

Abstract
PED (phosphoprotein enriched in diabetes) is a death-effector domain (DED) family member with a broad anti-apoptotic action. PED inhibits the assembly of the death-inducing signalling complex (DISC) of death receptors following stimulation. Recently, we reported that the expression of PED is increased in breast cancer cells and determines the refractoriness of these cells to anticancer therapy. In the present study, we focused on the role of PED in non-small cell lung cancer (NSCLC), a tumour frequently characterized by evasion of apoptosis and drug resistance. Immunohistochemical analysis of a tissue microarray, containing 160 lung cancer samples, indicated that PED was strongly expressed in different lung tumour types. Western blotting performed with specimens from NSCLC-affected patients showed that PED was strongly up-regulated (>6 fold) in the areas of tumour compared to adjacent normal tissue. Furthermore, PED expression levels in NSCLC cell lines correlated with their resistance to tumour necrosis factor related apoptosis-inducing ligand (TRAIL)-induced cell death. The involvement of PED in the refractoriness to TRAIL-induced cell death was investigated by silencing PED expression in TRAIL-resistant NSCLC cells with small interfering (si) RNAs: transfection with PED siRNA, but not with cFLIP siRNA, sensitized cells to TRAIL-induced cell death. In conclusion, PED is specifically overexpressed in lung tumour tissue and contributes to TRAIL resistance.
Keywords: lung cancer, apoptosis, AKT
Introduction
Lung cancer is the leading cause of cancer-related mortality in western countries [1]. The late stage at diagnosis in many patients, and the high rate of relapse for those initially diagnosed in earlier stages, contribute to its lethality. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for approximately 80% of cases [2]. One of the most important issues that affects survival rate, is resistance to therapeutic drugs. Only 20–30% of treated NSCLC patients have clinical evidence of a response. Therefore, the development of new therapeutic strategies is necessary for the treatment of this type of tumour.
Apo2L/TNF-related apoptosis-inducing ligand (TRAIL) is a relatively new member of the tumour necrosis factor (TNF) ligand family, which induces apoptosis in a variety of cancers. Four cognate receptors have been identified: the death receptors, TRAIL-R1/DR4 and TRAIL-R2/DR5; and the decoy receptors, TRAIL-R3/DcR1 and TRAIL-R4/DcR2 [3]. The decoy receptors have been proposed to competitively inhibit TRAIL-induced apoptosis by acting as non-functional receptors. All four TRAIL receptors are highly expressed in a wide variety of normal cells, but the expression of decoy receptors is substantially limited in tumour cells [4, 5].
Treatment with TRAIL induces programmed cell death in a wide range of transformed cells, both in vitro and in vivo, without producing significant effects in normal cells [6, 7]. This unique property makes TRAIL an attractive candidate for cancer therapy. However, a significant proportion of cancer cell lines is resistant to TRAIL-induced apoptosis [8]. Apoptotic signalling could be opposed by enhanced expression of intracellular molecules acting at multiple levels [9, 10]. Among these molecules is PED (phosphoprotein enriched in diabetes) (known also as PED/PEA-15), a death-effector domain (DED) family member of 15 kD involved in cell growth and metabolism [9, 11, 12]. PED inhibits the formation of a functional death-inducing signalling complex (DISC) and the activation of caspase 8, which take place following treatment with different apoptotic cytokines including CD95/FasL, TNF-α and TRAIL [9, 13–16]. The anti-apoptotic action of PED is accomplished, at least in part, through its DED domain, which acts as a competitive inhibitor for pro-apoptotic molecules during the assembly of the DISC [9, 15]. PED is over-expressed in a number of different tumours, including human glioma [13], squamous carcinoma [17], breast cancer [18] and B-cell lymphocytic leukaemia [19]. We recently reported that human breast cancer cells express high levels of PED and that AKT/PKB activity regulates PED protein levels [18, 19]. Furthermore, high PED expression levels determine resistance of breast cancer cells to chemotherapy-induced cell death.
In this study, we focused on the involvement of PED in determining the ‘TRAIL-resistant phenotype’ in NSCLC. We analysed PED expression in specimens from 27 NSCLC-affected patients and in a large human tissue microarray containing 160 lung tumour samples. The effect of silencing PED expression in NSCLC cell lines was also investigated.
Methods
Materials
Media, sera and antibiotics for cell culture were from Life Technologies, Inc. (Grand Island, NY, USA). Protein electrophoresis reagents were from Bio-Rad (Richmond, VA, USA), and Western blotting and ECL reagents were from Amersham (Aringhton Heights, IL, USA). All other chemicals were from Sigma (St. Louis, MO, USA) unless otherwise stated. The following primary antibodies were used: anti-PED antibody [11]; anti-β-actin antibody (Ab-1, mouse IgM, Oncogene, Darmstadt, Germany); anti-caspase 8 antibody (1C12, Cell Signaling Technology, Inc., Danvers, MA, USA), anti-caspase 3 antibody (Abcam, Cambridge, USA); anti-caspase 10 antibody (StressGene, Victoria, BC Canada); anti-cFLIP (NF6) antibody (Alexis, Lausen, Switzerland); anti-FADD antibody (BD Transduction Laboratories, San Jose, CA, USA); anti-TRAIL receptor antibodies (R&D Systems, Minneapolis, MN, USA) and anti PARP antibody (SC7150 Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
Cell culture
Human CALU-1 and A459 NSCLC cell lines were grown in DMEM; H460 and A549 cell lines were grown in RPMI. Media were supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine and 100 U/ml penicillin-streptomycin.
Protein isolation and Western blotting
Lung tissue specimens (neoplastic and adjacent normal tissue) were collected during surgical intervention on 27 patients affected with lung tumour, in accordance with the ethical standards of the institutional responsible committee on human experimentation. The clinical and pathological characterization (including tumor, node, metastasis (TNM) staging) of these patients, which were not receiving medical treatment at the time of the operation, is shown in Table 1. The collected samples were homogenized in 1 ml PBS (0.14M NaCl, 2.7 mM KCl, 8 mM Na2HPO4, 1.5 mM KH2PO4) containing 1% Triton X-100 and proteinase inhibitor cocktail, with a tissue homogenator. Cultured cells were pelleted, washed twice with cold PBS and lysed in the same harvest buffer. Solubilized proteins were incubated for 30 min. on ice, and after centrifugation at 10,000 ×g for 30 min. at 4°C, supernatants were collected. Fifty micrograms of sample extract were resolved on 12% SDS-polyacrylamide gels using a mini-gel apparatus and transferred to Hybond-C extra nitrocellulose. Membranes were blocked for 1 hr with 5% non-fat dry milk in Tris Buffered Saline (TBS) containing 0.05% Tween-20, incubated for 2 hrs with primary antibody, washed and incubated with secondary antibody, and visualized by chemiluminescence.
Table 1.
Clinical features of the patients
| P # | Sex | Age | Histology | TNM |
|---|---|---|---|---|
| 1 | M | 49 | ADENO | T1N0MX |
| 2 | M | 61 | ADENO | T2N2MX |
| 3 | M | 68 | ADENO | T3N0MX |
| 4 | M | 64 | ADENO | T1N0MX |
| 5 | M | 63 | ADENO | T2N1MX |
| 6 | M | 55 | ADENO | T2N2MX |
| 7 | M | 56 | ADENO | T2N0MX |
| 8 | M | 56 | SQUAM | T1N1MX |
| 9 | M | 63 | ADENO | T3N2MX |
| 10 | M | 66 | ADENO | T1N1MX |
| 11 | M | 77 | ADENO | T1N0MX |
| 12 | M | 69 | SQUAM | T2N0MX |
| 13 | M | 61 | SQUAM | T2N0MX |
| 14 | F | 52 | ADENO | T2N0MX |
| 15 | M | 57 | ADENO | T2N2M1 |
| 16 | M | 80 | SQUAM | T2N0MX |
| 17 | M | 63 | ADENO | T1N2MX |
| 18 | M | 55 | ADENO | T1N2MX |
| 19 | M | 63 | SQUAM | T2N0MX |
| 20 | M | 67 | SQUAM | T1N0MX |
| 21 | M | 63 | SQUAM | T1N0MX |
| 22 | M | 70 | SQUAM | T3N0MX |
| 23 | M | 68 | SQUAM | T1N0MX |
| 24 | M | 55 | ADENO | T1N0MX |
| 25 | M | 64 | ADENO | T1N0MX |
| 26 | M | 69 | SQUAM | T2N0MX |
| 27 | M | 68 | ADENO | T1N0MX |
Age (years), sex and TNM (tumour, node, metastasis) staging [30] of patients (P) are reported. All patients were smokers. Histology of the tumours indicated that most were adenocarcinoma (ADENO) and the remaining squamous cell carcinomas (SQUAM).
Cell death and cell proliferation quantification
Cells were plated in 96-well plates in triplicate and incubated at 37°C in a 5% CO2 incubator. To induce apoptosis, Superkiller TRAIL (Alexis Biochemicals) was used for 24 hrs at 10 ng/ml. Cell viability was evaluated with the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI, USA), according to the manufacturer's protocol. Metabolically active cells were detected by adding 20 μl of MTS to each well. After 2 hrs of incubation, the plates were analysed in a Multilabel Counter (Bio-Rad). Apoptosis was also assessed using annexin V-FITC Apoptosis Detection Kits followed by flow cytometric analysis. Cells were seeded at 1.8 × 106 cells per 100-mm dish, grown overnight in 10% Fetal Bovine Serum (FBS)/RPMI, washed with PBS, then treated for 24 hrs with 200 ng TRAIL. Following incubation, cells were washed with cold PBS and removed from the plates by very mild trypsinization conditions (0.01% trypsin/ethylenediaminetetraacetic acid). The resuspended cells were washed with cold PBS and stained with FITC-conjugated annexin V antibody and propidium iodide according to the instructions provided by the manufacturer (Roche Applied Science, Indianapolis, IN, USA). Cells (50,000 per sample) were then subjected to flow cytometric analysis. Propidium iodide staining and flow cytometry analysis were done as described [19].
Flow cytometry
The relative level of surface TRAIL receptors was assessed by FACS analysis. To this end, 1 million cells were collected and washed twice in PBS, incubated with PE-conjugated purified monoclonal antibodies against TRAIL receptors or with mouse isotype control phycoerythrin IgG2b for 1 hr on ice, and then washed once with 3 ml PBS. After centrifugation, the cell pellet was resuspended in 1 ml PBS and analysed with a FACSsort (Becton Dickinson, Franklin Lakes, NJ, USA).
Small interfering (si) RNAs
The Dharmacon siDesign Center software program was used to design a duplex siRNA targeting PED mRNA (siPED).This duplex consisted of a 21 nt double-stranded RNA, comprised 19 base pairs with two T, 3' overhanging ends, synthesized by Invitrogen (Invitrogen Corporation, Carlsbad, CA, USA) (UCACUAUGGUGGUUGACUATT), c-FLIP siRNA was purchased from Santa Cruz Biotechnology, Inc. (sc-35388). siCONTROL Non-Targeting siRNA Pool #2 (D-001206-14-05) was from Dhamarcon (Lafayette, CO, USA) and comprised four siCONTROL non-targeting siRNAs. Each individual siRNA within this pool was characterized by genome-wide microarray analysis and found to have minimal off-target signatures.
Transfection experiments
CALU-1 or H460 cells were cultured to 80% confluence in p60 plates. Control, PED or cFLIP siRNA (100 nM), PED or c-FLIP siRNA were transiently transfected in cells kept in antibiotic-free, serum-containing medium, using LIPOFECTAMINE 2000, according to the manufacturer's instructions. Cells were incubated with siRNAs for the indicated times. PED protein levels were up-regulated where indicated by transfecting cells with 5 μg of pcDNA3-Myc PED [12].
Tissue microarray construction and immunohistochemistry
Cylinders of 0.6-mm diameter were punched from donor blocks in areas identified as neoplastic after analysis of haematoxylin and eosin stained sections. The tissue cylinders were inserted into a recipient paraffin block using a precision instrument (Beecher Instruments, Sun Prairie, WI, USA) [20]. Standard indirect staining procedures were used for immunohisto-chemistry (ABC-Elite-Kit, Vector Laboratories, Burlingame, CA, USA). After heat-induced pre-treatment (in citrate buffer, pH6, water bath at 90°C for 30 min.) for antigen retrieval, a rabbit polyclonal anti-PED antibody [11] was applied for 2 hrs at a dilution of 1:5000 at 37°C. The slides were then incubated with the secondary, biotinylated antibody. Osmium-enhanced diaminobenzidine was used as the chromogen. Counterstaining was carried out with Harris' haematoxylin. Only fresh cut sections were stained to minimize the influence of slide ageing and maximize repeatability and reproducibility of the experiment. For negative controls, the primary antibody was omitted; as positive control, normal tissue with known PED positivity was used. For each sample, percentage of positive tumour cells and staining intensity (0, faint, moderate or intense) were recorded. A case was graded: (i) negative, if no cells were stained; (ii) weakly positive (mild), if up to 33% of cells were stained; (iii) moderately positive if 34–66% of cells were stained or (iv) strongly positive if 67–100% of cells were positive. Staining intensity was not used for correlation with clinical findings, as it can vary depending on the manner of tissue fixation. The slides were all evaluated in 1 day by an experienced pathologist (DB).
Results
PED expression in NSCLC tissue
Specimens were collected from 27 NSCLC-affected patients (clinical features are summarized in Table 1) and processed for Western blotting. PED was increased in the tumour tissue of all the patients analysed and only rarely expressed in the adjacent normal tissue (Fig.1A). The mean PED expression level was sixfold higher in the transformed areas compared to normal tissue (Fig.1B). Furthermore, analysis of TNM staging revealed that the expression of PED was greater during the initial stages of the disease (T1 and N0) than in T2 and N1 lesions (Fig.1C). We also analysed PED expression on a tissue microarray constructed from 160 different histological lung cancer samples. As shown in Table 2, PED expression was considered high in the majority of cases and in all the types of lung tumour present, with only 4% of the tumours analysed resulting negative for PED staining. Immunohistochemical analysis of PED expression is depicted in Fig.2 where it is possible to evaluate the different PED staining intensity. Part (A) represents an example of PED negative staining; part (B), of mild; part (C) of moderate and part (D) of strong PED staining. The majority of samples showed strong staining for PED.
Figure 1.
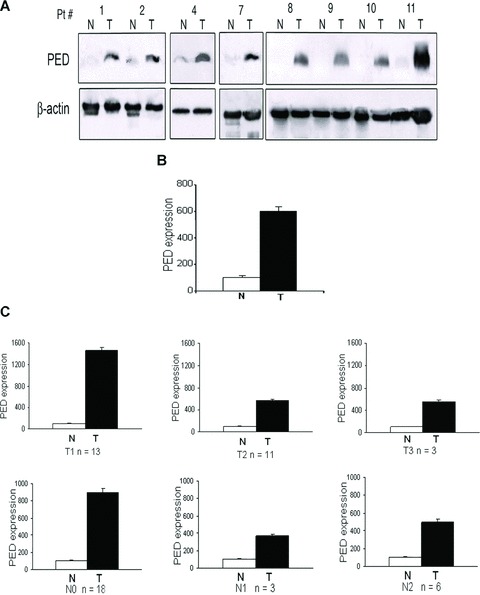
PED expression is increased in human lung cancer. (A) Western blots showing expression of PED in tumour (T) and adjacent normal (N) lung tissue from some of the 27 NSCLC-affected patients (Pt). β-Actin was used for the loading control. (B) Graph of densitometric analysis. Mean ± SD PED expression of all the tumour samples (T) normalized to β-actin and expressed as percentage respect to the adjacent normal tissue (N). PED was >6-fold higher in cancer tissue compared to normal areas. (C) PED expression during T1, T2, T3 and N0, N1 and N2 stages of the disease. PED expression is greater during the initial stages (T1 and N0) of the disease compared with the T2 and N1 lesions.
Table 2.
PED expression analysed in the lung cancer tissue microarray
| Histological type | PED expression | Total | |||
|---|---|---|---|---|---|
| Negative | sssMild | Moderate | Strong | ||
| Adenocarcinoma | 1 (2.3%) | 8 (18.6%) | 10 (23.2%) | 24 (55.8%) | 43 |
| Large cell carcinoma | 1 (2.4%) | 2 (4.8%) | 15 (36.5%) | 23 (56.1%) | 41 |
| Small cell carcinoma | 2 (6.6%) | 5 (16.6%) | 7 (23.3%) | 16 (53.3%) | 30 |
| Squamous cell carcinoma | 2 (4.3%) | 11 (23.9%) | 13 (28.2%) | 20 (43.4%) | 46 |
Top number indicates number of cases present in the tissue microarray; the bottom number indicates its relative percentage. PED expression was graded: negative: no staining; mild: 0–33% positively staining cells; moderate: 34–66% positively staining cells and strong: 67–100% positively staining cells.
Figure 2.
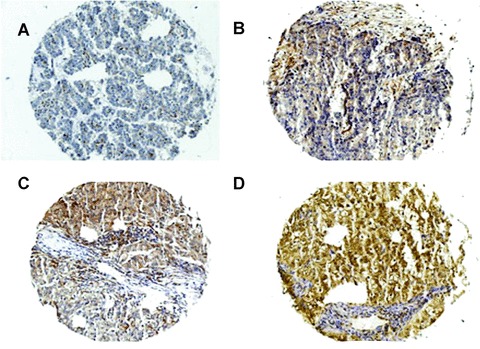
PED expression levels in NSCLC cancer samples. Immunohistochemical analysis of paraffin-embedded NSCLC sections labelled with anti-PED antibody (1:5000) and revealed by secondary, biotinylated antibody. (A) Negative; (B) mild; (C) moderate and (D) strong staining.
PED expression correlates with resistance to TRAIL in NSCLC cell lines
PED is a DED-containing protein that inhibits the formation of a functional DISC following treatment with different apoptotic stimuli, including TRAIL, in several cell types [9, 15, 21]. We therefore, investigated whether PED expression correlated with resistance to TRAIL in different NSCLC cell lines. As shown in Fig.3A, PED is expressed at higher levels in CALU-1 cells compared to H460 cells. Intermediate levels of expression were observed in A459 and A549 cells. When exposed to TRAIL, H460 cells underwent TRAIL-induced cell death, whereas CALU-1 cells were completely resistant (Fig.3B and C). A459 and A549 cells exhibited intermediate sensitivities to TRAIL (Fig.3B and C). Therefore, PED expression levels correlated with TRAIL resistance in the NSCLC cell lines analysed. We also studied several components of the extrinsic cell death signalling pathway. As shown in Fig.3A, the expression of these TRAIL signalling molecules was comparable in the four cell lines, although c-FLIP expression was slightly lower in H460 cells.
Figure 3.
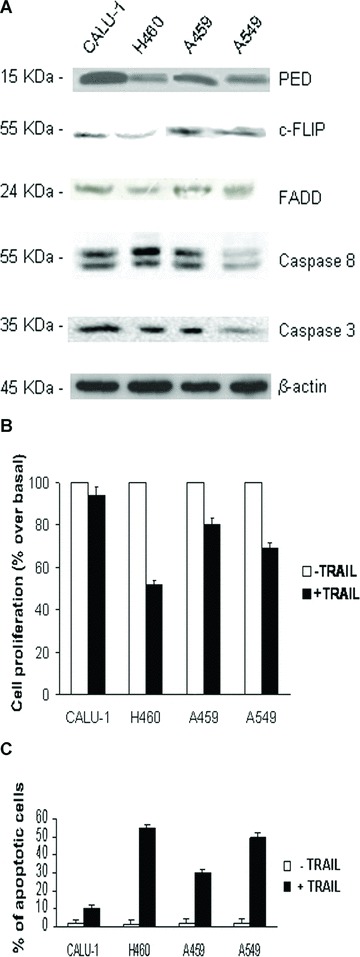
PED expression correlates with TRAIL resistance in NSCLC cell lines. (A) Fifty micrograms of total cell extract from CALU-1, H460, A459 and A549 cells were analysed by Western blotting for the expression of PED and other TRAIL signalling molecules. β-Actin was used as the loading control. Representative blots are shown. (B) Viability after treatment with TRAIL. Cells were incubated with superkiller TRAIL (10 ng/ml) for 24 hrs and viability evaluated as described in the methods section. Mean ± SD of three independent experiments in triplicate. (C) Annexin V and propidium iodide staining of NSCLC cells after TRAIL treatment. Mean ± SD of four independent experiments in duplicate.
TRAIL resistance in NSCLC cells does not depend on the expression of TRAIL receptors
TRAIL resistance could be correlated with different expression levels of TRAIL receptors. To exclude this possibility, we investigated the cell surface expression of all four TRAIL receptor subtypes in CALU-1 and H460 cells. As shown in Fig.4, expression of functional and decoy TRAIL receptors was comparable in the two cell lines.
Figure 4.
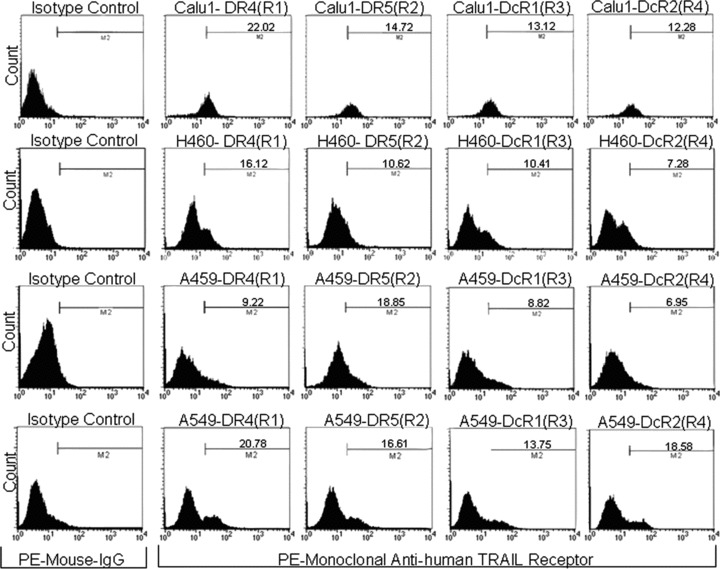
Surface expression of TRAIL receptors does not differ in TRAIL-resistant and TRAIL-sensitive cells. Surface expression of the four TRAIL receptor isotopes (R1, R2, R3 and R4) was analysed by flow cytometry with specific PE-conjugated antibodies. Isotype-matched antibodies were used as control for unspecific binding. Receptor expression levels were comparable in CALU-1 and H460 cells.
Effect of the down-regulation of PED and c-FLIP on insensitivity to TRAIL in CALU-1 cells
To better clarify the role of PED in TRAIL resistance in lung cancer cells, we transfected CALU-1 cells with PED siRNA. PED siRNA specificity was tested in CALU-1 cells by the co-transfection of PED siRNA with Myc-tagged PED cDNA [12]. The PED siRNA duplex suppressed both endogenous (15 kD) and exogenous (23 kD) PED expression, whereas a control siRNA was not effective in reducing PED protein levels (Fig.5A). This silencing effect was evident at 48 hrs and more marked at 72 hrs. We found that also other in cell lines (HeLa, Human embryonic kidney 293) PED siRNA was able to knock-down PED expression (data not shown). Transfected cells were then exposed to TRAIL. As expected, siRNA-mediated knock-down of PED was responsible for sensitization of CALU-1 cells to TRAIL-induced cell death (Fig.5B). Interestingly, comparable results were obtained when we knocked-down PED expression in A459 and A549 cells (Fig.5B).
Figure 5.
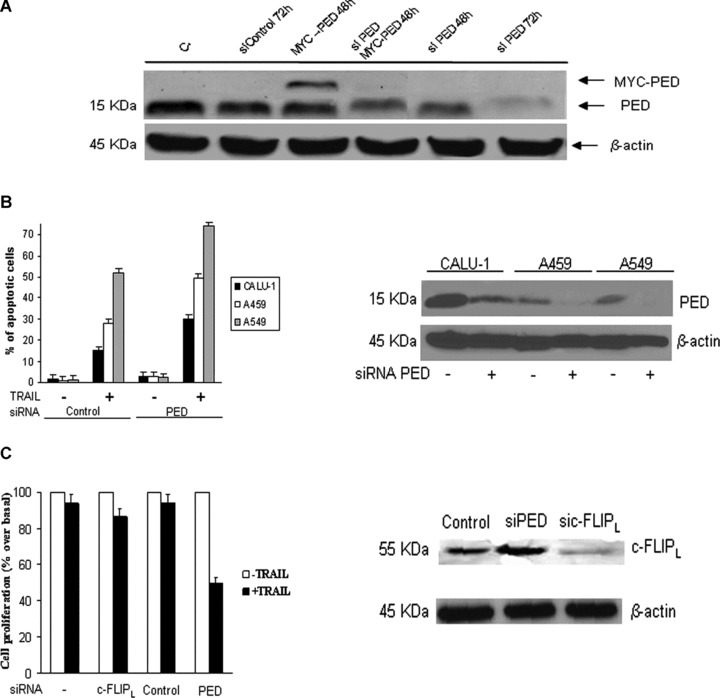
Down-regulation of PED restores TRAIL sensitivity in CALU-1 cells. (A) PED siRNA or a control oligo were transiently transfected in CALU-1 cells in the presence or absence of PED-Myc cDNA. Cells were incubated for 48 or 72 hrs and analysed by Western blotting. The PED siRNA duplex suppressed both exogenous and endogenous PED expression, whereas control siRNA had no effects. (B) PED siRNA effects in A459 and A549 cells. PEDsi RNA, transfected in NSCLC cells was able to reduce PED expression levels (right panel) and induce an increase in TRAIL sensitivity (left panel), as assessed by flow cytometry. Mean ± SD of two independent experiments in duplicate. (C) c-FLIPL siRNA or PED siRNA were transfected as described in Methods. Cells were analysed for c-FLIP expression after 72 hrs incubation. c-FLIPL siRNA but not PED siRNA was able to reduce c-FLIPL expression Effects of silencing PED and c-FLIPL on TRAIL-induced cell death: CALU-1 cells were transfected with siRNA for PED, c-FLIPL or control for 48 hrs, after which cells were trypsinized, plated in 96-well plates in triplicate and further incubated with superkiller TRAIL for 24 hrs. Metabolically active cells were then detected as indicated in the Methods. Mean ± SD of four independent experiments in duplicate. Down-regulation of PED, but not cFLIPL, was responsible for increased sensitivity of CALU-1 cells to TRAIL-mediated cell death.
The treatment of CALU-1 cells with a specific c-FLIPL siRNA down-regulated c-FLIPL expression but did not increase sensitivity to TRAIL-induced cell death (Fig.5C). These data further confirm that PED protects lung cancer cells from TRAIL-induced apoptosis, and that inhibiting PED expression results in increased TRAIL sensitivity.
Effects of silencing PED on caspase activation in CALU-1 cells
We then examined the activation of caspase 8 and PARP upon exposure to TRAIL in CALU-1 cells treated with PED siRNA. As shown in Fig.6A, TRAIL-induced caspase 8 and PARP activation was greater in CALU-1 cells following PED siRNA transfection.
Figure 6.
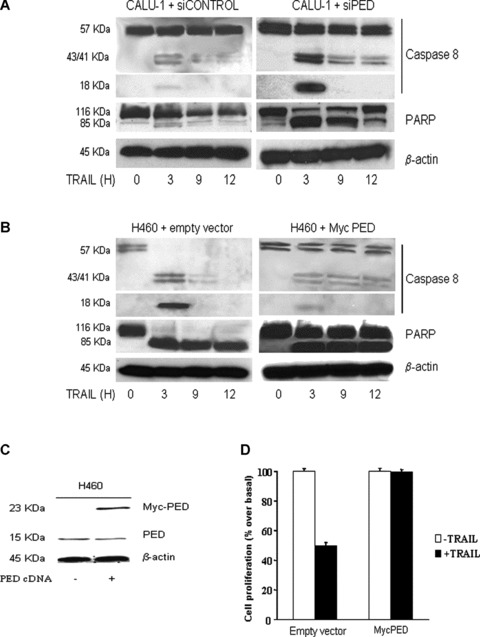
Effects of PED on caspase activation. (A) CALU-1 cells were transfected with PED or control siRNA for 72 hrs and then treated with superkiller TRAIL for the indicated times. Lysates were examined by Western blotting with anti-caspase 8 or anti-PARP antibodies. Cleavage of caspase 8 and PARP was detected at a greater amount in CALU-1 cells transfected with PED siRNA. β-Actin was used as the loading control. (B) PED cDNA (PED-Myc) was transiently transfected in H460 cells and cells were analysed for caspase 8 and PARP activation as previously described or for or for cell viability (D) as indicated. (C) Western blot analysis of PED expression revealed that transfection increased PED expression levels in H460 cells. β-Actin was used as the loading control. Representative blots are shown.
Effect of increasing PED expression on TRAIL sensitivity in H460 cells
To further evaluate the role of PED in apoptosis resistance, TRAIL-sensitive H460 cells were transfected with PED cDNA to up-regulate PED protein levels and then analysed for their susceptibility to TRAIL-induced cell death. Increasing the expression of PED in these cells (Fig.6C) rendered them resistant to TRAIL, as assessed by Western blotting for caspase 8 and PARP (Fig.6B) or by a cell viability assay (Fig.6D).
Discussion
Increased expression of DED family members with anti-apoptotic functions, such as PED, represents a possible mechanism by which tumour cells escape apoptosis. PED acts primarily by preventing the interaction between the adaptor molecule, FADD, and procaspase-8 [9, 15]. We have reported that PED inhibits the anti-apoptotic signal of death receptors in many different cell types, including breast carcinoma and glioma [9, 13–16], in which it is overexpressed. Lung tumours are among the most aggressive types of cancer and are frequently resistant to drug-induced cell death [1]. Eighty percentage of lung cancers are of the NSCLC type. Advances in standard treatment for this tumour, such as surgery, radiotherapy and chemotherapy, have not significantly increased patient survival. Thus, novel treatment strategies are urgently needed to improve the clinical management of this serious disease. Recent studies have demonstrated that targeting TNF superfamily death receptors is a promising strategy for the treatment of cancer [22]. Apoptosis-based anti-cancer therapies are designed to achieve tumour eradication through the use of death-inducing molecules capable of activating the apoptotic program selectively in neoplastic cells. Due to its specific toxicity for transformed cells, recombinant forms of TRAIL are among the most promising apoptosis-based anti-tumour agents [7, 23–25]. In fact, a number of biotech and pharmaceutical companies developed recombinant TRAIL/Apo2L as well as humanized agonistic mAb targeting TRAIL-R1 or TRAIL-R2 that are currently being evaluated in phase I and phase II clinical trails [7]. However, in a number of patients, tumour cells evade death signals generated by drugs through the activation of effective antiapoptotic mechanisms [26–28]. In the present report, we demonstrate that PED is overexpressed in lung cancer tissue obtained from 27 NSCLC-affected patients. Interestingly, TNM staging revealed that PED expression is greater during the first phase of the disease (T1) than in T2 lesions. The analysis of PED expression in pre-malignant lesions, such as metaplastic bronchial lesions, could shed light on the role of PED in tumour initiation, progression and invasiveness. This study shows that PED regulates the susceptibility to TRAIL-induced death in lung cancer cells. In fact, although the levels of PED differ significantly in CALU-1 (TRAIL-insensitive) and H460 (TRAIL-sensitive) cells, the levels of the other signalling components (such as TRAIL receptors caspases, and FADD) were comparable. Modulation of the sensitivity to TRAIL by PED was confirmed by down regulating PED expression with PED siRNA: when transfected, the otherwise TRAIL-insensitive CALU-1 cells became susceptible to this cytotoxic cytokine. Moreover, PED overexpression in H460 cells was able to change their phenotype from TRAIL-sensitive to TRAIL insensitive.
Because resistance of different tumours may be mediated by diverse survival mechanisms, we investigated the role of c-FLIP in TRAIL resistance by down-regulation of c-FLIP expression with a specific siRNA. As already observed in other tumour types such as glioma [13] and B-cell chronic leukaemia [19], the effects of PED are prevalent over those mediated by c-FLIP for sensitivity to TRAIL. Thus, it is possible that c-FLIP and PED contribute differently, depending to the cell type or tumour type to induce resistance to TRAIL. Our data thus demonstrate that PED plays a major role in TRAIL resistance in NSCLC. Although PED expression was completely and specifically inhibited by the transfection with PED siRNA duplex, TRAIL sensitivity was not fully recovered. These results suggest that it is likely that there might be alternative mechanisms for TRAIL resistance. We have recently addressed this point by a microRNAs screening of TRAIL resistance in NSCLC and found several new microRNAs and targets involved in TRAIL resistance (Garofalo et al., in press).
Mice that are genetically deficient in the TRAIL gene, exhibit increased susceptibility to experimental and spontaneous tumours [29], suggesting an important role of endogenous TRAIL in tumour surveillance. Thus, the overexpression of a gene involved in resistance to TRAIL could be important for responsiveness to chemotherapy. Because expression levels of PED are a focal point for the regulation of apoptosis, PED represents a key target for treatment of cancer.
Acknowledgments
This work was partially supported by funds from: Associazione Italiana Ricerca sul Cancro, AIRC (G.C.), MIUR-FIRB (RBIN04J4J7), and EU grant EMIL (European Molecular Imaging Laboratories Network) contract no. 503569. M.A. is a recipient of the FIRC fellowship. G.R. is a recipient of the Fondazione SDN fellowship. C.Q. is a recipient of the Clinica Mediterranea-Federico II Training programme. We wish to thank Michael Latronico for paper revision.
References
- 1.Parker SL, Tong T, Bolden S, Wingo PA. Cancer statistics. CA Cancer J Clin. 1997;47:5–27. doi: 10.3322/canjclin.47.1.5. [DOI] [PubMed] [Google Scholar]
- 2.Travis WD, Travis LB, Devessa SS. Lung Cancer. Cancer. 1995;75:191–202. doi: 10.1002/1097-0142(19950101)75:1+<191::aid-cncr2820751307>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 3.Marsters SA, Sheridan JP, Pitti RM, Huang A, Skubatch M, Baldwin D, Yuan J, Gurney A, Goddard AD, Godowski P,Ashkenazi A. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr Biol. 1997;7:1003–6. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 4.Pan G, Ni J, Wei F, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 5.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 6.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–75. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Schaefer U, Voloshanenko O, Willen D, Walczak H. TRAIL: a multifunctional cytokine. Front Biosci. 2007;12:3813–24. doi: 10.2741/2354. [DOI] [PubMed] [Google Scholar]
- 8.Zhang L, Fang B. Mechanisms of resistance to TRAIL-induced apoptosis in cancer. Cancer Gene Ther. 2005;12:228–37. doi: 10.1038/sj.cgt.7700792. [DOI] [PubMed] [Google Scholar]
- 9.Condorelli G, Vigliotta G, Cafieri A, Trencia A, Andalo P, Oriente F, Miele C, Caruso M, Formisano P, Beguinot F. PED/PEA-15: an antiapoptotic molecule that regulates FAS/TNFR1-induced apoptosis. Oncogene. 1999;18:4409–15. doi: 10.1038/sj.onc.1202831. [DOI] [PubMed] [Google Scholar]
- 10.Reed JC. Apoptosis-targeted therapies for cancer. Cancer Cell. 2003;3:17–22. doi: 10.1016/s1535-6108(02)00241-6. [DOI] [PubMed] [Google Scholar]
- 11.Condorelli G, Vigliotta G, Iavarone C, Caruso M, Tocchetti CG, Andreozzi F, Cafieri A, Tecce MF, Formisano P, Beguinot L, Beguinot F. PED/PEA-15 gene controls glucose transport and is overex-pressed in type 2 diabetes mellitus. EMBO J. 1998;17:3858–66. doi: 10.1093/emboj/17.14.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condorelli G, Trencia A, Vigliotta G, Perfetti A, Goglia U, Cassese A, Musti AM, Miele C, Santopietro S, Formisano P, Beguinot F. Multiple members of the mitogen-activated protein kinase family are necessary for PED/PEA-15 anti-apoptotic function. J Biol Chem. 2002;277:11013–8. doi: 10.1074/jbc.M110934200. [DOI] [PubMed] [Google Scholar]
- 13.Hao C, Beguinot F, Condorelli G, Trencia A, Van Meir EG, Yong VW, Parney IF, Roa WH, Petruk KC. Induction and intracellular regulation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) mediated apotosis in human malignant glioma cells. Cancer Res. 2001;61:1162–70. [PubMed] [Google Scholar]
- 14.Kitsberg D, Formstecher E, Fauquet M, Kubes M, Cordier J, Canton B, Pan G, Rolli M, Glowinski J, Chneiweiss H. Knock-out of the neural death effector domain protein PEA-15 demonstrates that its expression protects astrocytes from TNFalpha-induced apoptosis. J Neurosci. 1999;19:8244–51. doi: 10.1523/JNEUROSCI.19-19-08244.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao C, Yang BF, Asadi N, Beguinot F, Hao C. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J Biol Chem. 2002;277:25020–5. doi: 10.1074/jbc.M202946200. [DOI] [PubMed] [Google Scholar]
- 16.Ricci-Vitiani L, Pedini F, Mollinari C, Condorelli G, Bonci D, Bez A, Colombo A, Parati E, Peschle C, De Maria R. Absence of caspase 8 and high expression of PED protect primitive neural cells from cell death. J Exp Med. 2004;200:1257–66. doi: 10.1084/jem.20040921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong G, Loukinova E, Chen Z, Gangi L, Chanturita TI, Liu ET, Van Waes C. Molecular profiling of transformed and metastatic murine squamous carcinoma cells by differential display and cDNA microarray reveals altered expression of multiple genes related to growth, apoptosis, angiogenesis, and the NF-kappaB signal pathway. Cancer Res. 2001;61:4797–808. [PubMed] [Google Scholar]
- 18.Stassi G, Garofalo M, Zerilli M, Ricci-Vitiani L, Zanca C, Todaro M, Aragona F, Limite G, Petrella G, Condorelli G. PED mediates AKT-dependent chemoresistance in human breast cancer cells. Cancer Res. 2005;65:6668–75. doi: 10.1158/0008-5472.CAN-04-4009. [DOI] [PubMed] [Google Scholar]
- 19.Garofalo M, Romano G, Quintavalle C, Romano MF, Chiurazzi F, Zanca C, Condorelli G. Selective inhibition of PED protein expression sensitizes B-cell chronic lymphocytic leukaemia cells to TRAIL-induced apoptosis. Int J Cancer. 2007;120:1215–22. doi: 10.1002/ijc.22495. [DOI] [PubMed] [Google Scholar]
- 20.Kononen J, Bubendorf L, Kallioniemi A, Barlund M, Schraml P, Leighton S, Torhorst J, Mihatsch MJ, Sauter G, Kallioniemi OP. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nat Med. 1998;4:844–7. doi: 10.1038/nm0798-844. [DOI] [PubMed] [Google Scholar]
- 21.Trencia A, Perfetti A, Cassese A, Vigliotta G, Miele C, Oriente F, Santopietro S, Giacco F, Condorelli G, Formisano P, Beguinot F. Protein kinase B/Akt binds and phosphorylates PED/PEA-15, stabilizing its antiapoptotic action. Mol Cell Biol. 2003;23:4511–21. doi: 10.1128/MCB.23.13.4511-4521.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamada K, Chen L. Renewed interest in cancer immunotherapy with the tumor necrosis factor superfamily molecules. Cancer Immunol Immunother. 2006;55:355–62. doi: 10.1007/s00262-005-0081-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carlo-Stella C, Lavazza C, Locatelli A, Viganò L, Gianni AM, Gianni L. Targeting TRAIL agonistic receptors for cancer therapy. Clin Cancer Res. 2007;13:2313–7. doi: 10.1158/1078-0432.CCR-06-2774. [DOI] [PubMed] [Google Scholar]
- 24.Cretney E, Takeda K, Smyth MJ. Cancer: novel therapeutic strategies that exploit the TNF-related apoptosis-inducing ligand (TRAIL)/TRAIL receptor pathway. Int J Biochem Cell Biol. 2007;39:280–6. doi: 10.1016/j.biocel.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Lee JY, Huerta-Yepez S, Vega M, Baritaki S, Spandidos DA, Bonavida B. 2007;31:685–91. The NO TRAIL to YES TRAIL in cancer therapy Int J Oncol. [PubMed] [Google Scholar]
- 26.Brown JM, Attardi LD. The role of apoptosis in cancer development and treatment response. Nat Rev Cancer. 2005;5:231–7. doi: 10.1038/nrc1560. [DOI] [PubMed] [Google Scholar]
- 27.Kaufmann SH, Vaux DL. Alterations in the apoptotic machinery and their potential role in anticancer drug resistance. Oncogene. 2003;22:7414–30. doi: 10.1038/sj.onc.1206945. [DOI] [PubMed] [Google Scholar]
- 28.Ghobrial IM, Witzig TE, Adjei AA. Targeting apoptosis pathways in cancer therapy. CA Cancer J Clin. 2005;55:178–94. doi: 10.3322/canjclin.55.3.178. [DOI] [PubMed] [Google Scholar]
- 29.Cretney E, Takeda K, Yagita H, Glaccum M, Peschon JJ, Smyth MJ. Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing lig-and-deficient mice. J Immunol. 2002;168:1356–61. doi: 10.4049/jimmunol.168.3.1356. [DOI] [PubMed] [Google Scholar]
- 30.Kang J, Kisenge RR, Toyoda H, Tanaka S, Bu J, Azuma E, Komada Y. Chemical sensitization and regulation of TRAIL-induced apoptosis in a panel of B-lymphocytic leukaemia cell lines. Br J Haematol. 2003;123:921–32. doi: 10.1046/j.1365-2141.2003.04699.x. [DOI] [PubMed] [Google Scholar]