

Abstract
p97/VCP, a member of the AAA-ATPase super family, has been associated with a wide variety of essential cellular protein pathways com prising: (i) nuclear envelope reconstruction, (ii) cell cycle, (iii) Golgi reassembly, (iv) suppression of apoptosis and (v) DNA-damage response [1-6]. In addition, vasolin-containing protein (VCP) dislodges the ubiquitinated proteins from the endoplasmic reticulum (ER) and chaperones them to the cytosol for proteasomal degradation by endoplasmic reticulum-associated degradation (ERAD) [7]. The interactions of VCP in the endoplasmic reticulum-associated degradation (ERAD) pathway determine the substrate selection for proteasomal degradation. Moreover, the interaction with VCP is also required for the ubiquitination of substrate. VCP is phosphorylated by the master cellular kinase, Akt as a mechanism to regulate ERAD [8]. These multiple interactions in protein degradation pathways points to central role of VCP in misfolded protein degradation. VCP has a poly-glutamine and ubiquitin-binding capacity and is involved in proteasomal degradation, cytosolic aggregation and processing of polyQ and polyUb aggregates in neurodegenerative and other misfolded protein diseases [9, 10]. Mutations in VCP gene are also linked to a protein deposition disorder, IBMFD [11]. We propose VCP as a therapeutic target for diseases caused by cytosolic protein aggregation or degradation of misfolded protein. We predict that selective interference of VCP interaction(s) with aberrant protein or its ERAD function will be an effective therapeutic site to rescue functional misfolded protein in diseases like cystic fibrosis and alpha-1-trypsin deficiency. The control of VCP expression is also proposed to be a potential therapeutic target in ex-polyQ-induced neurodegenerative diseases [12]. The further functional characterization of VCP and associated proteins in these diseases will help in designing of selective therapeutics.
Keywords: AAA ATPase, p97/VCP, ERAD, ubiquitination and therapeutics
Introduction
The AAA+ gene family (ATPase associated with diverse cellular activities) is characterized by conserved 200–250 residues that include Walker A and B motifs. The most common function of AAA+ domain is to catalyse protein folding or unfolding in an ATP-dependent manner. All known AAA+ proteins contain either 1 or 2 AAA+ domains, and the family can be divided into 2 groups on this basis. p97/VCP (valosin-containing protein), a member of the AAA-ATPase family with 2 AAA+ domains, is associated with diverse cellular functions comprising nuclear envelope recon-struction, cell cycle, post-mitotic Golgi reassembly, suppression of apoptosis, DNA damage response and endoplasmic reticulum-associated degradation (ERAD) [1–6].
In the eukaryotic cell secretory pathway, a significant proportion of unwanted proteins that enter the endoplasmic reticulum (ER) are specifically extracted from the ER and targeted to the cytosol, where they are degraded by the ERAD. In ERAD, protein ubiquitylation plays a role in both protein extraction from the ER and proteasome-mediated protein degradation. This crucial protein modification is mediated by a set of three enzymes: a ubiquitin-activating enzyme (E1), a ubiquitin-conjugating enzyme (E2) and a ubiquitin ligase (E3) [13]. VCP is known to associate with E3/E4 ubiquitin ligases like Dorfin [12], gp78/AMFR (autocrine motility factor receptor) [14, 15] and Rma1 (Ring finger protein with membrane anchor 1) [16, 17] to promote ERAD. Dorfin is a RING-IBR type ubiquitin ligase which ubiquitilates the superoxide dismutase (SOD1) in amyotrophic lateral sclerosis [12]. This interaction is required for the ubiquitin ligase function of Dorfin. While gp78 and Rma1 have been described to be involved in degradation of phenylalanine mutant of cystic fibrosis transmembrane regulator (ΔF508-CFTR) by our group and more recently by other groups [16–18]. The gp78-VCP complex is also known to be involved in degradation of other misfolded proteins like CD3delta, Z variant of alpha-1-trypsin and HMG CoA reductase, a rate-limiting enzyme in cholesterol synthesis [14, 19, 20]. VCP-gp78 interaction is known to enhance both ubiquitination and VCP-polyubiquitin binding during ERAD [14].
In addition to its pivotal role in ubiquitin-dependent protein degradation [7], VCP binds to expanded polyglutamine protein aggregates [9] and has been shown to have this polyglutamine protein binding capacity in Huntington's disease and spinocerebellar ataxia type III (SCA III) [9, 10]. Recently, Schroder et al.[11] presented a report of a patient with a VCP mutation leading to an inclusion body myopathy with frontotemporal dementia (IBMFD), a protein deposition disorder. While Klein et al.[8] demonstrated that VCP is phosphorylated by the anti-apoptotic affinity-regulating kinase (Akt) that regulates ERAD pathway. More recently, small VCP interacting protein (SVIP) has been identified as an endogenous inhibitor of VCP ERAD function [21, 22]. Taken together, (1) VCP has a key function in protein ubiquitilation in the ERAD pathway, thereby, being regulated through phosphorylation by Akt, endogenous regulator SVIP or through interaction with Dorfin/gp78/Rma1. These multiple interactions in protein degradation pathways points to an important role of VCP in protein degradation disorders such as amyotrophic lateral sclerosis and inclusion body myopathies. (2) VCP mutations can be responsible for a nuclear inclusion body type of frontotemporal dementia. (3) We and others recently showed that VCP specifically binds to mutant-CFTR protein as compared to wt-CFTR followed by its proteasome-mediated degradation [15, 23].
There is emerging evidence on association of VCP expression with chronic inflammatory pathology in diseases such as cystic fibrosis, alpha-1-antitrypsin deficiency and idiopathic pulmonary fibrosis (IPF) [24, 25]. The most common phenotype presenting with clinical evidence of alpha1-antitrypsin deficiency (AATD) is the Z phenotype (ATZ), with decreased levels of circulating AAT due to retention of the aberrantly folded protein in ER. Recent studies show that ATZ induces VCP and C/EBP-homologous protein (CHOP), components of ERAD [24]. Thus, it is tempting to speculate that VCP plays a pathogenic role in conformational or misfolded protein diseases like cystic fibrosis (CF) and AATD by inducing ERAD that may be an ER stress response to presence of misfolded protein. VCP is known to induce the NFkB-mediated chronic inflammatory signalling by presenting its endogenous inhibitor, IκB for proteasomal degradation [26]. We recently proposed that VCP is a novel target for not only rescuing misfolded protein from proteasomal degradation but also IkB, endogenous inhibitor of NFi<B-mediated chronic inflammation associated with chronic pathophysiology of these diseases [15]. Several recent reviews discuss the role of VCP in ERAD, apoptotic signalling and neurodegenerative disorders [1, 27, 28]. We discuss and provide here the perspective on cellular functions of VCP, its association with the disease and the proposed therapeutic potential.
Role of VCP in inflammatory signalling and ER stress
We observed overexpression of VCP protein in CF primary epithelial cells obtained at bronchoscopy from AF508 CF subjects as compared to non-CF controls [15]. VCP is known to be implicated in inflammation, with its mRNA expression elevated in type II A549 lung pneumocyte in response to Pseudomonas aeruginosa PAK strain (Geoprofile database, NCBI) infection. VCP expression is also proposed to be a marker for cancer metastasis. Overall prognosis of pancreatic, hepatocellular, gastric and prostrate cancer is proposed to exhibit metastatic function by VCP-mediated NFkB activation [29, 30]. Cell lines overex-pressing VCP demonstrate resistance to apoptosis, and increased metastatic potential [31]. The anti-apoptotic and pro-metatstatic effects of VCP occur via activation of the NFkB signalling pathway [31]; this pathway has seminal importance in pancreatic cancer and numerous reports confirm activation of NFkB signalling in the majority of cases [32–34]. We have preliminary data demonstrating that VCP inhibition not only rescues IkB (the endogenous inhibitor of NFkB) from proteasomal degradation but also independently suppresses NFkB activation [15, 35]. This ability of VCP inhibition to ameliorate NFKB-asso-ciated cell survival and proliferation signals harbours considerable promise for pancreatic and other forms of cancer.
The VCP expression in human CF bronchial epithelial cells, IB3–1, is also mediated by elevated NFkB levels and ER stress associated with the presence of misfolded protein (our unpublished data). We found that specific NFkB inhibitor, caffeic acid phenethyl ester (CAPE), significantly inhibits VCP protein levels. These results indicate that VCP expression is regulated by NFkB and on the contrary NFkB levels are regulated by VCP-dependent IkB degradation [26]. VCP is also involved in ubiquitylated body inclusions (UBIs) formation in neurodegenerative disorders, such as amyotrophic lateral sclerosis (ALS) and Parkinson's disease [12].
The VCP overexpression in these neurological disorders is again believed to be associated with chronic inflammation initiated by environmental toxins and genetic factors. The control of VCP expression is proposed to be a potential therapeutic target in ex-polyQ-induced neurodegenerative diseases [12]. The functional characterization of VCP and associated proteins in these diseases will help in defining the role of VCP-mediated NFkB regulation. Nonetheless, selective interference of VCP-IκB interaction to inhibit (not block) IkB-NFkB signalling has a potential to be translated as a therapeutic intervention.
Role of VCP in ERAD and protein aggregation
In addition to its association with Derlin1 complex on ER membrane [36], VCP is also found in aggresomes containing ΔF508-CFTR [37] in association with cytosolic histone deacetylase, HDAC6. These AF508-CFTR containing aggresome bodies are detectable following proteasome inhibition. It was recently reported that finely tuned balance of the associations of HDAC6 and VCP with ΔF508-CFTR determines its fate [38]. This study used the ΔF508-CFTR mutant that lacked its C-terminal ATPase domain (deletion of 1057–1480 region). Recently, we used the N-terminal truncated form of CFTR (Δ264-CFTR) and ΔF508-CFTR to demonstrate that small molecule inhibition of HDAC6 induces the proteasome inhibitor-mediated CFTR rescue from ERAD [39]. Presence of VCP in both ER-associated complex and aggresome as well as its direct association with proteasome indicate that VCP is involved in both retrograde translocation from ER and presentation of client protein to proteasome. We observed the association of VCP with immune complexes pulled down with anti-CFTR, Hsp40, Hsp70, Hsp90, gp78, ataxin-3 (aggresome marker) and HDAC6 antibodies indicating interacting partnerships between these proteins. These results indicated the functional relationship of VCP with both ERAD (Hsp40-Hsp70 complex) and aggresome formation (Ataxin3-HDAC6 complex) pathways [18]. We confirmed the functional association of VCP with ERAD and aggresome formation using small molecule inhibitor of aggresome formation (tubacin) and proteasome inhibition (Fig.1, our unpublished data).
Figure 1.
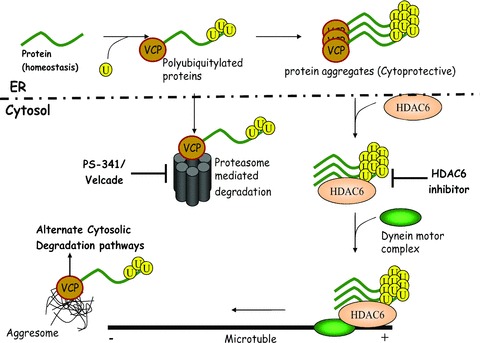
Schematic of therapeutic potential of selective VCP inhibition. VCP is required for misfolded protein recognition and proteasome-mediated degradation. In case of ER stress, misfolded protein overload or inflammation, VCP-aberrant protein interaction is dissociated by HDAC6 that direct the poly-ubiquinated-protein to aggresome bodies with the help of dyenin motor complex. VCP dissociates HDAC6 interaction in aggresome. Finely tuned balance of VCP and HDAC6 determine the fate of aberrant protein. These aggresomes are formed by mechanisms that require VCP activity. Available chemical inhibitors of proteasome (PS-341/Velcade) and aggresome formation (tubacin/HDAC6 inhibitor) inhibit protein degradation but will not help in rescue of polytopic membrane proteins from ER-associated degradation. Selective inhibition of VCP functional activity on ER membrane disrupts both protein degradation and aggresome formation and is a novel site for therapeutic intervention to rescue polytopic membrane proteins from degradation and induce forward trafficking.
We previously reported that modulating proteasomal degradation by bortezomib or VCP inhibition not only rescues the functional mutant CFTR but also repressed NFKB-mediated IL-8 activation. We observed that VCP has higher affinity for ΔF508-CFTR as compared to wt-CFTR [15]. Recently, it was demonstrated, using the endogenous expression system, that VCP is selectively associated with AF508-CFTR but not wt-CFTR. Association of VCP with wt-CFTR was only observed by CFTR overexpression or proteasome inhibition [23]. We also observed association of other misfolded protein, ATZ with VCP in HEK293 cells transiently transfected with ATZ. Moreover, we observed that ATZ expression induces VCP protein levels and observed further induction of VCP protein levels by proteasome inhibition in presence of ATZ (our unpublished data). Based on our preliminary data and available literature we anticipate that inhibiting the binding of VCP to the aberrant protein can lead to development of effective therapeutic or prophylactic drugs for conformational diseases such as CF and AATD.
We would like to point out here that misfolded protein disorders are of two types: (i) loss of functional protein by ubiquitin-proteasome-mediated ERAD and (ii) aggregation of misfolded protein in cystosol (aggresomes) or ER (Russel bodies). The misfolded protein disorders that fall under these two categories are summarized in Fig.2. In most of the misfolded protein disorders in first category, protein rescued from proteasomal degradation are functional and can rescue the disease pathophysiology. One approach is to use proteasome inhibitors but that result in cytosolic aggregates (aggresomes) as shown in Fig.1. Several chemical chaperones have been tested in these diseases to both avoid degradation and aggregation of misfolded protein (Table 1) but none of them could be developed as an effective therapeutic strategy due to their inability to interact specifically with the molecular targets. We propose here that identification and therapeutic targeting of molecular chaperones like VCP is a selective alternate approach. The selective interference of VCP interaction with aberrant protein or lkB can be developed as a therapeutic approach to not only rescue functional misfolded protein but also inhibiting chronic kB-NFKB-mediated inflammatory signalling.
Figure 2.
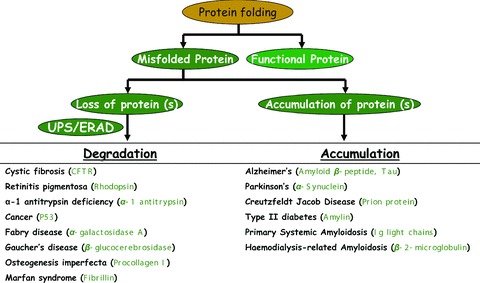
Summary of misfolded protein disorders. Misfolded protein disorders can be divided into two categories based on (1) loss of misfolded protein by ubiquitin proteasome system (UPS) or endoplasmic reticulum-associated degradation (ERAD) or (2) aggregation of misfolded protein in cytosol (aggresomes) or ER (Russell bodies). Misfolded protein associated with the disease are shown in bracket.
Table 1.
Misfolded protein disorders and tested chemical chaperone therapies
| Disease | Chemical chaperone |
|---|---|
| Cystic fibrosis | Benzoflavone/4-Phenylbutyric acid |
| Gaucher's disease | N-alkyalted deoxynorjirimycins |
| Retinitis Pigmentosa | Retinal-based ligands |
| Fabry disease | 1-deoxy-galactanojirimycin |
| α1-anti-trypsin deficiency | 4-Phenylbutyric acid |
| Gonatotropin-hormone receptor deficiency | Indoles and quinolines |
| Nephrogenic diabetes insipidus | SR11463A, VPA985 |
Selective inhibition of VCP ERAD function by targeting its tyrosine phosphorylation [8] or inducing the activity of endogenous inhibitor SVIP (small VCP interacting protein) [40] is a promising cancer therapeutic target as it will not only selectively rescue cancer cells from NFi<B-induced anti-apoptotic signalling but also suppress the retrograde translocation of proteins from ER to cytosol leading to added induction of apoptotic signalling. We propose that this selective therapeutic strategy has an added potential as a novel cancer therapeutics over FDA-approved proteasome-inhibiting drug, bortezomib/PS-341 [41], in suppressing both proteasome function and aggresome formation (Fig.1).
VCP is an essential protein with diverse cellular function hence we do not anticipate that modulating its expression will be an effective therapeutic strategy and hence we propose inhibiting its interaction with aberrant protein or its specific function as discussed above can be translated into a therapeutic intervention. VCP function in protein aggregation disorders that fall under category two needs to be carefully examined, if VCP is responsible for both aggresome formation and promoting degradation of these aggregates as recently proposed by Kobayashi et al. [42]. Inhibiting its interaction with aberrant protein will be effective in inhibiting aggresome formation and will be sufficient to rescue the disease pathology. In disorders where VCP is not required for aggresome formation and is exclusively involved in degradation of these aggregates, methods to enhance AAA ATPase activity or unfoldase function of VCP can be developed as a therapeutic approach to help with the clearance of these aggregates.
Role of VCP in ER and post-ER compartments
Although the mechanism of substrate delivery to the ER membrane-associated retrograde-translocation Derlin1-VCP protein complex is still unclear, there is recent evidence that N-linked glycans play a role in the selective targeting of misfolded glycoproteins to ERAD through interaction with the lectin EDEM (ER degradation-enhancing α-mannosidase-like protein; Htm1/Mnl1p in yeast) [43] and VCP regulates this process. The proposed mechanism elucidates that the selection of misfolded glycoproteins is based on the length of time glycoprotein spends in the ER. In the case of prolonged acquisition of a folded conformation, the glycoprotein GlcNAc2-Man9-(Glc1) oligosaccharide becomes a substrate of ER mannosidase I that specifically removes the terminal mannose from the middle branch (branch B), transforming it into GlcNAc2-Man8-Glc1 (also called Man8B-isomer), which functions as the predicted proximal signal for VCP-mediated translocon delivery and ERAD [44]. Accordingly, EDEM and VCP have been shown to accelerate the degradation of misfolded CFTR in yeast. Complementation of the HTM1 (EDEM) or Cdc48 (VCP) deficiency in yeast cells by the mammalian orthologue EDEM or VCP underlines the necessity of these proteins in CFTR degradation and highlights the similarity of quality control and ERAD in yeast and mammals [45]. A novel function of VCP in the control of N-glycosylation of proteins in the ER was recently demonstrated [46]. Mass spectrometric analysis of cellular N-linked glycans revealed that depletion of VCP decreases the level of high-mannose glycoproteins, increases the levels of truncated low-mannose glycoproteins and induces changes in the abundance of complex glycans assembled in post-ER compartments. Since proteasome inhibition was unable to mimic those changes, they cannot be regarded as a simple consequence of inhibited ERAD but represent a complex effect of VCP on the function of the ER.
The further examination of the role of VCP in ER protein-glyco-sylation is required to elucidate the exact function of VCP in selection of misfolded protein for ERAD, aggregation of misfolded proteins in ER (Russell bodies) and trafficking of membrane proteins. In addition, mammalian VCP and its yeast counterpart Cdc48p are also known to participate in the formation of organelles, including the ER, Golgi apparatus and nuclear envelope [5, 47–50]. VCP is also known to interact with proteins involved in membrane trafficking [51] as summarized in Table 2 but the functional roles of these interactions are poorly elucidated. The negative regulation of VCP's ERAD function by its endogenous inhibitor SVIP [40] needs further investigation as a therapeutic approach to induce membrane transport and forward trafficking in membrane transport disorders and diseases with both ERAD, and membrane transport dysfunction like C F.
Table 2.
Classification of VCP functions, interacting proteins and disease
| Cellular function | Interacting proteins | Disease |
|---|---|---|
| Protein degradation | Ub, Ufd1/2, Ufd3, Proteasome (PSMC1/PSMD2), Npl4, VCIP, p47, Ubx2 | Cystic Fibrosis, Alpha-1-trypsin deficiency |
| Mitosis/nuclear refor-mation | Ase1, Cdc5, Plx1, AuroraB | Cancer metastasis |
| Membrane trafficking | SVIP, Syt I, II, Clathrin | Membrane transport disorders |
| DNA/RNA repair | BRCA1, DUF, TB-RBP, WRN | Cancer metastasis |
| Aggresome | HDAC6, Ataxin-3, ex-polyQ | Neurodegenerative disorders |
| Inflammation | IkB-NFkB, Akt | Inflammation, cancer metastasis |
Molecular genetics of VCP
Watts et al.[52] identified missense mutations in VCP as the cause of inclusion body myopathy with Paget disease of bone and fron-totemporal dementia (OMIM, IBMPFD; 167320). Ten of 13 families with this disorder had an amino acid change at arginine-155, either to histidine, proline, or cysteine. Arginine-155 of VCP was conserved in homologues through all species examined except in two C. elegans homologues, which had glutamine at that position. Arginine-191 was invariant in all species examined, and arginine-95 was substituted by histidine in only two species. They proposed that since patients with IBMPFD are viable with relatively late onset of disease, the mutations identified do not disrupt the cell cycle or apoptosis pathways. They suggested that mutations in VCP cause Paget disease of bone by compromising ubiquitin binding and target similar cellular pathways or proteins. They concluded that the progressive neuronal degeneration involve protein quality control and ubiquitin protein degradation pathways. Interestingly, postmortem studies have confirmed that the IBMPFD associated with VCP mutations is indeed a ubiquitinopathy with widespread intranuclear ubiquitin inclusions in addition to the more classical cytoplasmic FTLD-U pathology. Transfection of mutant VCP impairs degradation of ubiquitinated proteins normally handled by the ERAD pathway [53]. The role of VCP in IBMFD and neurodegenerative disorders has been recently reviewed in detail [54].
Although IBMFD is a relatively uncommon disorder, its importance lies in providing a clear molecular link between a specific VCP mutation and ubiquitin-associated conformational disease pathology. While disruption of ERAD and/or UPS function is an attractive hypothesis, mutations in VCP may affect other cellular activities regulated by VCP. Alternatively, mutations in VCP may lead to a gain of function that is not otherwise predicted based on its known functions. The extensive molecular studies are required to elucidate the exact mechanism(s) leading to the IBMFD disease pathology. It is not clear if the IBMFD pathology is indeed a direct consequence of ERAD dysfunction or if VCP mutation disrupts the finely tuned balance of VCP and HDAC6 interactions that promotes aggresome formation (Fig.1). Selection of appropriate method for selective targeting of VCP ERAD or aggresome formation functions requires dissection of molecular mechanisms leading to the disease pathology in these genetic disorders. Decreasing aggregate formation activity and/or increasing unfoldase activity of VCP, is expected to be a novel therapeutic target for the treatment of IBMPFD and other neurodegenerative disorders with intracellular protein aggregation.
VCP structure and biochemical function
Mechanistically, misfolded protein substrates could associate with the N-terminal end of some VCP protomers through their exposed hydrophobic surfaces, whereas adaptors could still occupy other protomers. Although this may still hold, the recent study [55] surprisingly demonstrates the involvement of the D2 ring in substrate binding and propose an in-house mechanism for substrate processing. DeLaBarre and colleagues [55] recently introduced the ‘denaturation-collar’ model in which secondary structures are unfolded through the guanidyl-rich denaturing milieu of the arginine double ring (Arg586/Arg599) that guards the D2 pore [55]. Subsequently, substrates are either threaded out through transient grooves at the D1/D2 interphase or reversibly channelled out from the entry port. In contrast to other AAA proteins, which thread substrates through their symmetry axis, VCP has a narrow D1 pore that is unlikely able to accommodate a polypeptide chain. In this case, the D1 ring function seems to be limited to correct hexameric assembly and proper interprotomer communication. Furthermore, this model does not rule out the mechanistic involvement of the N-terminal domain and adaptor proteins in substrate recruitment to the C-terminal pore or substrate release from the D2 ring. Further structural, molecular and biochemical analysis is required to determine the exact sites and mechanisms of misfolded substrate or ubiquitin binding to identify highly specific small molecule inhibitors to dissociate VCP-aberrant protein interaction. It still remains an open question if different substrates like misfolded protein and lkB are recognized and chaperoned by VCP similar mechanisms.
VCP and molecular purgatory
A number of recent functional studies now establish a novel paradigm by which VCP plays a purgatorial (deciding) role in regulating the ubiquitylation status of protein substrates, which is consistent with the apparently differing biochemical functions previously observed. The fact that VCP is considerably more abundant (1% of total cytosolic protein) than its partners suggests that cofactor expression, stability or localization may all be important determinants of the identity of the assembled complexes. Alternatively, VCP and its cofactors may be subject to a dynamic post-translational modification process that determines the identity of the assembled complex within pockets of cellular microdomains and according to specific metabolic demands. This would not be surprising considering that the cell cycle-dependent phosphorylation of p47 by Cdc2 regulates its localization on the ER and Golgi membranes and may subsequently affect its interaction with VCP [56]. Similarly, Akt-mediated phosphorylation regulates VCP interaction with ubiquitylated substrates, although it is unclear whether this is a direct effect on substrate binding or a mere consequence of abolishing cofactor binding [8], VCP may also assume the role of a protein-proofing chaperone that is coevolved to alleviate the metabolic pressures of protein synthesis and degradation. Moreover, as a cellular home-ostatic process VCP's ERAD function is negatively regulated by SVIP [40]. VCP directly interacts with proteins involved in (i) protein degradation, (ii) mitosis, (iii) nuclear reformation, (iv) membrane trafficking, (v) DNA/RNA repair, (vi) aggresome formation and (vii) inflammatory signalling. The association of these VCP interactions and functional pathways with various human diseases is shown in Table 2. To summarize, VCP is an essential cellular protein involved in diverse cellular functions and disease pathologies hence selective intervention of VCP function is required for the development of novel therapeutic with minimal side effects.
Perspective
Depending on the conformational recoverability, a crucial decision must be made to either ubiquitylate and degrade or deubiquitylate and recycle target substrates. A better mechanistic understanding of VCP functions, especially its potential role in protein unfolding or aggregation, is evermore pressing considering the scope and complexity of its interaction with the aberrant proteins involved in conformational diseases. Better understanding of these molecular mechanisms will lead to the development of methods for selective intervention of VCP as a novel therapeutic. The therapeutic approaches we are presently testing include chemical or small molecule interference of VCP-misfolded protein interaction, VCP-ubiquitin interaction, VCP-cofactor interaction, VCP phos-phorylation and VCP ATPase activity.
Acknowledgments
The author is thankful for financial support from Cystic Fibrosis Foundation (CFF RDP and VIJ07I0) USA.
References
- 1.Braun RJ, Zischka H. Mechanisms of Cdc48/VCP-mediated cell death – from yeast apoptosis to human disease. Biochim Biophys Acta. 2008;1783:1418–35. doi: 10.1016/j.bbamcr.2008.01.015. [DOI] [PubMed] [Google Scholar]
- 2.Partridge JJ, Lopreiato JO, Jr, Latterich M, Indig FE. DNA damage modulates nucleolar interaction of the Werner protein with the AAA ATPase p97/VCP. Mol Biol Cell. 2003;14:4221–9. doi: 10.1091/mbc.E03-02-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kondo H, Rabouille C, Newman R, Levine TP, Pappin D, Freemont P, Warren G. p47 is a cofactor for p97-mediated membrane fusion. Nature. 1997;388:75–8. doi: 10.1038/40411. [DOI] [PubMed] [Google Scholar]
- 4.Meyer HH, Shorter JG, Seemann J, Pappin D, Warren G. A complex of mammalian ufd1 and npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. EMBO J. 2000;19:2181–92. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hetzer M, Meyer HH, Walther TC, Bilbao-Cortes D, Warren G, Mattaj IW. Distinct AAA-ATPase p97 complexes function in discrete steps of nuclear assembly. Nat Cell Biol. 2001;3:1086–91. doi: 10.1038/ncb1201-1086. [DOI] [PubMed] [Google Scholar]
- 6.Rabinovich E, Kerem A, Frohlich KU, Diamant N, Bar-Nun S. AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol Cell Biol. 2002;22:626–34. doi: 10.1128/MCB.22.2.626-634.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001;3:740–4. doi: 10.1038/35087056. [DOI] [PubMed] [Google Scholar]
- 8.Klein JB, Barati MT, Wu R, Gozal D, Sachleben LR, Jr, Kausar H, Trent JO, Gozal E, Rane MJ. Akt-mediated valosin-containing protein 97 phosphorylation regulates its association with ubiquitinated proteins. J Biol Chem. 2005;280:31870–81. doi: 10.1074/jbc.M501802200. [DOI] [PubMed] [Google Scholar]
- 9.Hirabayashi M, Inoue K, Tanaka K, Nakadate K, Ohsawa Y, Kamei Y, Popiel AH, Sinohara A, Iwamatsu A, Kimura Y, Uchiyama Y, Hori S, Kakizuka A. VCP/p97 in abnormal protein aggregates, cytoplasmic vacuoles, and cell death, phenotypes relevant to neurodegeneration. Cell Death Differ. 2001;8:977–84. doi: 10.1038/sj.cdd.4400907. [DOI] [PubMed] [Google Scholar]
- 10.Higashiyama H, Hirose F, Yamaguchi M, Inoue YH, Fujikake N, Matsukage A, Kakizuka A. Identification of ter94, Drosophila VCP, as a modulator of polyglu-tamine-induced neurodegeneration. Cell Death Differ. 2002;9:264–73. doi: 10.1038/sj.cdd.4400955. [DOI] [PubMed] [Google Scholar]
- 11.Schroder R, Watts GD, Mehta SG, Evert BO, Broich P, Fliessbach K, Pauls K, Hans VH, Kimonis V, Thal DR. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol. 2005;57:457–61. doi: 10.1002/ana.20407. [DOI] [PubMed] [Google Scholar]
- 12.Ishigaki S, Hishikawa N, Niwa J, Iemura S, Natsume T, Hori S, Kakizuka A, Tanaka K, Sobue G. Physical and functional interaction between Dorfin and Valosin-containing protein that are colocalized in ubiquitylated inclusions in neurodegenerative disorders. J Biol Chem. 2004;279:51376–85. doi: 10.1074/jbc.M406683200. [DOI] [PubMed] [Google Scholar]
- 13.Meusser B, Hirsch C, Jarosch E, Sommer T. ERAD: the long road to destruction. Nat Cell Biol. 2005;7:766–72. doi: 10.1038/ncb0805-766. [DOI] [PubMed] [Google Scholar]
- 14.Zhong X, Shen Y, Ballar P, Apostolou A, Agami R, Fang S. AAA ATPase p97/valosin-containing protein interacts with gp78, a ubiquitin ligase for endoplas-mic reticulum-associated degradation. J Biol Chem. 2004;279:45676–84. doi: 10.1074/jbc.M409034200. [DOI] [PubMed] [Google Scholar]
- 15.Vij N, Fang S, Zeitlin PL. Selective inhibition of endoplasmic reticulum-associated degradation rescues {Delta}F508-cystic fibrosis transmembrane regulator and suppresses interleukin-8 levels: THERAPEUTIC IMPLICATIONS. J Biol Chem. 2006;281:17369–78. doi: 10.1074/jbc.M600509200. [DOI] [PubMed] [Google Scholar]
- 16.Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K, Nagata AK. Gp78 cooperates with RMA1 in endoplasmic reticulum-associated degradation of CFTR{Delta}F508. Mol Biol Cell. 2008;19:1328–36. doi: 10.1091/mbc.E07-06-0601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C, Cyr DM. Sequential quality-control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–82. doi: 10.1016/j.cell.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 18.Vij N, Mazur S, Zeitlin PL. VCP is involved in ERAD and aggresome formation of ΔF508-CFTR. Ped. Pulmonol. 2006;41:209. [Google Scholar]
- 19.Shen Y, Ballar P, Fang S. Ubiquitin ligase gp78 increases solubility and facilitates degradation of the Z variant of alpha-1-antitrypsin. Biochem Biophys Res Commun. 2006;349:1285–93. doi: 10.1016/j.bbrc.2006.08.173. [DOI] [PubMed] [Google Scholar]
- 20.Song BL, Sever N, DeBose-Boyd RA. Gp78 a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19:829–40. doi: 10.1016/j.molcel.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 21.Ballar P, Zhong Y, Nagahama M, Tagaya M, Shen Y, Fang S. Identification of SVIP as an endogenous inhibitor of endoplasmic reticulum-associated degradation. J Biol Chem. 2007;282:33908–14. doi: 10.1074/jbc.M704446200. [DOI] [PubMed] [Google Scholar]
- 22.Nagahama M, Suzuki M, Hamada Y, Hatsuzawa K, Tani K, Yamamoto A, Tagaya M. SVIP is a novel VCP/p97-inter-acting protein whose expression causes cell vacuolation. Mol Biol Cell. 2003;14:262–73. doi: 10.1091/mbc.02-07-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein RF, Niraj A, Sanderson TP. Wilson LS, Rab A, Kim H, Bebok Z, Collawn JF. VCP/p97 AAA-ATPase does not interact with the endogenous wild-type CFTR. Am J Respir Cell Mol Biol. 2007;36:706–14. doi: 10.1165/rcmb.2006-0365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carroll T, Greene CM, Taggart CC. O’Neill SJ, McElvaney NG. Z Alpha-1 antitrypsin induces the unfolded protein response (UPR), an ER stress pathway in bronchial epithelial cells. Proc Am Thorac Soc. 2007:A742. . ATS conference special issue. [Google Scholar]
- 25.Korfei M, Ruppert C, Mahavadi P, Henneke I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, Weaver TE, Guenther A. Epithelial endoplasmic reticulum stress and apoptosis in sporadic idiopathic pulmonary fibrosis. Am J Resp Crit Care Med. 2008;178:838–46. doi: 10.1164/rccm.200802-313OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dai RM, Chen E, Longo DL, Gorbea CM, Li CC. Involvement of valosincontaining protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J Biol Chem. 1998;273:3562–73. doi: 10.1074/jbc.273.6.3562. [DOI] [PubMed] [Google Scholar]
- 27.Ye Y, Shibata Y, Kikkert M, van Voorden S, Wiertz E, Rapoport TA. Inaugural article: recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc Natl Acad Sci. 2005;102:14132–8. doi: 10.1073/pnas.0505006102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kakizuka A. Protein precipitation: a common etiology in neurodegenerative disorders? Trends Genet. 1998;14:396–402. doi: 10.1016/s0168-9525(98)01559-5. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto S, Tomita Y, Hoshida Y, Iizuka N, Monden M, Yamamoto S, Iuchi K, Aozasa K. Expression level of valosin-containing protein (p97) is correlated with progression and prognosis of non-small-cell lung carcinoma. Ann Surg Oncol. 2004;11:697–704. doi: 10.1245/ASO.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 30.Yamamoto S, Tomita Y, Hoshida Y, Nagano H, Dono K, Umeshita K, Sakon M, Ishikawa O, Ohigashi H, Nakamori S, Monden M, Aozasa K. Increased expression of valosin-containing protein (p97) is associated with lymph node metastasis and prognosis of pancreatic ductal adenocarcinoma. Ann Surg Oncol. 2004;11:165–172. doi: 10.1245/aso.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 31.Asai T, Tomita Y, Nakatsuka S, Hoshida Y, Myoui A, Yoshikawa H, Aozasa K. VCP (p97) regulates NFkappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J Cancer Res. 2002;93:296–304. doi: 10.1111/j.1349-7006.2002.tb02172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fujioka S, Sclabas GM, Schmidt C, Frederick WA, Dong QG, Abbruzzese JL, Evans DB, Baker C, Chiao PJ. Function of nuclear factor kappaB in pancreatic cancer metastasis. Clin Cancer Res. 2003;9:346–54. [PubMed] [Google Scholar]
- 33.Khanbolooki S, Nawrocki ST, Arumugam T, Andtbacka R, Pino MS, Kurzrock R, Logsdon CD, Abbruzzese JL, McConkey DJ. Nuclear factor-kappaB maintains TRAIL resistance in human pancreatic cancer cells. Mol Cancer Ther. 2006;5:2251–60. doi: 10.1158/1535-7163.MCT-06-0075. [DOI] [PubMed] [Google Scholar]
- 34.Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ. Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2002;1:1243–53. [PubMed] [Google Scholar]
- 35.Vij N, Amoako MO, Mazur S, Zeitlin PL. CHOP transcription factor mediates IL-8 signaling in cystic fibrosis bronchial epithelial cells. Am J Respir Cell Mol Biol. 2008;38:176–84. doi: 10.1165/rcmb.2007-0197OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ye Y, Meyer HH, Rapoport TA. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature. 2001;414:652–6. doi: 10.1038/414652a. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–38. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 38.Boyault C, Gilquin B, Zhang Y, Rybin V, Garman E, Meyer-Klaucke W, Matthias P, Muller CW, Khochbin S. HDAC6-p97/VCP controlled polyubiquitin chain turnover. EMBO J. 2006;25:3357–66. doi: 10.1038/sj.emboj.7601210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cebotaru L, Vij N, Ciobanu I, Wright J, Flotte T, Guggino WB. The cystic fibrosis transmembrane regulator missing the first four transmembrane segments increases wild type and uF508 processing. J Biol Chem. 2008;283:21926–33. doi: 10.1074/jbc.M709156200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ballar P, Zhong Y, Nagahama M, Tagaya M, Shen Y, Fang S. Identification of SVIP as an endogenous inhibitor of ER-associated degradation. J Biol Chem. 2007;282:33908–14. doi: 10.1074/jbc.M704446200. [DOI] [PubMed] [Google Scholar]
- 41.Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Andtbacka RH, Dunner K, Jr, Pal A, Bornmann WG, Chiao PJ, Huang P, Xiong H, Abbruzzese JL, McConkey DJ. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–81. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi T, Manno A, Kakizuka A. Involvement of valosin-containing protein (VCP)/p97 in the formation and clearance of abnormal protein aggregates. Genes Cells. 2007;12:889–901. doi: 10.1111/j.1365-2443.2007.01099.x. [DOI] [PubMed] [Google Scholar]
- 43.Jakob CA, Bodmer D, Spirig U, Battig P, Marcil A, Dignard D, Bergeron JJ, Thomas DY, Aebi M. Htm1p, a mannosidase-like protein, is involved in glycoprotein degradation in yeast. EMBO Rep. 2001;2:423–30. doi: 10.1093/embo-reports/kve089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cabral CM, Liu Y, Sifers RN. Dissecting glycoprotein quality control in the secretory pathway. Trends Biochem Sci. 2001;26:619–24. doi: 10.1016/s0968-0004(01)01942-9. [DOI] [PubMed] [Google Scholar]
- 45.Gnann A, Riordan JR, Wolf DH. Cystic fibrosis transmembrane conductance regulator degradation depends on the lectins Htm1p/EDEM and the Cdc48 protein complex in yeast. Mol Biol Cell. 2004;15:4125–35. doi: 10.1091/mbc.E04-01-0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lass A, McConnell E, Nowis D, Mechref Y, Kang P, Novotny MV, Wojcik C. A novel function of VCP (valosin-containing protein; p97) in the control of N-glycosylation of proteins in the endoplasmic reticulum. Arch Biochem Biophys. 2007;462:62–73. doi: 10.1016/j.abb.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy L, Bergeron JJ, Lavoie C, Hendriks R, Gushue J, Fazel A, Pelletier A, Morre DJ, Subramaniam VN, Hong W, Paiement J. Role of p97 and syntaxin 5 in the assembly of transitional endoplasmic reticulum. Mol Biol Cell. 2000;11:2529–42. doi: 10.1091/mbc.11.8.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lavoie C, Chevet E, Roy L, Tonks NK, Fazel A, Posner BI, Paiement J, Bergeron JJ. Tyrosine phosphorylation of p97 regulates transitional endoplasmic reticulum assembly in vitro. Proc Natl Acad Sci. 2000;97:13637–42. doi: 10.1073/pnas.240278097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rabouille C, Kondo H, Newman R, Hui N, Freemont P, Warren G. Syntaxin 5 is a common component of the NSF- and p97-mediated reassembly pathways of Golgi cisternae from mitotic Golgi fragments in vitro. Cell. 1998;92:603–10. doi: 10.1016/s0092-8674(00)81128-9. [DOI] [PubMed] [Google Scholar]
- 50.Rabouille C, Levine TP, Peters JM, Warren G. An NSF-like ATPase, p97, and NSF mediate cisternal regrowth from mitotic Golgi fragments. Cell. 1995;82:905–14. doi: 10.1016/0092-8674(95)90270-8. [DOI] [PubMed] [Google Scholar]
- 51.Pleasure IT, Black MM, Keen JH. Valosin-containing protein, VCP, is a ubiquitous clathrin-binding protein. Nature. 1993;365:459–62. doi: 10.1038/365459a0. [DOI] [PubMed] [Google Scholar]
- 52.Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, Kimonis VE. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 53.Kitami MI, Kitami T, Nagahama M, Tagaya M, Hori S, Kakizuka A, Mizuno Y, Hattori N. Dominant-negative effect of mutant valosin-containing protein in aggresome formation. FEBS Lett. 2006;580:474–8. doi: 10.1016/j.febslet.2005.12.044. [DOI] [PubMed] [Google Scholar]
- 54.Kakizuka A. Roles of VCP in human neurodegenerative disorders. Biochem Soc Trans. 2008;36:105–8. doi: 10.1042/BST0360105. [DOI] [PubMed] [Google Scholar]
- 55.DeLaBarre B, Christianson JC, Kopito RR, Brunger AT. Central pore residues mediate the p97/VCP activity required for ERAD. Mol Cell. 2006;22:451–62. doi: 10.1016/j.molcel.2006.03.036. [DOI] [PubMed] [Google Scholar]
- 56.Uchiyama K, Kondo H. p97/p47-mediated biogenesis of Golgi and ER. J Biochem. 2005;137:115–9. doi: 10.1093/jb/mvi028. [DOI] [PubMed] [Google Scholar]