuropathologica. 1997;93:105–8. doi: 10.1007/s004010050590. [DOI] [PubMed] [Google Scholar]
- 84.Wullner U, Kornhuber J, Weller M, Schulz JB, Loschmann PA, Riederer P, Klockgether T. Cell death and apoptosis regulating proteins in Parkinson's disease-a cautionary note. Ada Neuropathologica. 1999;97:408–12. doi: 10.1007/s004010051005. [DOI] [PubMed] [Google Scholar]
- 85.Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cere-brospinal fluid from Parkinsonian patients. Neurosci Lett. 1994;165:208–10. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 86.Mogi M, Harada M, Kondo T, Mizuno Y, Narabayashi H, Riederer P, Nagatsu T. The soluble form of Fas molecule is elevated in Parkinsonian brain tissues. Neurosci Lett. 1996;220:195–8. doi: 10.1016/s0304-3940(96)13257-2. [DOI] [PubMed] [Google Scholar]
- 87.Hartmann A, Mouatt-Prigent A, Faucheux BA, Agid Y, Hirsch EC. FADD: a link between TNF family receptors and cas-pases in Parkinson's disease. Neurology. 2002;58:308–10. doi: 10.1212/wnl.58.2.308. [DOI] [PubMed] [Google Scholar]
- 88.Hartmann A, Troadec JD, Hunot S, Kikly K, Faucheux BA, Mouatt-Prigent A, Ruberg M, Agid Y, Hirsch EC. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson's disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–55. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Viswanath V, Wu Y, Boonplueang R, Chen S, Stevenson FF, Yantiri F, Yang L, Beal MF, Andersen JK. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson's disease. J Neurosci. 2001;21:9519–28. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Quigney DJ, Gorman AM, Samali A. Meat shock protects PC12 cells against MPP+ toxicity. Brain Res. 2003;993:133–9. doi: 10.1016/j.brainres.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 91.Mochizuki H, Nakamura N, Nishi K, Mizuno Y. Apoptosis is induced by 1-methyl-4-phenylpyridinium ion (MPP+) in ventral mesencephalic-striatal co-culture in rat. Neurosci Lett. 1994;170:191–4. doi: 10.1016/0304-3940(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 92.Ochu EE, Rothwell NJ, Waters CM. Caspases mediate 6-hydroxydopamine-induced apoptosis but not necrosis in PC12 cells. JNeurochem. 1998;70:2637–40. doi: 10.1046/j.1471-4159.1998.70062637.x. [DOI] [PubMed] [Google Scholar]
- 93.Ahmadi FA, Linseman DA, Grammatopoulos TN, Jones SM, Bouchard RJ, Freed CR, Heidenreich KA, Zawada WM. The pesticide rotenone induces caspase-3-mediated apoptosis in ventral mesencephalic dopaminergic neurons. JNeurochem. 2003;87:914–21. doi: 10.1046/j.1471-4159.2003.02068.x. [DOI] [PubMed] [Google Scholar]
- 94.Tanaka Y, Engelender S, Igarashi S, Rao RK, Wanner T, Tanzi RE, Sawa A, V LD, Dawson TM, Ross CA. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet. 2001;10:919–26. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 95.Kirik D, Rosenblad C, Burger C, Lundberg C, Johansen TE, Muzyczka N, Mandel RJ, Bjorklund A. Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. JNeurosci. 2002;22:2780–91. doi: 10.1523/JNEUROSCI.22-07-02780.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mochizuki H, Yamada M, Mizuno Y. Alpha-synuclein overexpression model. J Neural Transm Suppl. 2006:281–4. [PubMed] [Google Scholar]
- 97.Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxic-ity in Parkinson's disease: a target for neuro-protection. Ann Neurol. 1998;44:S175–88. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- 98.Beal MF. Excitotoxicity and nitric oxide in Parkinson's disease pathogenesis. Ann Neurol. 1998;44:S110–4. doi: 10.1002/ana.410440716. [DOI] [PubMed] [Google Scholar]
- 99.Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol. 1997;12:25–31. [PubMed] [Google Scholar]
- 100.Forno LS, Norville RL. Ultrastructure of Lewy bodies in the stellate ganglion. Ada Neuropathologica. 1976;34:183–97. doi: 10.1007/BF00688674. [DOI] [PubMed] [Google Scholar]
- 101.Sulzer D, Bogulavsky J, Larsen KE, Behr G, Karatekin E, Kleinman MH, Turro N, Krantz D, Edwards RH, Greene LA, Zecca L. Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci USA. 2000;97:11869–74. doi: 10.1073/pnas.97.22.11869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma SH-SY5Y cells. J Neurosci Res. 2003;73:341–50. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 103.Zhu JH, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am JPathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 105.Graveland GA, Williams RS, DiFiglia M. Evidence for degenerative and regenerative changes in neostriatal spiny neurons in Huntington's disease. Science. 1985;227:770–3. doi: 10.1126/science.3155875. [DOI] [PubMed] [Google Scholar]
- 106.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neu-rites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 107.Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–18. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic tox-icity. Nat Genet. 2000;25:385–9. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 109.Solans A, Zambrano A, Rodriguez M, Barrientos A. Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III. Hum Mol Genet. 2006;15:3063–81. doi: 10.1093/hmg/ddl248. [DOI] [PubMed] [Google Scholar]
- 110.Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P, ten Donkelaar HJ, Kremer B. Expression pattern of apoptosis-related markers in Huntington's disease. Ada Neuropathologica. 2005;109:321–8. doi: 10.1007/s00401-004-0957-5. [DOI] [PubMed] [Google Scholar]
- 111.Kiechle T, Dedeoglu A, Kubilus J, Kowall NW, Beal MF, Friedlander RM, Hersch SM, Ferrante RJ. Cytochrome C and cas-pase-9 expression in Huntington's disease. Neuromolecular Med. 2002;1:183–95. doi: 10.1385/NMM:1:3:183. [DOI] [PubMed] [Google Scholar]
- 112.Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J Neurosci. 2002;22:7862–72. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. JBiolChem. 2000;275:19831–8. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 114.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polygluta-mine tract. J Biol Chem. 1998;273:9158–67. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 115.McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–9. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- 116.Guidetti P, Luthi-Carter RE, Augood SJ, Schwarcz R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington's disease. Neurobiol Dis. 2004;17:455–61. doi: 10.1016/j.nbd.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 117.Estrada Sanchez AM, Mejia-Toiber J, Massieu L. Excitotoxic neuronal death and the pathogenesis of Huntington's disease. Arch Med Res. 2008;39:265–76. doi: 10.1016/j.arcmed.2007.11.011. [DOI] [PubMed] [Google Scholar]
- 118.Zeron MM, Fernandes HB, Krebs C, Shehadeh J, Wellington CL, Leavitt BR, Baimbridge KG, Hayden MR, Raymond LA. Potentiation of NMDA receptor-mediated excitotoxicity linked with intrinsic apoptotic pathway in YAC transgenic mouse model of Huntington's disease. Mol Cell Neurosci. 2004;25:469–79. doi: 10.1016/j.mcn.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 119.Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA. 1999;96:8727–32. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morton AJ, Leavens W. Mice transgenic for the human Huntington's disease mutation have reduced sensitivity to kainic acid toxicity. Brain Res Bull. 2000;52:51–9. doi: 10.1016/s0361-9230(00)00238-0. [DOI] [PubMed] [Google Scholar]
- 121.Kegel KB, Kim M, Sapp E, Mclntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endoso-mal-lysosomal activity, endosome tabulation, and autophagy. J Neurosci. 2000;20:7268–78. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zuchner T, Brundin P. Mutant huntingtin can paradoxically protect neurons from death. Cell Death Differ. 2008;15:435–42. doi: 10.1038/sj.cdd.4402261. [DOI] [PubMed] [Google Scholar]
- 123.Nitatori T, Sato N, Waguri S, Karasawa Y, Araki H, Shibanai K, Kominami E, Uchiyama Y. Delayed neuronal death in the CA1 pyramidal cell layer of the gerbil hippocampus following transient ischaemia is apoptosis. J Neurosci. 1995;15:1001–11. doi: 10.1523/JNEUROSCI.15-02-01001.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischaemia: a role for apoptosis. J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Sauer D, Allegrini PR, Cosenti A, Pataki A, Amacker H, Fagg GE. Characterization of the cerebroprotective efficacy of the competitive NMDA receptor antagonist CGP40116 in a rat model of focal cerebral ischaemia: an In vivo magnetic resonance imaging study. J Cereb Blood Flow Metab. 1993;13:595–602. doi: 10.1038/jcbfm.1993.77. [DOI] [PubMed] [Google Scholar]
- 126.Rami A. Upregulation of Beclin 1 in the ischemic penumbra. Autophagy. 2008;4:227–9. doi: 10.4161/auto.5339. [DOI] [PubMed] [Google Scholar]
- 127.Rami A, Langhagen A, Steiger S. Focal cerebral ischaemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132–41. doi: 10.1016/j.nbd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 128.Uchiyama Y, Koike M, Shibata M. Autophagic neuron death in neonatal brain ischaemia/hypoxia. Autophagy. 2008;4:404–8. doi: 10.4161/auto.5598. [DOI] [PubMed] [Google Scholar]
- 129.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic retic-ulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Bio! 2004;14:20–8. doi: 10.1016/j.tcb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 131.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 132.Saleh M, Mathison JC, Wolinski MK, Bensinger SJ, Fitzgerald P, Droin N, Ulevitch RJ, Green DR, Nicholson DW. Enhanced bacterial clearance and sepsis resistance in caspase-12-deficient mice. Nature. 2006;440:1064–8. doi: 10.1038/nature04656. [DOI] [PubMed] [Google Scholar]
- 133.Szegezdi E, Herbert KR, Kavanagh ET, Samali A, Gorman AM. Nerve growth factor blocks thapsigargin-induced apoptosis at the level of the mitochondrion via regulation of Bim. J Cell Mol Med. 2008:2482–96. doi: 10.1111/j.1582-4934.2008.00268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Smith MI, Deshmukh M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007;14:1011–9. doi: 10.1038/sj.cdd.4402089. [DOI] [PubMed] [Google Scholar]
- 135.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 136.Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signalling affects cell fate during the unfolded protein response. Science. 2007;318:944–9. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer's disease. Ada Neuropathologica. 2005;110:165–72. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 139.Mattson MP, Chan SL, Camandola S. Presenilin mutations and calcium signalling defects in the nervous and immune systems. Bioessays. 2001;23:733–44. doi: 10.1002/bies.1103. [DOI] [PubMed] [Google Scholar]
- 140.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–11. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 141.Holtz WA, O'Malley KL. Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem. 2003;278:19367–77. doi: 10.1074/jbc.M211821200. [DOI] [PubMed] [Google Scholar]
- 142.Ryu EJ, Harding HP, Angelastro JM, Vitolo OV, Ron D, Greene LA. Endoplasmic reticulum stress and the unfolded protein response in cellular models of Parkinson's disease. J Neurosci. 2002;22:10690–8. doi: 10.1523/JNEUROSCI.22-24-10690.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Suva RM, Ries V, Oo TF, Yarygina O, Jackson-Lewis V, Ryu EJ, Lu PD, Marciniak SJ, Ron D, Przedborski S, Kholodilov N, Greene LA, Burke RE. CH0P/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neuro-toxin model of Parkinsonism. J Neurochem. 2005;95:974–86. doi: 10.1111/j.1471-4159.2005.03428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–55. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science. 2001;292:1552–5. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- 146.Reijonen S, Putkonen N, Norremolle A, Lindholm D, Korhonen L. Inhibition of endoplasmic reticulum stress counteracts neuronal cell death and protein aggregation caused by N-terminal mutant hunt-ingtin proteins. Exp Cell Res. 2008;314:950–60. doi: 10.1016/j.yexcr.2007.12.025. [DOI] [PubMed] [Google Scholar]
- 147.Paschen W, Aufenberg C, Hotop S, Mengesdorf T. Transient cerebral ischaemia activates processing of xbpl messenger RNA indicative of endoplasmic reticulum stress. J Cereb Blood Flow Metab. 2003;23:449–61. doi: 10.1097/01.WCB.0000054216.21675.AC. [DOI] [PubMed] [Google Scholar]
- 148.Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischaemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–15. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- 149.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40:427–46. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 150.Hegde AN. Ubiquitin-proteasome-medi-ated local protein degradation and synaptic plasticity. Prog Neurobiol. 2004;73:311–57. doi: 10.1016/j.pneurobio.2004.05.005. [DOI] [PubMed] [Google Scholar]
- 151.Upadhya SC, Hegde AN. Ubiquitin-proteasome pathway components as therapeutic targets for CNS maladies. Curr Pharma Des. 2005;11:3807–28. doi: 10.2174/138161205774580651. [DOI] [PubMed] [Google Scholar]
- 152.Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP. HDAC6 rescues neurode-generation and provides an essential link between autophagy and the UPS. Nature. 2007;447:859–63. doi: 10.1038/nature05853. [DOI] [PubMed] [Google Scholar]
- 153.Chen Q, Ding Q, Thorpe J, Dohmen RJ, Keller JN. RNA interference toward UMP1 induces proteasome inhibition in Saccharomyces cerevisiae: evidence for protein oxidation and autophagic cell death. Free Radic Biol Med. 2005;38:226–34. doi: 10.1016/j.freeradbiomed.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 154.Lennox G, Lowe J, Morrell K, Landon M, Mayer RJ. Ubiquitin is a component of neurofibrillary tangles in a variety of neurodegenerative diseases. Neurosci Lett. 1988;94:211–7. doi: 10.1016/0304-3940(88)90297-2. [DOI] [PubMed] [Google Scholar]
- 155.Lowe J, Blanchard A, Morrell K, Lennox G, Reynolds L, Billett M, Landon M, Mayer RJ. Ubiquitin is a common factor in intermediate filament inclusion bodies of diverse type in man, including those of Parkinson's disease, Pick's disease, and Alzheimer's disease, as well as Rosenthal fibres in cerebellar astrocytomas, cyto-plasmic bodies in muscle, and mallory bodies in alcoholic liver disease. J Pathol. 1988;155:9–15. doi: 10.1002/path.1711550105. [DOI] [PubMed] [Google Scholar]
- 156.Keller JN, Hannl KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000;75:436–9. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- 157.van Leeuwen FW, de Kleijn DP, Van Den Hurk HH, Neubauer A, Sonnemans MA, Sluijs JA, Koycu S, Ramdjielal RD, Salehi A, Martens GJ, Grosveld FG, Peter J, Burbach H, Hoi EM. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer's and Down patients. Science. 1998;279:242–7. doi: 10.1126/science.279.5348.242. [DOI] [PubMed] [Google Scholar]
- 158.Lam YA, Plckart CM, Alban A, Landon M, Jamleson C, Ramage R, Mayer RJ, Layfleld R. Inhibition of the ubiquitin-pro-teasome system in Alzheimer's disease. Proc Natl Acad Sci USA. 2000;97:9902–6. doi: 10.1073/pnas.170173897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Imai Y, Takahashl R. How do Parkin mutations result in neurodegeneration. Curr Opin Neurobiol. 2004;14:384–9. doi: 10.1016/j.conb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 160.Shlmura H, Hattorl N, Kubo S, Mlzuno Y, Asakawa S, Mlnoshlma S, Shlmlzu N, Iwal K, Chlba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet. 2000;25:302–5. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 161.Kltada T, Asakawa S, Hattorl N, Matsumine H, Yamamura Y, Mlnoshlma S, Yokochl M, Mlzuno Y, Shlmlzu N. Mutations in the parkin gene cause auto-somal recessive juvenile parkinsonism. Nature. 1998;392:605–8. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 162.Ishlkawa A, Tsujl S. Clinical analysis of 17 patients in 12 Japanese families with autosomal-recessive type juvenile Parkinsonism. Neurology. 1996;47:160–6. doi: 10.1212/wnl.47.1.160. [DOI] [PubMed] [Google Scholar]
- 163.Imal Y, Soda M, Inoue H, Hattorl N, Mlzuno Y, Takahashl R. An unfolded putative trans-membrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell. 2001;105:891–902. doi: 10.1016/s0092-8674(01)00407-x. [DOI] [PubMed] [Google Scholar]
- 164.Imal Y, Soda M, Takahashl R. Parkin suppresses unfolded protein stress-induced cell death through its E3 ubiquitin-protein ligase activity. J Biol Chem. 2000;275:35661–4. doi: 10.1074/jbc.C000447200. [DOI] [PubMed] [Google Scholar]
- 165.Wilkinson KD, Lee KM, Deshpande S, Duerksen-Hughes P, Boss JM, Pohl J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl-terminal hydrolase. Science. 1989;246:670–3. doi: 10.1126/science.2530630. [DOI] [PubMed] [Google Scholar]
- 166.Liu Y, Fallen L, Lashuel HA, Liu Z, Lansbury PT., Jr The UCH-L1 gene encodes two opposing enzymatic activities that affect alpha-synuclein degradation and Parkinson's disease susceptibility. Cell. 2002;111:209–18. doi: 10.1016/s0092-8674(02)01012-7. [DOI] [PubMed] [Google Scholar]
- 167.Osaka H, Wang YL, Takada K, Taklzawa S, Setsule R, LI H, Sato Y, Nlshlkawa K, Sun YJ, Sakural M, Harada T, Hara Y, Klmura I, Chlba S, Namlkawa K, Klyama H, Noda M, Aokl S, Wada K. Ubiquitin carboxy-terminal hydrolase L1 binds to and stabilizes monoubiquitin in neuron. Hum Mole Genet. 2003;12:1945–58. doi: 10.1093/hmg/ddg211. [DOI] [PubMed] [Google Scholar]
- 168.Kahle PJ, Haass C. How does parkin lig-ate ubiquitin to Parkinson's disease. EMBORep. 2004;5:681–5. doi: 10.1038/sj.embor.7400188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bennett MC, Bishop JF, Leng Y, Chock PB, Chase TN, Mouradlan MM. Degradation of alpha-synuclein by proteasome. J Biol Chem. 1999;274:33855–8. doi: 10.1074/jbc.274.48.33855. [DOI] [PubMed] [Google Scholar]
- 170.Tofarls GK, Layfleld R, Spillantini MG. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001;509:22–6. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 171.McNaught KS, Olanow CW, Halllwell B, Isacson O, Jenner P. Failure of the ubiqui-tin-proteasome system in Parkinson's disease. Nat RevNeurosci. 2001;2:589–94. doi: 10.1038/35086067. [DOI] [PubMed] [Google Scholar]
- 172.McNaught KS, Perl DP, Brownell AL, Olanow CW. Systemic exposure to proteasome inhibitors causes a progressive model of Parkinson's disease. Ann Neuroi. 2004;56:149–62. doi: 10.1002/ana.20186. [DOI] [PubMed] [Google Scholar]
- 173.McNaught KS, Olanow CW. Proteasome inhibitor-induced model of Parkinson's disease. Ann Neuroi. 2006;60:243–7. doi: 10.1002/ana.20936. [DOI] [PubMed] [Google Scholar]
- 174.Kordower JH, Kanaan NM, Chu Y, Suresh Babu R, Stansell J, 3rd, Terpstra BT, Sortwell CE, Steece-Colller K, Collier TJ. Failure of proteasome inhibitor administration to provide a model of Parkinson's disease in rats and monkeys. Ann Neuroi. 2006;60:264–8. doi: 10.1002/ana.20935. [DOI] [PubMed] [Google Scholar]
- 175.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neuroi. 2007;6:352–61. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 176.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cuervo AM, Dice JF. Lysosomes, a meeting point of proteins, chaperones, and proteases. JMolMed. 1998;76:6–12. doi: 10.1007/s001090050185. [DOI] [PubMed] [Google Scholar]
- 178.Rubinsztein DC, DIFIglla M, Helntz N, Nixon RA, Qln ZH, Ravlkumar B, Stefanls L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy. 2005;1:11–22. doi: 10.4161/auto.1.1.1513. [DOI] [PubMed] [Google Scholar]
- 179.Yu WH, Kumar A, Peterhoff C, Shapiro Kulnane L, Uchlyama Y, Lamb BT, Cuervo AM, Nixon RA. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer's disease. Int J Biochem Cell Biol. 2004;36:2531–40. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 180.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem. 2003;278:25009–13. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 181.Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant alpha-synuclein by chaper-one-mediated autophagy. Science. 2004;305:1292–5. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 182.Qin ZH, Wang Y, Kegel KB, Kazantsev A, Apostol BL, Thompson LM, Yoder J, Aronin N, DiFiglia M. Autophagy regulates the processing of amino terminal huntingtin fragments. Hum Mol Genet. 2003;12:3231–44. doi: 10.1093/hmg/ddg346. [DOI] [PubMed] [Google Scholar]
- 183.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 184.Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature. 2006;443:780–6. doi: 10.1038/nature05291. [DOI] [PubMed] [Google Scholar]
- 185.Ding WX, Ni HM, Gao W, Yoshimori T, Stolz DB, Ron D, Yin XM. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–24. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Pandey UB, Batlevi Y, Baehrecke EH, Taylor JP. HDAC6 at the intersection of autophagy, the ubiquitin-proteasome system and neurodegeneration. Autophagy. 2007;3:643–5. doi: 10.4161/auto.5050. [DOI] [PubMed] [Google Scholar]
- 187.Ravikumar B, Berger Z, Vacher C, O'Kane CJ, Rubinsztein DC. Rapamycin pre-treat-ment protects against apoptosis. Hum Mol Genet. 2006;15:1209–16. doi: 10.1093/hmg/ddl036. [DOI] [PubMed] [Google Scholar]
- 188.Lansbury PT., Jr Evolution of amyloid: what normal protein folding may tell us about fibrillogenesis and disease. Proc Natl Acad Sci USA. 1999;96:3342–4. doi: 10.1073/pnas.96.7.3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 190.Iwai A, Yoshimoto M, Masliah E, Saitoh T. Non-A beta component of Alzheimer's disease amyloid (NAC) is amyloidogenic. Biochemistry. 1995;34:10139–45. doi: 10.1021/bi00032a006. [DOI] [PubMed] [Google Scholar]
- 191.Han H, Weinreb PH, Lansbury PT., Jr The core Alzheimer's peptide NAC forms amyloid fibrils which seed and are seeded by beta-amyloid: is NAC a common trigger or target in neurodegenerative disease? Chem Biol. 1995;2:163–9. doi: 10.1016/1074-5521(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 192.Bingol B, Schuman EM. Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol. 2005;15:536–41. doi: 10.1016/j.conb.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 193.Ravikumar B, Vacher C, Berger Z, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–95. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 194.Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J BiolChem. 2007;282:5641–52. doi: 10.1074/jbc.M609532200. [DOI] [PubMed] [Google Scholar]
- 195.Fitzgerald U, Gorman AM, Samali A. 2006 Heat shock proteins and the regulation of apoptosis. In: Richter-Landsberg C, editor. Heat shock proteins in neural cells. 2006. pp. 53–66. . In:, editor.. Texas: USA:; Landes Bioscience. [Google Scholar]
- 196.Wagstaff MJ, Collaco-Moraes Y, Aspey BS, Coffin RS, Harrison MJ, Latchman DS, de Belleroche JS. Focal cerebral ischaemia increases the levels of several classes of heat shock proteins and their corresponding mRNAs. Brain Res Mol Brain Res. 1996;42:236–44. doi: 10.1016/s0169-328x(96)00127-1. [DOI] [PubMed] [Google Scholar]
- 197.Fargnoli J, Kunisada T, Fornace AJ, Jr, Schneider EL, Holbrook NJ. Decreased expression of heat shock protein 70 mRNA and protein after heat treatment in cells of aged rats. Proc Natl Acad Sci USA. 1990;87:846–50. doi: 10.1073/pnas.87.2.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Concannon CG, Gorman AM, Samali A. On the role of Hsp27 in regulating apoptosis. Apoptosis. 2003;8:61–70. doi: 10.1023/a:1021601103096. [DOI] [PubMed] [Google Scholar]
- 199.Hartl FU, Hayer-Hartl M. Molecular chap-erones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 200.Pandey P, Farber R, Nakazawa A, Kumar S, Bharti A, Nalin C, Weichselbaum R, Kufe D, Kharbanda S. Hsp27 functions as a negative regulator of cytochrome c-dependent activation of procaspase-3. Oncogene. 2000;19:1975–81. doi: 10.1038/sj.onc.1203531. [DOI] [PubMed] [Google Scholar]
- 201.Concannon CG, Orrenius S, Samali A. Hsp27 inhibits cytochrome c-mediated caspase activation by sequestering both pro-caspase-3 and cytochrome c. Gene Expr. 2001;9:195–201. doi: 10.3727/000000001783992605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Samali A, Robertson JD, Peterson E, Manero F, van Zeijl L, Paul C, Cotgreave IA, Arrigo AP, Orrenius S. Hsp27 protects mitochondria of thermotolerant cells against apoptotic stimuli. Cell Stress Chaperones. 2001;6:49–58. doi: 10.1379/1466-1268(2001)006<0049:hpmotc>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Catley L, Tai YT, Hayashi T, Shringarpure R, Burger R, Munshi N, Ohtake Y, Saxena S, Anderson KC. Hsp27 inhibits release of mitochondr-ial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood. 2003;102:3379–86. doi: 10.1182/blood-2003-05-1417. [DOI] [PubMed] [Google Scholar]
- 204.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–75. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 205.Zourlidou A, Payne Smith MD, Latchman DS. HSP27 but not HSP70 has a potent protective effect against alpha-synuclein-induced cell death in mammalian neuronal cells. J Neurochem. 2004;88:1439–48. doi: 10.1046/j.1471-4159.2003.02273.x. [DOI] [PubMed] [Google Scholar]
- 206.Dong Z, Wolfer DP, Lipp HP, Bueler H. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of Parkinson disease. Mol Ther. 2005;11:80–8. doi: 10.1016/j.ymthe.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 207.Wyttenbach A, Sauvageot O, Carmichael J, Diaz-Latoud C, Arrigo AP, Rubinsztein DC. Heat shock protein 27 prevents cellular polyglutamine toxicity and suppresses the increase of reactive oxygen species caused by huntingtin. Hum Mol Genet. 2002;11:1137–51. doi: 10.1093/hmg/11.9.1137. [DOI] [PubMed] [Google Scholar]
- 208.Kang BS, Ahn JY, Kim MK, Kim HJ, Kang L, Lim HC, Park KS, Lee JS, Seo JS, Cha CI, Kim SU, Park YJ, Kim M. Heat shock protein 70 alters the endosome-lysosomal localization of huntingtin. Exp Mol Med. 2007;39:38–46. doi: 10.1038/emm.2007.5. [DOI] [PubMed] [Google Scholar]
- 209.Van Der Weerd L, Lythgoe MF, Badin RA, Valentim LM, Akbar MT, de Belleroche JS, Latchman DS, Gadian DG. Neuropro-tective effects of HSP70 overexpression after cerebral ischaemia-an MRI study. Exp Neurol. 2005;195:257–66. doi: 10.1016/j.expneurol.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 210.Waldmeier P, Bozyczko-Coyne D, Williams M, Vaught JL. Recent clinical failures in Parkinson's disease with apoptosis inhibitors underline the need for a paradigm shift in drug discovery for neu-rodegenerative diseases. Biochem Pharmacol. 2006;72:1197–206. doi: 10.1016/j.bcp.2006.06.031. [DOI] [PubMed] [Google Scholar]