

Abstract
This study examined how the neurotrophin, nerve growth factor (NGF), protects PC12 cells against endoplasmic reticulum (ER) stress-induced apoptosis. ER stress was induced using thapsigargin (TG) that inhibits the sarcoplasmic/ER Ca2+-ATPase pump (SERCA) and depletes ER Ca2+ stores. NGF pre-treatment inhibited translocation of Bax to the mitochondria, loss of mitochondrial transmembrane potential, cytochrome c release, activation of caspases (−3, −7 and −9) and apoptosis induction by TG. Notably, TG also caused a marked induction of Bimel mRNA and protein, and knockdown of Bim with siRNA protected cells against TG-induced apoptosis. NGF delayed the induction and increased the phosphorylation of Bimel. NGF-mediated protection was dependent on phosphatidylinositol-3 kinase (PI3K) signalling since all above apoptotic events, including expression and phosphorylation status of Bimel protein, could be reverted by the PI3K inhibitor LY294002. In contrast, NGF had no effect on the TG-mediated induction of the unfolded protein response (increased expression of Grp78, GADD34, splicing of XBP1 mRNA) or ER stress-associated pro-apoptotic responses (induction of C/EBP homologous protein [CHOP], induction and processing of caspase-12). These data indicate that NGF-mediated protection against ER stress-induced apoptosis occurs at the level of the mitochondria by regulating induction and activation of Bim and mitochondrial translocation of Bax.
Keywords: Bimel, endoplasmic reticulum (ER), mitochondria, nerve growth factor (NGF), thapsigargin (TG)
Introduction
Endoplasmic reticulum (ER) stress is associated with cell death in a number of pathologies including ischaemia, Alzheimer's and Parkinson's diseases [1]. ER stress is caused by physiological and pathophysiological conditions that overwhelm the protein folding or impairs the Ca2+-storage capacity of the ER. Prolonged or severe ER stress leads to apoptotic cell death which is mediated by the activity of caspase proteases [2]. There have been conflicting reports concerning the mechanism of caspase activation during ER stress-induced apoptosis. Some evidence supports a role for caspase-12 as the apical caspase activated directly by the ER [3–5]. Other recent evidence points to involvement of the mitochondrial apoptotic pathway by showing that ER stress induces mitochondrial release of cytochrome c, assembly of the apoptosome and activation of caspase-9; leading to execution of death [6, 7].
Central to the regulation of apoptosis is the Bcl-2 family, which includes both pro- (e.g. Bax, Bak) and anti-apoptotic (e.g. Bcl-2, Bcl-xL) members [8]. The multi-domain members of the Bcl-2 family (which contain Bcl-2 homology domains, BH1, BH2 and BH3) act on intracellular membranes, including ER and mitochondrial membranes, affecting their permeability towards ions and/or proteins. Their best understood function is at the mitochondrial outer membrane, where different family members either promote or inhibit release of pro-apoptotic factors including cytochrome c [8]. BH3-only members of the family (e.g. Bad, Bim, PUMA, Noxa, Bid) regulate the function of the multi-domain Bcl-2 proteins and induce Bax/Bak-mediated cytochrome crelease [8–10]. BH3-only proteins are regulated transcriptionally (e.g. Bim, PUMA) and/or post-translationally (e.g. phosphorylation of Bim or Bad) [9].
Neurotrophins, such as nerve growth factor (NGF) act through tyrosine kinase (Trk) receptors to provide survival and differentiation signals for neuronal cells during development [11]. Deprivation of NGF in sympathetic neurons and differentiated PC12 cells induces apoptosis [12, 13]. In addition, NGF can also protect cells against oxidative stress or toxin-induced apoptosis [14–18]. NGF promotes survival largely through activation of the TrkA receptor and intracellular kinase pathways, including the phosphatidylinositol-3 kinase (PI3K)/Akt and mitogen-activated protein kinase (MAPK) pathways [14, 17, 19, 20]. NGF has also been reported to protect against ER stress-induced apoptosis, however, the molecular mechanism is unclear [15, 17].
The aim of this study was to identify the mechanism by which NGF protects PC12 cells against thapsigargin (TG)-induced ER stress. PC12 cells express TrkA receptors and are responsive to NGF [21]. TG inhibits the sarcoplasmic/ER Ca2+-ATPase pump (SERCA) and causes severe ER stress culminating in apoptosis [22]. We examined the induction by TG of the unfolded protein response (UPR) and activation of the apoptotic execution machinery, and investigated the effect of NGF on each of these TG-induced responses in order to identify its mechanism of protection against lethal ER stress.
Materials and methods
Materials
All chemicals were purchased from Sigma unless otherwise stated. Ac-Asp-Glu-Val-Asp-α-(4-methyl-coumaryl-7-amide) (DEVD-AMC) was from the Peptide Institute. Rabbit polyclonal antibodies against caspase-3, cas-pase-9, cleaved caspase-7, phospho-Bad (Ser136) and Bax were from Cell Signalling Technologies. Mouse monoclonal anti-Bcl-2, rat monoclonal anti-caspase-12 and rabbit polyclonal anti-actin antibodies were from Sigma. Rabbit polyclonal antibodies against Apaf-1, Grp78 and Bim were from StressGen Biotechnologies. Mouse monoclonal anti-Bcl-xL and rabbit polyclonal anti-CHOP antibodies were from Santa Cruz Biotechnology. Mouse monoclonal anti-cytochrome cantibody was from BD Pharmingen. Goat secondary antibodies conjugated to horseradish peroxidase were from Pierce. Rat pheochromocytoma cells (PC12 cells) were from the ECACC. Mouse nerve growth factor-2.5S Grade II (NGF) was from Alomone Laboratory. Plasmid construct encoding green fuorescent protein (GFP)-tagged Bax (Bax-GFP) was a kind gift from Prof. Jochen Prehn (Department of Physiology Royal College of Surgeons, Dublin, Ireland).
Culture and treatment of cells
PC12 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% horse serum, 5% foetal calf serum, 50 U/ml penicillin and 50 μg/ml streptomycin as previously described [18]. For experiments, dishes were coated with poly-L-lysine (10 μg/ml for 3 hrs to assist adherence of cells) and cells were seeded at 7 × 105 per cm2 24 hrs prior to treatments. Cells were treated with 1.5 μM TG for times indicated. For determining the effect of NGF, 100 ng/ml NGF was added 2 hrs prior to the addition of TG. Pre-treatment with kinase inhibitors was for 1 hr prior to other treatments.
Assessment of cell morphology
Cells were harvested by gentle trypsinization and 5 × 104 cells were cyto-centrifuged onto glass slides (using a Shandon Cytospin 3), air-dried and stained using haematoxylin and eosin. Cell morphology was examined using a Zeiss inverse phase microscope. Three fields for each sample and minimum 300 cells/sample from three different experiments were counted.
Detection of caspase-3-like activity
Caspase-3-like activity (DEVDase activity) was determined fluorometri-cally as previously described [23]. Cells were harvested by gentle scraping and washed once in ice-cold phosphate-buffered saline (PBS). Cell pellets were re-suspended in 25 μl PBS and lysates obtained by snap freezing in liquid nitrogen. Lysate and substrate (DEVD-AMC, 50 μM) were combined in reaction buffer (100 mM N-2-hydroxyethyl-piperazine-N-2-ethanesulphonic acid (HEPES) pH 7.25, 10% sucrose, 0.1% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate, 5 mM dithiothreitol (DTT), 10–4% Igepal-630) and added to a microtitre plate. Substrate cleavage leading to release of free 7-amino-4-methylcoumarin (AMC) was monitored at 37°C at 60 sec. intervals over a 30 min. period using a Wallac Victor Multilabel counter (excitation 355 nm, emission 460 nm). Enzyme activity was expressed as nmol AMC released per minute by 1 mg cellular protein.
Preparation of whole cell extracts for Western blotting
Cells were harvested by gentle scraping and washed once with ice-cold PBS. Pellets were re-suspended in 100 μl whole cell lysis buffer (20 mM HEPES pH 7.5, 350 mM NaCl, 1 mM MgCl2, 0.5 mM ethylenediaminetetraacetic acid [EDTA], 0.1 mM EGTA, 1% Igepal-630, 0.5 mM DTT, 100 μM phenylmethanesulfonyl fluoride (PMSF), 2 μg/ml pepstatin A, 25 μM ALLN, 2.5 μg/ml aprotinin and 10 μM leupeptin) and allowed to lyse on ice for 5 min. Cellular debris were removed by centrifugation at 21,000 ×g for 3 min. Samples were stored at −20°C until further analysis.
Western blotting
25 μg protein denatured in Laemmli's sample buffer (62.5 mM Tris-HCl pH 6.8, 2% SDS, 5%β-mercaptoethanol, 4% glycerol, 1 mM PMSF, 0.05% bromophenol blue) was separated by 10–12% SDS-PAGE and transferred onto nitrocellulose. Membranes were blocked for 1 hr in PBS containing 0.05% Tween 20 and 5% (w/v) non-fat dried milk. Membranes were then incubated with primary antibodies as follows: caspase-3 (1:500), cytochrome c (1:2000), caspase-9 (1:1000), cleaved caspase-7 (1:500), phospho-Bad (1:500), Apaf-1 (1:1000), Bcl-2 (1:500), Bcl-xL (1:200), CHOP (1:1000), caspase-12 (1:5000), Bax (1:1000) or Bim (1:1000) overnight at 4°C or Grp78 (1:1000) or actin (1:500) for 2 hrs at room temperature. This was followed by incubation with appropriate horseradish peroxidase-conjugated goat secondary antibody for 2 hrs at room temperature. Protein bands were visualized using Supersignal West Pico chemi-luminescent detection kit (Pierce) and detected on an X-ray film (Agfa). All data shown are representative of at least three separate experiments.
Isolation of cytosolic fractions for detection of cytochrome c release
Cells were harvested by gentle scraping and washed once with ice cold PBS. Cell pellets were re-suspended in 100 μl cell lysis and mitochondria intact (CLAMI) buffer (250 mM sucrose, 70 mM KCl, 0.5 mM DTT, 2.5 μg/ml pepstatin in PBS) containing 50 μg/ml digitonin and allowed to swell on ice for 5 min. The cell suspension was centrifuged at 20,000 × g for 5 min. at 4°C. The supernatant was kept as the cytosolic fraction and the pellet was re-suspended in 100 μl CLAMI buffer as the mitochondrial and nuclear fraction. Samples were stored at −20°C until further analysis by Western blotting.
Measurement of mitochondrial transmembrane potential (ΔΨm)
ΔΨm was measured using the fluorescent dye tetramethylrhodamine ethyl ester perchlorate (TMRE). Cells were harvested into the medium by trypsinization, and TMRE was added to a final concentration of 100 nM. Cells were incubated for 30 min. at room temperature in the dark followed by immediate analysis by flow cytometry (FacsCalibur flow cytometer, Beckton Dickinson). As a positive control for mitochondrial depolarization, cells were treated for 2 hrs with 10-μM carbonyl cyanide 3-chlorophenyl-hydrazone (CCCP).
Analysis of Bax subcellular distribution
PC12 cells were seeded at 70,000 cells/well in 24-well plates 24 hrs before transfection. Cells were transfected with 0.6 μg of Bax-GFP using Effectene transfection reagent (Qiagen, Crawley, West Sussex, England, at an Effectene:DNA ratio of 10:1. After 24 hrs of incubation, the culture medium was replaced and the cells exposed to experimental treatments. Cells were harvested by trypsinization, fixed with 3.7% formaldehyde for 10 min. at room temperature, washed with PBS, spun onto microscope slides and mounted with 4′-6-Diamidino-2-phenylindote (DAPI)-containing Vectashield (Vector Laboratories, Peterborough, England, to stain the nuclei. Analysis of Bax-GFP subcellular distribution was carried out using Image-Pro software with an Olympus BX51 fluorescent microscope at an overall magnification of 1000 ×.
RNA extraction and RT-PCR
Total RNA from cells was isolated using a GenElute Mammalian Total RNA Extraction kit, (Sigma-Aldrich Ireland Ltd., Dublin, Ireland). Reverse transcription was carried out with 2 μg total RNA and oligo(dT) (Invitrogen Bio Science Ltd., Dun Laoghaire, Ireland) using 20 U/25 μl reaction of avian myeloblastosis virus (AMV) reverse transcriptase (Sigma). cDNAs for genes of interest were amplified during 32 cycles of 30 sec. denaturing at 94°C, 30 sec. annealing at 56°C and 60 sec. extension at 72°C, with the following primers: XBP1 forward CAGACTACGTGCGCCTCTGC; XBP1 reverse CTTCTGGGTAGACCTCTGGG; sXBP1 forward TCTGCTGAGTCCGCAGCAGG; sXBP1 reverse CTCTAAGACTAGAGGCTTGG; GADD34 forward: TTTCTAGGCCAGACACATGG; GADD34 reverse: TGTTCCTTTTTC-CTCCGTGG. Bik forward: ACGGGTGTCAGAGGTATTTTCA Bik reverse:
AAGAAGACCAG-CAGCACCAT; Bim total forward: GCC CCT ACC TCC CTA CAG AC; Bim total reverse: TCA ATG CCT TCT CCA TAC CAG ACG. PUMA forward: CTC GGT CAC CAT GAG TCC TT; PUMA reverse: CCC TGG AGG GTC ATG TAT AA. GAPDH was used as a loading control, its cDNA was amplified during 26 cycles of 30 sec. denaturing at 94°C, 60 sec annealing at 56°C and 60 sec extension at 72°C, with the following primers: GAPDH forward ACCACAGTCCATGCCATC; GAPDH reverse TCCACCACCCTGTTGCTG.
Knockdown with Bim siRNA
PC12 cells were seeded with 200,000 cells/well in 6-well plates at the time of transfection. 50 nM siRNA was incubated for 10 min. at room temperature with 200 μl culture medium and 10.5 μl Lipofectamine2000 transfec-tion reagent (Invitrogen) before adding to the cells. Culture medium was replaced 16 hrs after transfection. 48 hrs after transfection, culture medium was changed again and cells were exposed to 1.5 μM TG for 24 hrs. The following siRNA sequences purchased from Ambion were used: Bim siRNA-1: 5′-AAGUCUCAUUGAACUCGUCTC-3′; Bim siRNA-2: 5′-CGUGUAAGUCUCAUUGAACTC-3′: Bim siRNA-3: 5′-CAGGCUGCAAUUGUCCACCTT-3′. An equal mixture of the Bim siRNAs (16.67 nM each) was used in the experiments. Scrambled siRNA sequence (50 nM of Silencer Negative Control no. 1, catalogue no. AM4611 from Ambion) was used as a negative control.
Phosphatase treatment of whole cell lysates
Whole cell extracts containing 40-μg protein (prepared as for Western blotting) were incubated with 400 units of lambda protein phosphatase (New England Biolabs) with or without 10 mM sodium orthovanadate, at 30°C for 30 min. Samples were then heated at 95°C for 5 min. in 1 × Laemmli's sample buffer and separated on 11% SDS-PAGE acrylamide gel.
Statistical analysis
Results are expressed as means ± S.E.M. All experiments were repeated at least three times. Statistical analysis was performed using repeated measures ANOVA followed by post hoc tests as described in the figure legends.
Results
NGF blocks TG-induced apoptosis, but not UPR or caspase-12 processing in PC12 cells
In agreement with other reports [15, 17], pre-treatment of PC12 cells with 100 ng/ml NGF for 2 hrs prior to exposure to TG (1.5 μm) inhibited development of apoptotic morphology, caspase (DEVDase) activity and activation of caspases-3 and -7 (Fig.1).
Figure 1.
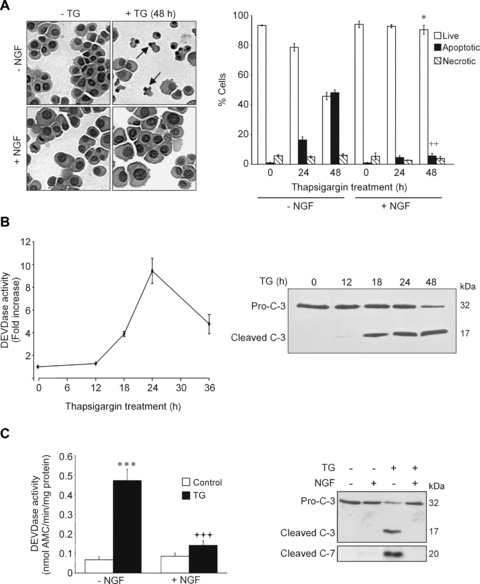
Pre-treatment with nerve growth factor (NGF) prevents thapsigargin (TG)-induced cell death in PC12 cells. (A), PC12 cells were treated with NGF (100 ng/ml) for 2 hrs prior to exposure to 1.5 μM TG for 24 and 48 hrs. Left hand panel: Cytocentrifuge preparations stained with haematoxylin and eosin. Arrows indicate apoptotic nuclei. Right hand panel: The proportion of apoptotic and necrotic cells was calculated as a percentage of the total number of cells. Values represent the mean ± SEM of three separate determinations. Statistical analysis was performed with repeated measures anova followed by Tukey—Kramer post hoc test ++P< 0.01 versus apoptosis at 48 hrs in the absence of NGF, *P< 0.05 versus live cells at 48 hrs in the absence of NGF. (B) PC12 cells were treated with 1.5 μM TG for 0–36 hrs and DEVD-AMC cleavage activity was measured in whole cell extracts (left hand graph). The fold increase in activity as compared with untreated cells is shown. Values are means ± SEM of three separate determinations. In the right hand panel the Western blot shows proteolytic processing of caspase-3. Pro-caspase-3 (Pro-C-3; 32 kD) and cleaved caspase-3 (17 kD) are indicated. (C) NGF blocks TG-mediated caspase activation. PC12 cells were treated with NGF (100 ng/ml) for 2 hrs prior to exposure to 1.5 μM TG for 24 hrs. DEVD-AMC cleavage activity (left hand panel) was measured. Values shown are means ± SEM of five separate determinations. Statistical analysis was performed with repeated measures anova followed by Tukey—Kramer multiple comparisons post hoc test. ***P < 0.001 versus control cells in the absence of NGF, +++P< 0.001 versus TG-treatment in the absence of NGF. Proteolytic processing of pro-caspase-3 and pro-caspase-7 was determined by Western blotting (right hand panel).
Since NGF has been reported to down-regulate the SERCA pump [24] and thus may alter the ability of TG to cause ER stress, we investigated whether NGF had any effect on the UPR. TG exposure caused a time-dependent induction of the ER chaperone Grp78/BiP (a hallmark of UPR activation) (Fig.2A, Supplementary Fig. 1A). In addition to Grp78, we chose specific target molecules for each of the three pathways of the UPR. XBP1 and spliced XBP1 (sXBP1) were examined to show the co-ordinated action of ATF6 and Ire1 and GADD34 was examined to show activation of PERK [2, 25] (Fig.2B and C, Supplementary Fig. 2A and B). The effect of NGF on the TG-mediated activation of these genes was examined during the onset of the UPR (1–4 hrs treatment) (Fig.2B, Supplementary Fig. 2A) as well as at later times during ER stress (3–24 hrs) (Fig.2C, Supplementary Fig. 2B). TG induced all of these UPR markers, however NGF pre-treatment exhibited no effect on the regulation of any of these UPR-specific genes eitherat the early or the late stages of the UPR (Fig.2A–C, Supplementary Fig. 2A and B).
Figure 2.
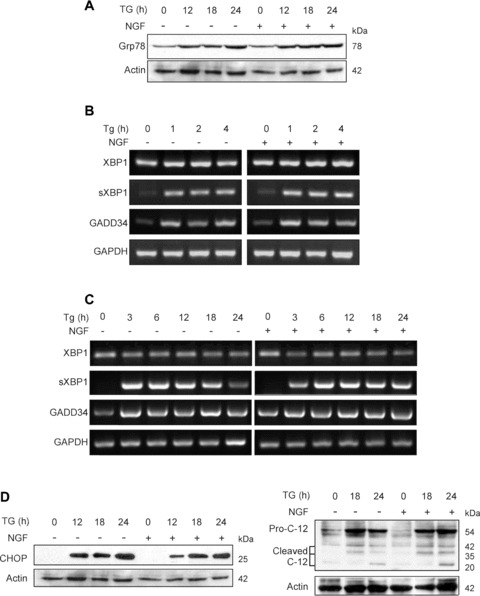
NGF does not affect the onset of unfolded protein response (UPR) induced by TG. PC12 cells were treated with NGF (100 ng/ml) for 2 hr prior to exposure to 1.5 μM TG for the times indicated. (A) Western blot analysis of Grp78 expression. The levels of actin expression were also analysed and used as loading control. (B and C) RT-PCR analysis of UPR markers. Total RNA was extracted, converted to cDNA and RT-PCR analysis of UPR markers (XBP1, spliced XBP1 [sXBP1] and GADD34) was performed. GAPDH signal was also determined and used as loading control. (D) Expression of proapoptotic endoplasmic reticulum (ER) stress markers CHOP and caspase-12 analysed by Western blotting. Procaspase-12 (Pro-C-12) and the cleavage products are indicated. The levels of actin expression were also analysed and used as loading control.
We next hypothesized that NGF may prevent ER stress-induced apoptosis by selectively blocking ER stress-related events that are linked to the induction of apoptosis. Two such events are CHOP induction and caspase-12 processing [2]. The transcription factor CHOP was strongly induced by TG, however this was unaffected by pre-treatment with NGF (Fig.2D, Supplementary Fig. 1A). Similarly, TG treatment caused induction and processing of pro-caspase-12 which was unaffected by NGF pre-treatment (Fig.2D). Taken together, these data suggest that NGF acts at a point downstream of the ER in TG-induced apoptosis of PC12 cells.
NGF blocks the mitochondrial pathway to intervene in TG-induced apoptosis through inhibition of Bax translocation to the mitochondria
The next possible point of interference with the ER stress-induced apoptotic pathway is the mitochondria [26–28]. TG treatment induced pro-caspase-9 processing between 12 and 18 hrs of treatment, which was markedly reduced by pre-treat-ment with NGF (Fig.3A). This was not accompanied by any changes in the expression of Apaf-1 (Fig.3B, Supplementary Fig. 1B), suggesting that inhibition of caspase-9 activation was not due to down-regulation of Apaf-1 which has been reported to occur during NGF-mediated differentiation of sympathetic neurons over 7 days [29], but instead upstream of apoptosome formation. To this end, the effect of NGF on TG-induced loss of mitochondrial transmembrane potential (ΔΨm) and release of cytochrome cfrom mitochondria were investigated. Exposure to TG for 24 hrs caused a decrease in ΔΨm, which was markedly reduced by pre-treatment of the cells with NGF (Fig.3C). Furthermore, loss of ΔPsi;m was associated with the release of cytochrome c, which was also prevented by NGF pre-treatment (Fig.3D), suggesting that NGF blocks pro-caspase-9 processing by blocking outer mitochondrial membrane permeabilization and thus, cytochrome c release.
Figure 3.
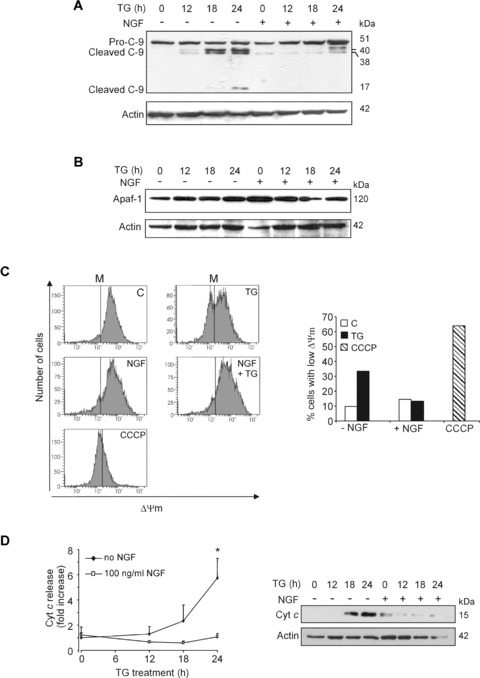
NGF blocks mitochondrial changes associated with TG-induced apoptosis. PC12 cells were treated with NGF (100 ng/ml) for 2 hrs prior to exposure to 1.5 μM TG for 0–24 hrs. (A) Processing of pro-caspase-9 and (B) levels of Apaf-1 determined by Western blotting. Pro-caspase-9 (51 kD) and the cleavage products (40, 38 and 17 kD) are indicated. The levels of actin expression were analysed for loading control. (C) Mitochondrial membrane potential (ΔΨm) measured with tetramethyl-rhodamine ethyl ester perchlorate (TMRE). The cells were exposed to TG for 24 hrs before harvesting. The mitochondrial uncoupler carbonyl cyanide 3-chlorophenylhydrazone (CCCP) (10 μM) was added to control cells 2 hrs prior to harvesting to act as a positive control for loss of ΔΨm. The vertical lines on the graphs (M) are markers to indicate cells with high and low ΔΨm. The right-hand graph shows the percentage of cells with low ΔΨm in each of the cell treatments. (D) Level of cytochrome c in the cytosolic cell fractions. Cells were lysed and the cytosolic fraction was isolated. The level of cytochrome c in the cytosols was analysed by Western blotting. The graph shows densitometric quantification normalized for expression levels of actin as averaged fold activation ± SD from three independent experiments. *P < 0.05 versus 100 ng/ml NGF. The right-hand side panel shows a representative Western blot.
Translocation of Bax to the mitochondria and oligomerization, causing formation of pores in the membrane that allow release of cytochrome c has been shown to be sufficient for commitment to apoptosis [30]. In order to study the effect of NGF on TG-mediated Bax translocation, PC12 cells were transfected with Bax-GFP and the subcellular localization of Bax was monitored in situ. All untreated cells displayed a diffuse Bax-GFP signal, indicative of cytoplasmic localization of Bax. After 24 hrs treatment with TG, 64 ± 3% of the Bax-GFP-positive cells displayed punctuate staining (indicating mitochondrial translocation of Bax) along with nuclear condensation and/or fragmentation (Fig.4A). This changed cellular distribution was specific to Bax; in cells transfected with pEGFP, TG treatment did not cause any change in the diffuse staining pattern of GFP (data not shown). Pre-treatment with NGF reduced the proportion of cells with punctate fluorescence staining and apoptotic nuclear morphology to 31 ± 3% (Fig.4A). Although the NGF-mediated protection was only partial, this was probably due to the fact that overexpression of Bax-GFP potentiated TG-induced apoptosis, reflected by a lower percentage of apoptotic morphology in the GFP-negative fraction of the same cultures (12 ± 2%, data not shown). Western blot analysis of Bax levels in TG ± NGF-treated mitochondrial cell fractions showed similar results (data not shown). These results indicate that NGF blocks TG-induced Bax translocation to the mitochondria and in this way prevents cytochrome c release and subsequent caspase activation.
Figure 4.
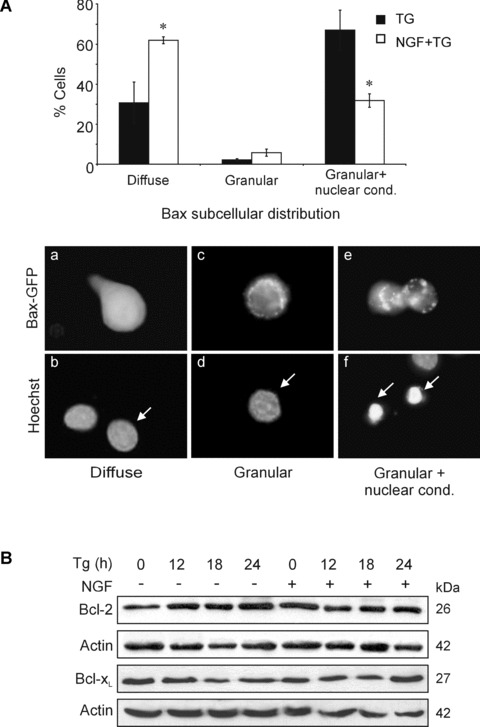
Regulation of anti-apoptotic and pro-apoptotic multi-domain Bcl-2 family members by TG and NGF. (A) Effect of NGF and TG on the subcellular localization of Bax-GFP. PC12 cells were transiently transfected with Bax-GFP. 24-hrs after transfection cells were treated with 1.5 μM TG for 24 hrs with or without 2 hrs pre-treatment with 100 ng/ml NGF. Cytospins of trypsinized and fixed cells were mounted with DAPI-contain-ing mounting medium to stain nuclei. The diffuse or granular pattern of GFP-positivity together with nuclear condensation/fragmentation (nuclear cond.) was examined and quantified. Values represent the mean ± SEM of three separate determinations. Statistical analysis was performed with repeated measures ANOVA followed by Student—Newman—Keuls post hoc test. *P< 0.05 versus absence of NGF. The panel of microscopic images under the graph shows representative cells displaying (a, b) diffuse Bax-GFP staining with no nuclear condensation/fragmentation, (c, d) granular Bax-GFP with no nuclear changes and (e, f) cells displaying granular Bax-GFP with nuclear condensation/ fragmentation. The arrows point to the nuclei of Bax-GFP positive cells. (B) PC12 cells were treated with NGF (100 ng/ml) for 2 hrs prior to exposure to 1.5 μM TG for 0–24 hrs. Whole cell lysates (25 μg/lane) were subjected to 12% SDS-PAGE followed by Western blotting and expression of Bcl-2 and Bcl-xL were determined. The levels of actin expression were also determined for loading control. The data demonstrated are representative of three separate experiments.
Since NGF treatment has been reported to promote survival through modulation of anti-apoptotic Bcl-2 family members [31, 32], we examined the expression of the two main anti-apoptotic Bcl-2 proteins, Bcl-2 and Bcl-xL as possible inhibitors of Bax-translocation. Immunoblotting showed that expression of Bcl-2 and Bcl-xL were not altered by TG treatment, either in the presence or absence of NGF, over the time course examined (0–24 hrs) (Fig.4B, Supplementary Fig. 1C).
NGF blocks TG-induced expression of Bim
Among the BH3-only members of the Bcl-2 family, Bad, Bik/Nbk, Bim and PUMA have been previously linked to ER stress [33], and therefore, their regulation by TG was examined. Exposure of PC12 cells to TG led to dephosphorylation of Bad on Ser136 detectable after 12 hrs (Fig.5A). However, pre-treatment with NGF did not prevent TG-induced dephosphorylation of Bad at Ser136 (Fig.5A ). Bim, PUMA and Bik are primarily regulated transcriptionally [9], therefore the effect of TG on their expression was examined using RT-PCR. Of the three genes, Bik expression was unaltered, PUMA mRNA levels were slightly increased and the three major splice variants of Bim: Bim extra long (Bimel), Bim long (BimL) and Bim short (BimS) were all strongly induced by TG treatment in a time-dependent manner (Fig.5B, Supplementary Fig. 2C).
Figure 5.
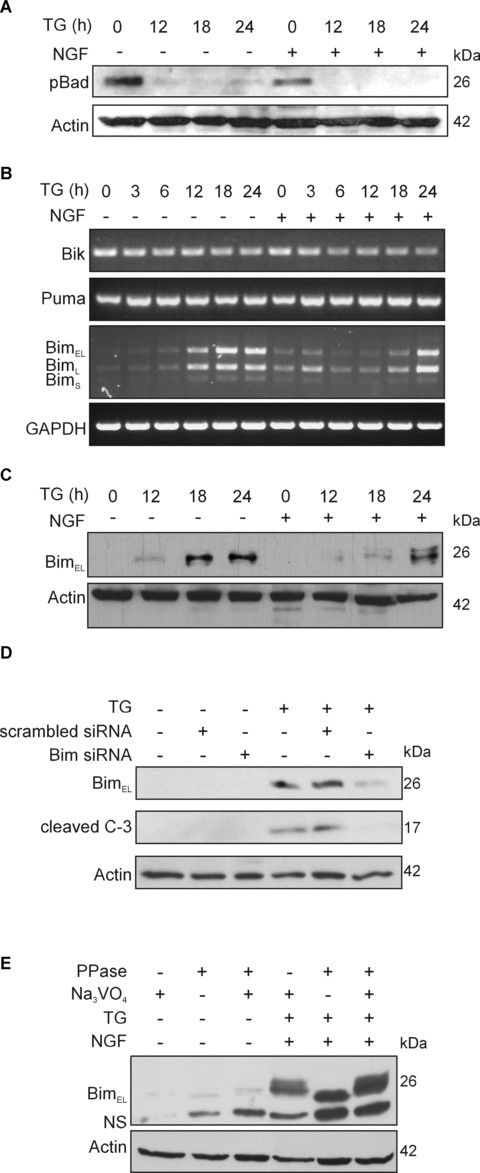
Regulation of BH3-only proteins by TG and NGF. PC12 cells were treated with NGF (100 ng/ml) for 2 hrs prior to exposure to 1.5 μM TG for 0–24 hrs. (A) Effect of TG and NGF on Bad phosphorylation. Phosphorylation of Bad on Ser136 (pBad) was determined in whole cell lysates (25 μg protein/lane) subjected to 12% SDS-PAGE followed by Western blotting. The levels of actin expression were also determined and used as loading control. (B) Expression of Bik, Puma and Bim mRNA after exposure to TG and NGF. Total RNA was isolated and converted to cDNA from which the Bik, Puma and Bim cDNA was amplified by PCR. Expression of GAPDH mRNA was detected for loading control. The three splice variants of Bim are indicated. (C) NGF blocks TG-mediated Bimel induction. Protein expression of Bim was determined in whole cell lysates (30 μg protein/lane) subjected to 12% SDS-PAGE followed by Western blotting. The levels of actin expression were also determined for loading control. (D) Bim is necessary for TG-induced caspase-3 activation. Bim expression was knocked down with siRNA and the cells were treated with 1.5 μM TG for 24 hrs. The effect of the knockdown on Bim expression and caspase-3 processing was analysed by Western blotting. Actin levels were detected to show equal protein loading. The figure shows a representative of two independent experiments. (E) NGF mediates Bimel phosphorylation. Control and NGF+TG-treated whole cell lysates were treated with 400 U lambda phosphatase (PPase) for 30 min. at 30°C in the presence or absence of sodium orthovanadate (10 mM) prior to detection of Bimel by Western blotting. The levels of actin in the samples were analysed as a loading control.
NGF did not affect TG-mediated induction of PUMA mRNA, but it significantly delayed the induction of all three Bim mRNA splice variants (Fig.5B, Supplementary Fig. 2C). The effect of NGF on Bim expression was confirmed by examining Bim protein levels using immunoblotting. Induction of Bimel protein by TG was detectable after 12 hrs of treatment (Fig.5C, Supplementary Fig. 1D). The other two splice variants of Bim were not detectable by immunoblotting. Furthermore, Bim was found to be necessary for TG-induced apoptosis, as knockdown of Bim with siRNA prevented TG-induced pro-caspase-3 processing (Fig.5D). Pre-treatment of the cells with NGF caused delayed and reduced induction of Bimel protein in response to TG and also elicited the appearance of a higher molecular weight form of the protein, suggestive of Bimel phosphorylation (Fig.5C). Treatment of cell lysates with λ-phosphatase prior to SDS-PAGE resulted in disappearance of the upper band that could be blocked by co-incubation with the phosphatase inhibitor sodium orthovanadate, demonstrating that this higher molecular weight band is in fact a phosphorylated form of Bimel (Fig.5E).
NGF-induced cytoprotection against TG is dependent on PI3K signalling
NGF is known to activate multiple kinase pathways [34]. In two separate publications, PI3K/Akt signalling [15], but not MAPK signalling [17], have been shown to be involved in NGF protection against TG. Using the MTT viability assay, we tested a range of kinase inhibitors, and found that the PI3K inhibitor LY294002, but not the MAPK inhibitor U0126, reversed the protective effects of NGF against TG, while inhibition of Jun N-terminal Kinase (JNK), protein kinase C or hexokinase translocation to the mitochondria had no effect (data not shown). In contrast, the non-specific kinase inhibitor staurosporine reversed NGF-dependent protection (Supplementary Fig. 3A and B).
We further examined whether PI3K signalling was involved in NGF-mediated inhibition of caspase activation. NGF-mediated inhibition of TG-induced DEVDase activity was reversed by LY294002 in a dose-dependent manner (Fig.6A). This was accompanied by reappearance of the p17 active fragment of caspase-3 upon treatment with LY294002 (Fig.6B). The effect of NGF on TG-induced loss of ΔΨm was also reversed by PI3K inhibition, demonstrating that mitochondrial changes were also dependent on PI3K signalling (Fig.6C). Furthermore, the inhibitory effect of NGF on TG-mediated Bimel induction was reduced by pre-treating the cells with LY294002 indicating that the effect of NGF on Bimel protein is also dependent on PI3K signalling (Fig.6D, Supplementary Fig. 1D). Notably, LY294002 alone caused a mild induction of Bimel protein, which is probably due to the reduction of basal PI3K/Akt activity and has previously been reported [35, 36]. At the same time, LY294002 treatment alone did not induce caspase activation or apoptotic morphology (Fig.6A and morphology data not shown). Staurosporine, which reversed NGF cytoprotection, also reversed the effect of NGF on Bimel protein (Supplementary Fig. 3C). In common with LY294002, stau-rosporine also reduced Akt phosphorylation (Supplementary Fig. 3D), which may be due to PI3K inhibition by staurosporine [37].
Figure 6.
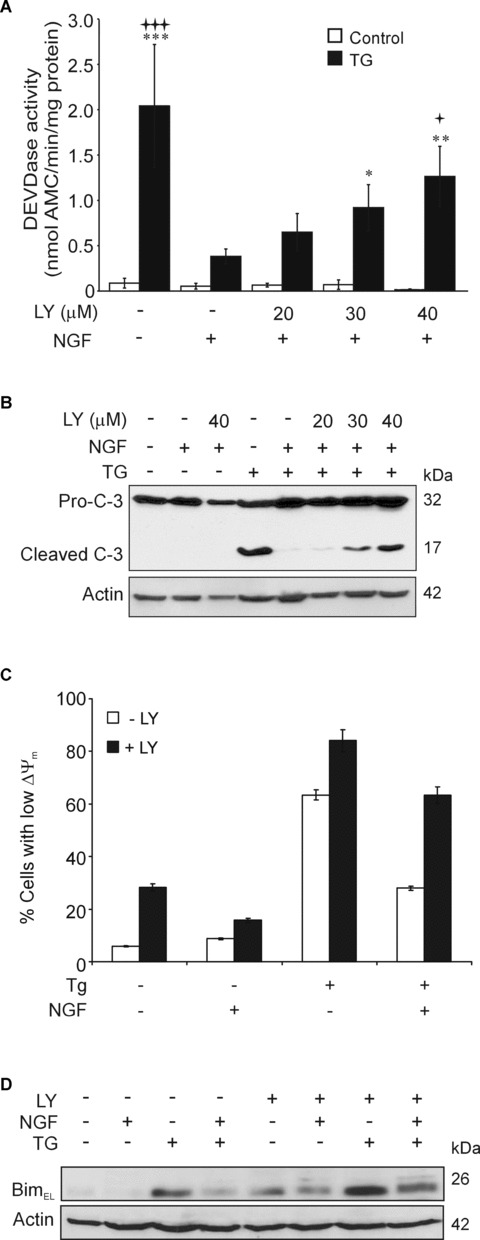
Role of PI3K signalling in NGF-mediated protection against TG. (A) Effect of LY294002 on NGF-mediated block of TG-induced caspase activation. DEVDase activity in PC12 cells pre-treated with 20, 30 or 40 μM LY294002 for 1 hr before treating with NGF (100 ng/ml) for 2 hrs and 1.5 μM TG for 24 hrs. Values are means ± SEM of three separate determinations. Statistical analysis was performed with repeated measures anova followed by Tukey—Kramer multiple comparisons post hoc test ***P< 0.001, **P<0.01, *P<0.05 versus control cells in the absence of NGF or LY294002, +++P< 0.001, +P< 0.05 versus TG treatment in the presence of NGF and absence of LY294002. (B) Effect of LY294002 on NGF-mediated block of TG-induced caspase-3 processing. Cells were pretreated with 20–40 μM LY294002 for 1 hr before treating with NGF (100 ng/ml) for 2 hrs and 1.5 μM TG for 24 hrs. Pro-caspase-3 processing was detected by Western blotting. Actin expression is shown as a loading control. (C) LY294002 reverts the NGF-mediated block of mitochondrial depolarization induced by TG. Cells were treated with 40 μM LY294002 for 1 hr prior to NGF for 2 hrs and TG for a further 36 hrs and the percentage of cells with depolarized mitochondria (low ΔΨm) was measured with TMRE. (D) Effect of the PI3K inhibitor LY294002 on Bimel expression. Cells were treated with 40 μM LY294002 for 1 hr prior to consecutive treatment with NGF (100 ng/ml, 2 hrs) and 1.5 μM TG for 18 hrs. The expression of Bimel protein was analysed in whole cell lysates by Western blotting. The levels of actin in the samples were analysed as a loading control.
Discussion
Impaired ER function is an important factor in a variety of neurodegenerative disorders including Alzheimer's disease, Parkinson's disease and ischaemia [1]. A number of recent reports show that NGF can protect PC12 cells from ER stress-induced apoptosis, however, the mechanism is not understood [15, 17, 38]. At least one report suggests that NGF blocks tunicamycin-induced apoptosis via reduced processing of caspase-12 [38]. This supports a number of studies that report an important role for caspase-12 in ER stress-induced apoptosis as the initiator caspase [3–5]. However, other studies have reported a requirement for the mitochondrial pathway and apoptosome formation for caspase activation and execution of death during ER stress [26–28, 39].
It is obvious from the present study that TG-induced apoptosis in PC12 cells involves the mitochondrial pathway (Fig.7). NGF protection does not affect activation or duration of the UPR or activation of pro-apoptotic responses that arise directly from the ER, that is, CHOP induction and caspase-12 induction and processing. These data support and extend an earlier ‘snapshot’ study that showed NGF does not affect TG-induced increase in Grp78 at 6 hrs, or CHOP and nuclear XBP1 at 24 hrs [40]. However, in contrast to our findings, the Mao study showed that NGF partly reversed TG-induced pro-caspase-12 processing [40]. In contrast, NGF protection against TG-induced apoptosis in PC12 cells involved inhibition of the mitochondrial apoptosis pathway. NGF exerted this effect by preventing Bax translocation, release of cytochrome c from the mitochondria and activation of caspases-9 and -3. These effects were linked to regulation of Bimel levels and phosphorylation that involved NGF-mediated activation of PI3K.
Figure 7.
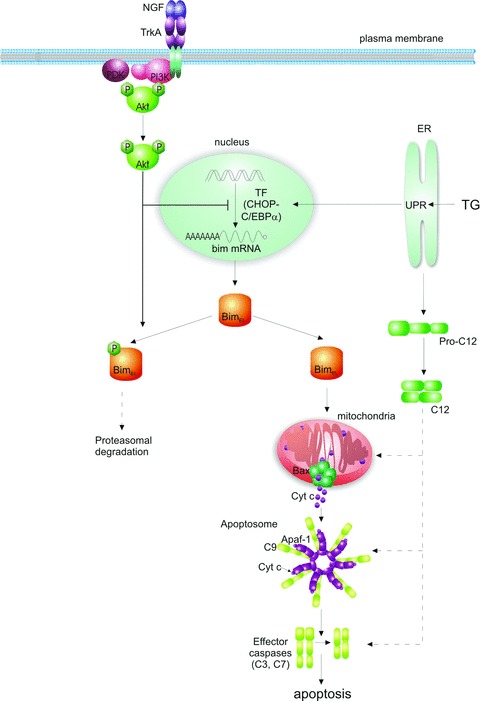
Schematic diagram showing proposed model of NGF inhibition of ER stress-induced apoptosis. Prolonged ER stress induces the proapoptotic transcription factor (TF) CHOP (C/EBP homologous protein), and processing of pro-caspase-12 (pro-C12). CHOP, together with its heterodimeric partner, C/EBPα (CCAAT/enhancer-binding protein), initiates the transcription of the bim gene. From the different splice mRNA variants, the extra long form (Bimel) is the predominantly translated. Bimel accumulation in the cell leads to translocation of Bax to the mitochondria where it forms oligomeric megachannels in the outer mitochondrial membrane, through which cytochrome c is released. In the cytosol, cytochrome c (Cyt c) triggers assembly of the apoptosome complex (comprising cytochrome c, Apaf-1, pro-caspase-9 and dATP). This results in activation of the initiator caspase-9 (C9), followed by effector caspases-3 and -7 (C3, C7) and execution of apoptosis. This pathway can be blocked by NGF, which binds to TrkA receptors on the cell surface, causing their dimerization and autophosphorylation. This leads to activation of the phosphatidylinositol 3-kinase (PI3K)/Akt kinase pathway. PI3K/Akt blocks the ER stress-mediated apoptotic-signalling pathway by regulating Bim expression in two ways. PI3K/Akt attenuates transcription of bim (without affecting CHOP induction). PI3K/Akt can also phosphorylate the Bimel protein, leading to its inactivation and possibly proteasomal degradation.
The pro-apoptotic effects of TG were linked to Bimel induction, and knockdown of Bim using siRNA reduced TG-induced apoptosis. Recently, induction of Bimel has been shown to be essential for ER stress-induced apoptosis in diverse cell types, including thymocytes, MCF-7 breast carcinoma and Vero African green monkey kidney epithelial cells [41], although this may be cell type-dependent [42]. NGF is known to regulate Bimel levels during trophic factor deprivation-induced apoptosis [43]. Recent studies of the proximal Bim promoter show that c-jun, FoxO and Mybs are all involved in NGF deprivation-induced bim transcription [44]. In contrast, induction of Bimel during ER stress has been shown to require CHOP, but whether NGF can control ER stress-mediated Bim induction has not been explored [41]. In our study, we show that NGF pre-treatment significantly reduced TG-induced Bimel mRNA and protein levels. However, it appeared to be independent of CHOP induction by TG. This can be explained by the finding that although CHOP is essential for ER stress-induced bim gene transcription, it is not sufficient [41]. In fact, heterodimeric CHOP-CCAAT/enhancer-binding protein α (C/EBPα) has been shown to up-regulate bim transcription [41]. It is noteworthy that PI3K/Akt signalling can inhibit C/EBPα transcriptional activity [45, 46] and that NGF protection against TG as well as regulation of Bim was dependent on PI3K activity [15] and present data). Thus, NGF may reduce Bim induction by PI3K/Akt-dependent inhibition of c/EBPα transactivation activity, rather than inhibition of CHOP induction.
TG induced mainly the dephosphorylated form of Bimel and NGF pre-treatment lead to a decrease in bim mRNA and Bimel protein levels, with mainly phosphorylated Bimel being expressed. These data suggest a dual effect of NGF on TG-induced Bimel, through reduction in bim transcription and promotion of the phosphorylation of Bimel protein. Phosphorylation of Bimel protein is known to affect the stability of the protein, as well as its pro-apoptotic function [43, 47]. ERK-dependent phosphorylation has been shown to target Bimel for ubiquitination and proteasomal degradation [43, 47]. A previous report showed that a deletion mutant of Bimel lacking ERK phosphorylation sites, Ser109 and Thr110, retained a mobility shift in response to NGF in PC12 cells [43]. In addition, there is also some recent evidence that Bimel can be directly phosphorylated by Akt [48]. Thus, NGF could lead to an Akt-dependent phosphorylation of Bimel. The effect of such phosphorylation is unknown, but an attractive hypothesis is that it targets Bimel for degradation in a manner similar to ERK-mediated phosphorylation, or that it at least reduces the pro-apoptotic potential of Bimel (Fig.7).
With regard to other Bcl-2 family members, the present study did not reveal any alteration in the expression of Bcl-2 or Bcl-xL in PC12 cells. This is in contrast to some [31, 32] and in agreement with other studies [49, 50]. TG was, however, found to activate the BH3-only protein Bad by initiating its dephosphorylation on Ser136. Along with Bim, Bad has been shown to sequester anti-apoptotic Bcl-2 proteins, and cause cytochrome c release [51]. Bad dephosphorylation may be mediated by calcineurin, activated by Ca2+ released from the ER [52]. However, in agreement with some other studies, NGF promotes survival independently of Bad phosphorylation on Ser136 [53]. Together, these results suggest that NGF-dependent survival signalling is downstream, or independent, of Bad dephosphorylation and point to Bimel as the key BH3-only protein in initiating apoptosis by TG.
In summary, these data indicate that TG-induced apoptosis in PC12 cells involves transcriptional induction of bim leading to translocation of Bax to the mitochondria and activation of the mito-chondrial pathway. NGF-dependent protection targets mitochondr-ial changes associated with apoptosis without inhibiting induction of the UPR by TG (Fig.7). NGF protection is dependent on PI3K signalling and involves attenuation of Bimel levels in the cell. This ability of NGF to regulate Bimel levels induced by ER stress, is in addition to its previously reported ability to block bim induction by trophic factor withdrawal. This study points to Bim as a key molecule in ER stress-induced apoptosis that can be regulated by the neurotrophin NGF. Thus, it may warrant further investigation in neurodegenerative diseases where ER stress is a factor in neuronal cell death. Markers of the UPR have been observed in post-mortem brain samples from patient's with Alzhemier's and Parkinson's diseases [54, 55]. In trinucleotide-repeat disorders such as Huntington's disease, ER stress-induced neuronal death is triggered by expanded poly-glutamine repeats [56]. The pro-survival abilities of neurotrophic factors in these diseases may be linked to their ability to reduce ER stress-induced cell death.
Acknowledgments
We would like to thank Prof. Jochen Prehn (Dept. Physiology, Royal College of Surgeons, Dublin, Ireland) for providing the GFP-tagged Bax expression vector and Aoife O’Reilly for technical assistance. This work was financially supported by the Higher Education Authority of Ireland, (PRTLI III), the Health Research Board of Ireland, the National University of Ireland, Galway Millenium Research Fund and Science Foundation of Ireland. Kate Reed Herbert was supported by an NUI, Galway fellowship. Edel Kavanagh is supported by EMBARK scholarship from the Irish Research Council for Science, Engineering and Technology.
Supporting Information
Figure S1. Densitometric quantification of proteininduction. The band intensities of proteins run on SDS-PAGE gelwere determined densitometrically. The band intensities werecorrected for background signal and then normalized to actin signalintensity of the same samples. Protein induction is represented asaverage foldinduction compared to the control ± S.E.M.(A) Induction of ER stress markers, CHOP and Grp78,following ER stress in the absence and presence of NGF. (B)Induction of Apaf-1 during ER stress in the absence and presence ofNGF. (C) Induction of antiapoptotic Bcl-2 proteins, Bcl-2and Bcl-xL, during ER stress in the absence and presenceof NGF. (D) Effect of LY 294002 on the induction of BimELduring ER stress in the absence and presence of NGF.
Figure S2. Densitometric quantification of geneinduction. The band intensities of RT-PCR products run on agarosegel were determined densitometrically. The band intensities werecorrected for background signal and then normalized to GAPDH signalintensity of the same samples. Gene induction is represented asaverage fold-induction compared to the control ± S.E.M.(A) Induction of UPR genes at initial stages of ER stress inthe absence and presence of NGF. (B) Induction of UPR genesat late stages of ER stress in the absence and presence of NGF.(C) Induction of BH3-only genes during ER stress in theabsence and presence of NGF.
Figure S3. STS reverts the protective action of NGF. A,DEVDase activity in PC12 cells that were pre-treated for 1 h with10 nM STS before treating with NGF (100 ng/ml) for 2 h and 1.5μM TG for 24 h. DEVD-AMC cleavage activity was measured in wholecell extracts. Values are means ± SEM of 3 separatedeterminations. B, Processing of pro-caspase-3 in the same samplesanalysed by Western blotting. The levels of actin in the samplesare shown as loading control. C, Effect of STS on BimEL expression.STS (10 nM) was added for 1 h prior to treatment with NGF (100ng/ml, 2 h) and 1.5 μM TG for 18 h. Expression of BimEL wasdetermined by Western blotting. The levels of actin in the sampleswere also analyzed as a loading control. D, Effect of LY294002 (LY)or STS (STS) on Akt phosphorylation. Cells were treated withLY294002 (40 μM) or STS (10 nM) for 1 h prior to addition of NGF(100 ng/ml, 24 h). Expression of p-Akt was determined by Westernblotting. The levels of actin in the samples were also detected forloading control.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
References
- 1.Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–92. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- 2.Szegezdi E, Logue SE, Gorman AM, Samali A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006;7:880–5. doi: 10.1038/sj.embor.7400779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–94. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- 4.Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, Bredesen DE, Ellerby HM. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–42. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- 5.Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357:127–30. doi: 10.1016/j.neulet.2003.12.080. [DOI] [PubMed] [Google Scholar]
- 6.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 7.Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates pro-caspase-9. J Biol Chem. 1999;274:11549–56. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]
- 8.Cory S, Adams JM. The Bcl2 family: regulators of the cellular life-or-death switch. Nat Rev Cancer. 2002;2:647–56. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 9.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–25. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galonek HL, Hardwick JM. Upgrading the BCL-2 Network. Nat Cell Biol. 2006;8:1317–9. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan DR, Miller FD. Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol. 1997;9:213–21. doi: 10.1016/s0955-0674(97)80065-8. [DOI] [PubMed] [Google Scholar]
- 12.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–22. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mesner PW, Winters TR, Green SH. Nerve growth factor withdrawal-induced cell death in neuronal PC12 cells resembles that in sympathetic neurons. J Cell Biol. 1992;119:1669–80. doi: 10.1083/jcb.119.6.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salinas M, Diaz R, Abraham NG, Ruiz de Galarreta CM, Cuadrado A. Nerve growth factor protects against 6-hydroxy-dopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-depend-ent manner. J Biol Chem. 2003;278:13898–904. doi: 10.1074/jbc.M209164200. [DOI] [PubMed] [Google Scholar]
- 15.Shimoke K, Kishi S, Utsumi T, Shimamura Y, Sasaya H, Oikawa T, Uesato S, Ikeuchi T. NGF-induced phos-phatidylinositol 3-kinase signaling pathway prevents thapsigargin-triggered ER stress-mediated apoptosis in PC12 cells. Neurosci Lett. 2005;389:124–8. doi: 10.1016/j.neulet.2005.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Shimoke K, Utsumi T, Kishi S, Nishimura M, Sasaya H, Kudo M, Ikeuchi T. Prevention of endoplasmic reticulum stress-induced cell death by brain-derived neurotrophic factor in cultured cerebral cortical neurons. Brain Res. 2004;1028:105–11. doi: 10.1016/j.brainres.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 17.Takadera T, Ohyashiki T. Apoptotic cell death and CPP32-like activation induced by thapsigargin and their prevention by nerve growth factor in PC12 cells. Biochim Biophys Acta. 1998;1401:63–71. doi: 10.1016/s0167-4889(97)00116-x. [DOI] [PubMed] [Google Scholar]
- 18.Kavanagh ET, Loughlin JP, Herbert KR, Dockery P, Samali A, Doyle KM, Gorman AM. Functionality of NGF-protected PC12 cells following exposure to 6-hydroxy-dopamine. Biochem Biophys Res Commun. 2006;351:890–5. doi: 10.1016/j.bbrc.2006.10.104. [DOI] [PubMed] [Google Scholar]
- 19.Yao R, Cooper GM. Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science. 1995;267:2003–6. doi: 10.1126/science.7701324. [DOI] [PubMed] [Google Scholar]
- 20.Crowder RJ, Freeman RS. Phosphatidylinositol 3-kinase and Akt protein kinase are necessary and sufficient for the survival of nerve growth factor-dependent sympathetic neurons. J Neurosci. 1998;18:2933–43. doi: 10.1523/JNEUROSCI.18-08-02933.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–60. doi: 10.1038/350158a0. [DOI] [PubMed] [Google Scholar]
- 22.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc Natl Acad Sci USA. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gorman AM, Szegezdi E, Quigney DJ, Samali A. Hsp27 inhibits 6-hydroxy-dopamine-induced cytochrome c release and apoptosis in PC12 cells. Biochem Biophys Res Commun. 2005;327:801–10. doi: 10.1016/j.bbrc.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 24.Keller D, Grover AK. Nerve growth factor treatment alters Ca2+ pump levels in PC12 cells. Neuroreport. 2000;11:65–8. doi: 10.1097/00001756-200001170-00013. [DOI] [PubMed] [Google Scholar]
- 25.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–91. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 26.Smith MI, Deshmukh M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007;14:1011–1019. doi: 10.1038/sj.cdd.4402089. [DOI] [PubMed] [Google Scholar]
- 27.Zhang D, Armstrong JS. Bax and the mito-chondrial permeability transition cooperate in the release of cytochrome c during endoplasmic reticulum-stress-induced apoptosis. Cell Death Differ. 2007;14:703–715. doi: 10.1038/sj.cdd.4402072. [DOI] [PubMed] [Google Scholar]
- 28.Boya P, Cohen I, Zamzami N, Vieira HL, Kroemer G. Endoplasmic reticulum stress-induced cell death requires mitochondrial membrane permeabilization. Cell Death Differ. 2002;9:465–7. doi: 10.1038/sj.cdd.4401006. [DOI] [PubMed] [Google Scholar]
- 29.Wright KM, Linhoff MW, Potts PR, Deshmukh M. Decreased apoptosome activity with neuronal differentiation sets the threshold for strict IAP regulation of apoptosis. J Cell Biol. 2004;167:303–13. doi: 10.1083/jcb.200406073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005;24:2096–103. doi: 10.1038/sj.emboj.7600675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Katoh S, Mitsui Y, Kitani K, Suzuki T. The rescuing effect of nerve growth factor is the result of up-regulation of bcl-2 in hyperoxia-induced apoptosis of a subclone of pheochromocytoma cells, PC12h. Neurosci Lett. 1997;232:71–4. doi: 10.1016/s0304-3940(97)00582-x. [DOI] [PubMed] [Google Scholar]
- 32.Bui NT, Livolsi A, Peyron JF, Prehn JH. Activation of nuclear factor kappaB and Bcl-x survival gene expression by nerve growth factor requires tyrosine phospho-rylation of IkappaBalpha. J Cell Biol. 2001;152:753–64. doi: 10.1083/jcb.152.4.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morishima N, Nakanishi K, Tsuchiya K, Shibata T, Seiwa E. Translocation of Bim to the Endoplasmic Reticulum (ER) Mediates ER Stress Signaling for Activation of Caspase-12 during ER Stress-induced Apoptosis. J Biol Chem. 2004;279:50375–81. doi: 10.1074/jbc.M408493200. [DOI] [PubMed] [Google Scholar]
- 34.Patapoutian A, Reichardt LF. Trk receptors: mediators of neurotrophin action. Curr Opin Neurobiol. 2001;11:272–80. doi: 10.1016/s0959-4388(00)00208-7. [DOI] [PubMed] [Google Scholar]
- 35.Stahl M, Dijkers PF, Kops GJ, Lens SM, Coffer PJ, Burgering BM, Medema RH. The forkhead transcription factor FoxO regulates transcription of p27Kip1 and Bim in response to IL-2. J Immunol. 2002;168:5024–31. doi: 10.4049/jimmunol.168.10.5024. [DOI] [PubMed] [Google Scholar]
- 36.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr Biol. 2000;10:1201–4. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 37.Walker EH, Pacold ME, Perisic O, Stephens L, Hawkins PT, Wymann MP, Williams RL. Structural determinants of phosphoinositide 3-kinase inhibition by wortmannin, LY294002, quercetin, myricetin, and staurosporine. Mol Cell. 2000;6:909–19. doi: 10.1016/s1097-2765(05)00089-4. [DOI] [PubMed] [Google Scholar]
- 38.Shimoke K, Amano H, Kishi S, Uchida H, Kudo M, Ikeuchi T. Nerve growth factor attenuates endoplasmic reticulum stress-mediated apoptosis via suppression of caspase-12 activity. J Biochem (Tokyo) 2004;135:439–46. doi: 10.1093/jb/mvh053. [DOI] [PubMed] [Google Scholar]
- 39.Elyaman W, Terro F, Suen KC, Yardin C, Chang RC, Hugon J. BAD and Bcl-2 regulation are early events linking neuronal endoplasmic reticulum stress to mitochondria-mediated apoptosis. Brain Res Mol Brain Res. 2002;109:233–8. doi: 10.1016/s0169-328x(02)00582-x. [DOI] [PubMed] [Google Scholar]
- 40.Mao W, Iwai C, Keng PC, Vulapalli R, Liang CS. Norepinephrine-induced oxidative stress causes PC-12 cell apoptosis by both endoplasmic reticulum stress and mitochondrial intrinsic pathway: inhibition of phosphatidylinositol 3-kinase survival pathway. Am J Physiol Cell Physiol. 2006;290:C1373–84. doi: 10.1152/ajpcell.00369.2005. [DOI] [PubMed] [Google Scholar]
- 41.Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND, Hughes PD, Michalak EM, McKimm-Breschkin J, Motoyama N, Gotoh T, Akira S, Bouillet P, Strasser A. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337–49. doi: 10.1016/j.cell.2007.04.027. [DOI] [PubMed] [Google Scholar]
- 42.Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–97. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Biswas SC, Greene LA. Nerve growth factor (NGF) down-regulates the Bcl-2 homology 3 (BH3) domain-only protein Bim and suppresses its proapoptotic activity by phosphorylation. J Biol Chem. 2002;277:49511–6. doi: 10.1074/jbc.M208086200. [DOI] [PubMed] [Google Scholar]
- 44.Biswas SC, Shi Y, Sproul A, Greene LA. Pro-apoptotic bim induction in response to NGF deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem. 2007;282:29368–74. doi: 10.1074/jbc.M702634200. [DOI] [PubMed] [Google Scholar]
- 45.Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA. Liver tumors escape negative control of proliferation via PI3K/Akt-mediated block of C/EBP alpha growth inhibitory activity. Genes Dev. 2004;18:912–25. doi: 10.1101/gad.1183304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Datta J, Majumder S, Kutay H, Motiwala T, Frankel W, Costa R, Cha HC, MacDougald OA, Jacob ST, Ghoshal K. Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3-kinase signaling cascade. Cancer Res. 2007;67:2736–46. doi: 10.1158/0008-5472.CAN-06-4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ley R, Ewings KE, Hadfield K, Cook SJ. Regulatory phosphorylation of Bim: sorting out the ERK from the JNK. Cell Death Differ. 2005;12:1008–14. doi: 10.1038/sj.cdd.4401688. [DOI] [PubMed] [Google Scholar]
- 48.Qi XJ, Wildey GM, Howe PH. Evidence that Ser87 of Bimel is phosphorylated by Akt and regulates Bimel apoptotic function. J Biol Chem. 2006;281:813–23. doi: 10.1074/jbc.M505546200. [DOI] [PubMed] [Google Scholar]
- 49.Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–94. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eves EM, Xiong W, Bellacosa A, Kennedy SG, Tsichlis PN, Rosner MR, Hay N. Akt, a target of phosphatidylinositol 3-kinase, inhibits apoptosis in a differentiating neu-ronal cell line. Mol Cell Biol. 1998;18:2143–52. doi: 10.1128/mcb.18.4.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–58. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 52.Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science. 1999;284:339–43. doi: 10.1126/science.284.5412.339. [DOI] [PubMed] [Google Scholar]
- 53.Kennedy SG, Kandel ES, Cross TK, Hay N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol Cell Biol. 1999;19:5800–10. doi: 10.1128/mcb.19.8.5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoozemans JJ, van Haastert ES, Eikelenboom P, de Vos RA, Rozemuller JM, Scheper W. Activation of the unfolded protein response in Parkinson's disease. Biochem Biophys Res Commun. 2007;354:707–11. doi: 10.1016/j.bbrc.2007.01.043. [DOI] [PubMed] [Google Scholar]
- 55.Hoozemans JJ, Veerhuis R, Van Haastert ES, Rozemuller JM, Baas F, Eikelenboom P, Scheper W. The unfolded protein response is activated in Alzheimer's disease. Acta Neuropathol. 2005;110:165–72. doi: 10.1007/s00401-005-1038-0. [DOI] [PubMed] [Google Scholar]
- 56.Nishitoh H, Matsuzawa A, Tobiume K, Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A, Ichijo H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–55. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item