

Abstract
The development and production of viral vaccines, in general, involve several steps that need the monitoring of viral load throughout the entire process. Applying a 2-step quantitative reverse transcription real time PCR assay (RT-qPCR), viral load can be measured and monitored in a few hours. In this context, the development, standardization and validation of a RT-qPCR test to quickly and efficiently quantify yellow fever virus (YFV) in all stages of vaccine production are extremely important. To serve this purpose we used a plasmid construction containing the NS5 region from 17DD YFV to generate the standard curve and to evaluate parameters such as linearity, precision and specificity against other flavivirus. Furthermore, we defined the limits of detection as 25 copies/reaction, and quantification as 100 copies/reaction for the test. To ensure the quality of the method, reference controls were established in order to avoid false negative results. The qRT-PCR technique based on the use of TaqMan probes herein standardized proved to be effective for determining yellow fever viral load both in vivo and in vitro, thus becoming a very important tool to assure the quality control for vaccine production and evaluation of viremia after vaccination or YF disease.
Keywords: molecular diagnosis, RT-qPCR, viral vaccines, viral load, yellow fever virus
Abbreviations
- PCR
polymerase chain reaction
- RT-qPCR
reverse transcriptase quantitative polymerase chain reaction
- YFV
yellow fever virus
- NS5
protein of the viral polyprotein, it is the largest and the most highly conserved among the flaviviral proteins
- 17DD
strain used to yellow fever vaccine
- YF
yellow fever
- RNA
ribonucleic acid
- C
capsid protein
- prM/M
membrane protein
- E
envelope protein
- NS
nonstructural protein
- ELISA
enzyme-linked immunosorbent assay
- DNA
deoxyribonucleic acid
- DL
detection limit
- QL
quantification limit
- ANVISA
Brazilian Health Surveillance Agency
- CV
coefficient of variation
- RNAse P
human constitutive gene
- Ct
cycle threshold
- EXO IPC
exogenous internal positive control
- PFU
plaque former unit
- FDA
food and drug administration agency
- JEV
japanese encephalitis virus
- DENV
dengue virus
- MuV
mumps virus
- MV
measles virus
- MOI
multiplicity of infection
- WNV
West Nile Virus
Introduction
The yellow fever virus (YF) belongs to the Flaviviridae family, a group constituted of about 60 closely related viruses, many of which cause serious human disease.1 The viruses of this family contain non-segmented, positive stranded RNA genome of 10862 nucleotides. The yellow fever virus genome encodes a polyprotein of 3411 amino acids which is cleaved by proteolytic processing into 11 viral polypeptides.2 The single open reading frame encodes the structural capsid (C), the membrane (prM/M), envelope (E) and nonstructural (NS) proteins, in the following order C-prM-E-NS1-NS2A-NS2B-NS3-NS4A-NS4B-NS5.3 The NS5 protein of flavivirus is located at the C-terminal portion of the viral polyprotein. It is the largest and the most highly conserved among the flaviviral proteins.2,4
Yellow fever infection in human can result in unapparent infection up to serious disease of high lethality (20 – 50%), provoking great public health concern for centuries.5 Due to the expansion of yellow fever virus circulation in Latin America and Africa, several countries have included the yellow fever vaccine (YFV) on their regular immunizations programs, or conducted vaccination campaigns. However, the number of yellow fever vaccine producers has decreased during the last 20 y. There are 10 producers of yellow fever vaccines, but only 3 are prequalified by the World Health Organization and supply vaccines to international agencies: Bio-Manguinhos/Fiocruz (Brazil), Sanofi-Pasteur (France) and Institute Pasteur in Dakar (Senegal).6
The development and production of viral vaccines, in general, involve several steps that need viral load monitoring throughout the process. These steps range from the production of antigens, purification, inactivation and lyophilization, up to preclinical testing and clinical trials, and once the product is licensed, a process of continuous drug surveillance is necessary.
Usually, the detection of yellow fever virus is carried out by virus cultivation on susceptible cells followed by immunofluorescence, ELISA, immunohistochemical examination or PCR.7-10 For quantifying the load of infectious particles in either, cell culture or serum samples, plaque assay is still the commonly used method. However, plaque titration is cumbersome and time-consuming technique that takes at least 5 d to get the result.11 With the recent development of quantitative Real-Time PCR, a rapid and accurate alternative to quantify viral load is available.12
The newly established method should be standardized in order to maintain high quality laboratory performance. Laboratory diagnostics must be tailored to a specific laboratory environment, to the objectives of clinical needs and availability of clinical specimens. Indeed, the speed and accuracy of diagnosis should be balanced against the test cost and its availability, as well as its adaptation to international standardized guidelines with the intention of improving quality assurance of these advanced laboratory tests.13
In this study, specificity, precision, reportable range detection and analytical sensitivity of a RT-qPCR directed to the NS5 region from yellow fever virus were evaluated, according to previous described parameters required to validate bioanalytical methods in Brazil.14 In addition, analytical specificity and reference controls were determined and standardized to assure reliability of the results.
Results
Standardization of RT-qPCR for yellow fever virus (YFV)
The assay specificity was confirmed as no fluorogenic signal was detected for any other virus, except for the yellow fever virus (Table 1). As shown in Figure 1, there were no significant differences between Ct values generated by the YFV standard curve (plasmid containing the NS5 region of 17DD yellow fever virus serially diluted in negative human serum), compared with the YFV standard curve diluted in serum containing together Mumps virus, Dengue 1, 2, 3 and Measles (YFV standard curve + viral pool) [P-value = 0.9950]. The results demonstrate that the presence of other viruses do not interfere with the quantification of yellow fever virus.
Table 1.
Specificity analysis of the Yellow Fever Virus RT-qPCR
| Sample | Threshold Cycle (Ct) |
|---|---|
| Yellow fever virus 106 copies/reaction | 19.31 |
| Yellow fever virus 105 copies/reaction | 22.76 |
| Yellow fever virus 104 copies/reaction | 25.78 |
| Yellow fever virus 103 copies/reaction | 29.60 |
| Yellow fever virus 102 copies/reaction | 34.54 |
| Dengue virus 1 105 copies/reaction | ND |
| Dengue virus 2 105 copies/reaction | ND |
| Dengue virus 3 105 copies/reaction | ND |
| Japanese encephalitis virus 105 copies/reaction | ND |
| Negative controls (human serum of healthy individual and non-template controls - NTC) | ND |
ND, Not Detected.
Figure 1.
Comparative analysis of Ct values generated by distinct concentrations of recombinant plasmid containing the 83 bp fragment from the NS5 region of the 17DD yellow fever virus (YFV) diluted in negative human serum (YFV standard curve) and in serum containing together Mumps virus, Dengue 1, 2 and 3 and Measles (YFV standard curve + viral pool).
The experiments conducted to infer the analytical sensitivity demonstrated that the lowest concentration of plasmid DNA containing the NS5 region of 17DD-YFV, detected in at least 50 % of the replicates, was 25 copies/reaction and this value was established as the detection limit (DL) of the test, although the test was able to detect up to 12.5 copies. Concentration below 12.5 copies/reaction was not able to be detected in both independent assays (Table 2).
Table 2.
Linearity between the number of plasmid copies/reaction and Ct values to estimate the limits of detection and quantification for the Yellow Fever Virus RT-qPCR
| Plasmid Copy Number (copies/reaction) | Ct mean | Standard Deviation | Number of positive replicates/Number of replicates |
|---|---|---|---|
| 107 | 16.93 | 0.16 | 8/8 |
| 106 | 20.59 | 0.06 | 8/8 |
| 105 | 24.08 | 0.11 | 8/8 |
| 104 | 27.50 | 0.13 | 8/8 |
| 103 | 31.10 | 0.41 | 8/8 |
| 102 | 34.17 | 0.49 | 8/8 QL |
| 50 | 34.88 | 18.07 | 5/8 |
| 25 | 35.53 | 18.40 | 5/8 DL |
| 12.5 | 36.13 | 18.70 | 3/8 |
| 6.2 | ND | - | 0/8 |
Ct = Threshold Cycle; ND = Not Detected; QL = Quantification Limit; DL = Detection Limit.
The assay linearity was established indicating linear variation between dilutions (r2 = 0.99) up to the point representing 102 copies/reaction, which indicates the reportable quantifiable range of the method (Fig. 2). Up to this point, the variation between the 8 tested replicates was less than one Ct. Thus, the quantification limit (QL) was set as 102 copies/reaction, being the lowest concentration at which the method allows the detection with precision of all tested replicates within the reportable range (Table 2), as preconized by ANVISA (Brazilian Health Surveillance Agency).14
Figure 2.

Reportable amplification range to evaluate the performance of the TaqMan YFV RT-qPCR. The standard curve parameters were: slope = - 3.68; intercept = 45.08; coefficient of determination (r2) = 0.99 and amplification efficiency (E) = 86.2 %. Each point of the curve was tested in 8 replicates in the same run.
The results for estimating the precision of the method are shown in Figure 3. For both panel samples represented in 3 distinct concentrations of yellow fever virus, being serological samples spiked with purified 17DD-YFV (Fig. 3A) and clinical samples from 17DD-YFV vaccinated individuals (Fig. 3B), the test repetitions did not reveal statistical differences (p ≥ 0.05) regarding the operator or the day of performing the test. The coefficient of variation (CV) was calculated between replicas for each viral concentration in both panels, and the values found were in agreement with the ANVISA acceptance criteria requirements that preconize CV values below 20%.14
Figure 3.

Precision evaluation of the YFV RT-qPCR. (A) Reconstituted serological samples spiked with purified 17DD-YFV: high viral load - 104 copies/reaction, average viral load - 103 copies/reaction and low viral load - 102 copies/reaction. Each viral concentration was assayed in 8 replicates. The experiments were performed by 2 different operators and repeated in 4 separate assays; 2 experiments were conducted on the same day and the other 2 were carried out on different days (standard deviations are indicated). (B) Clinical samples from 17DD-YFV vaccinated individuals represented in 3 distinct concentrations: high viral load - 103 copies/reaction, average load - 102 copies/reaction and low viral load - 50 copies/reaction. Each viral concentration was assayed in 10 replicates. The assays were conducted by 2 different operators and repeated in 3 independent tests; 2 experiments were performed on the same day by different operators and one test was conducted on a different day by one operator (standard deviations are indicated).
Reference Controls
The results concerning the use of human RNase P gene as an endogenous internal amplification control in the TaqMan multiplex YFV RT-qPCR are summarized on Table 3. It can be observed that the Ct values for the RNase P gene quantification remained quite constant, independent of the viral load measure that revealed distinct Cts in the tested samples. Similarly, our data support using the exogenous internal positive control (EXO IPC) for the in vitro free-serum samples or those which are not in optimal conditions for analysis (for example, samples which have undergone hemolysis). Data analysis showed that regardless virus concentration, the EXO IPC showed minimal variation between Ct values (Fig. 4).
Table 3.
Evaluation of the human RNase P gene as an endogenous internal amplification control in the TaqMan multiplex YFV RT-qPCR. Ct values for both RNAse P and NS5 region from yellow fever virus are indicated, as well as the standard deviation (SD). The samples were assayed in 8 replicates each
| Sample | Mean Ct (Yellow Fever Virus) | ± SD | Mean Ct (human RNase P) | ±SD |
|---|---|---|---|---|
| *Clinical sample (Low viral load) | 36.40 | 0.96 | 31.30 | 0.16 |
| *Clinical sample (Average viral load) | 35.84 | 0.83 | 31.41 | 0.02 |
| *Clinical sample (High viral load) | 34.95 | 0.83 | 31.93 | 0.02 |
| **Serological panel (Low viral load) | 35.94 | 0.23 | 30.22 | 0.11 |
| **Serological panel (High viral load) | 30.96 | 0.15 | 30.43 | 0.30 |
| Negative serum | ND | 30.86 |
Ct = Threshold Cycle; ND = Not Detected.
Pool of samples constituted by human sera from 17DD yellow fever virus vaccinated individuals.
Serological panel generated by spiking non-infected serum from a healthy individual (Negative Quality Control Serum) with purified 17DD yellow fever virus.
Figure 4.

In vitro samples of 17DD-YFV propagated in serum-free medium from 2 bioreactor vases analyzed by RT-qPCR for the NS5 viral region and for the EXO IPC (exogenous control), in separate reactions on the same plate. The experiments were performed during the first 4 d of viral propagation. Dotted lines indicate the results for EXO IPC, showing minimal Ct variation between samples.
Correlation between YFV RT-qPCR and virus titration
RT-qPCR data (viral copies/mL) obtained from 38 samples (sera from vaccinated individuals and in vitro propagated virus) were compared with the conventional quantification method by virus titration (PFU/mL). Data analysis revealed a linear relationship between viral copy number obtained by RT-qPCR and PFU, represented by the equation: Log10 PFU/mL = [0.974 × Log10 copies/mL] – 2.807. A high coefficient of linearity was observed (r2 = 0.96) between both quantification methods. By using this equation, it was possible to estimate values in PFU/mL from a result in copies/mL (Fig. 5).
Figure 5.
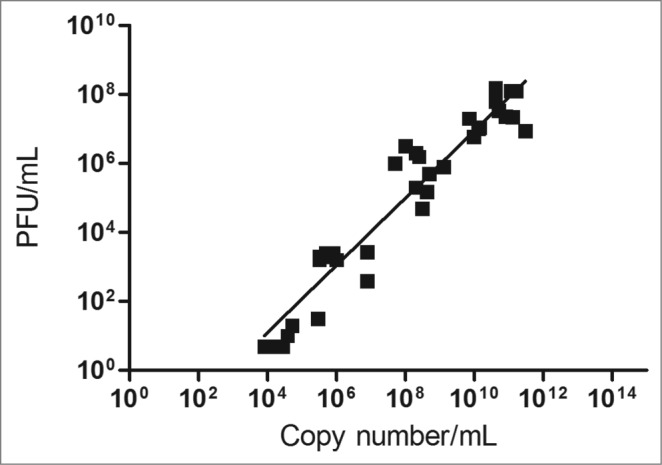
Correlation between the YFV RT-qPCR method (copies/mL) and virus titration (PFU/mL), from 38 analyzed samples (clinical samples from YFV vaccinated individuals and in vitro propagated virus), indicated by the equation: Log10 PFU/mL = [0.974 × Log10 copies/mL] – 2.807.
Discussion
Nowadays, the standardization and validation of Real-Time qPCR assay are extremely necessary not only for rapid monitoring viral load throughout the entire process of vaccine production, as well as to evaluate vaccine lots and for analyzing viremia in clinical studies or in individuals with adverse events to vaccination.6,15 During antigens production in vitro, the method can provides the best interval to interrupt growth and subsequent virus harvesting. Differently from the plaque lysis method, that is usually time consuming (in general, needs 7 d to get the result), qPCR monitors in real time the amounts of viral RNA/DNA, thus allowing an effective result much faster than the traditional method of virus titration (PFU/mL).16
In the present work we describe the standardization and validation of a Real-Time reverse transcriptase qPCR assay for yellow fever virus (YFV), following the Brazilian Health Surveillance Agency guidelines to validate bioanalytical methods in Brazil.14 Using primers and TaqMan probe directed for the YFV NS5 region, previously described by Mantel et al.,17 no cross-reaction was observed when using other flaviviruses, as dengue virus (DENV1, DENV2 and DENV3) and Japanese encephalitis virus (JEV), confirming the assay specificity for yellow fever virus detection. According to Gurukumar et al.18 similar experiments demonstrated the specificity of a Real-Time qPCR for Dengue virus, where 2 flaviviruses (JEV and WNV) and the alphavirus Chikungunya were assayed, achieving 100% specificity for Dengue virus.
For the accurate quantification of YFV load in clinical specimens, it is very important to assure the specificity of the method in areas where co-infection with other flavivirus is common. For instance, the state of Rio de Janeiro is considered an endemic area for dengue virus infection.19 Some authors emphasized the importance to evaluate the analytical specificity of the test, as also recommended by FDA.20,21 By our evaluation, no significant difference inferred by Pearson's correlation coefficient (p = 0.9950) was observed between the Ct values generated by distinct concentrations of plasmid containing the NS5 region of 17DD-YFV, diluted in negative human serum and in serum containing together Mumps virus, Dengue 1, 2 and 3 and Measles (Fig. 1). It is noteworthy that in these experiments, each dilution from both standard curves was independently processed for recovering the genetic material to be dosed, a fact that occasionally may have influenced minor variation of Ct values for some points of the curves.
The linearity of the method was established for the range 109 to 102 plasmid copies/reaction (r2 = 0.997), and this interval may be considered as the reportable range of our assay (Fig. 2). For concentrations minor than 102 copies/reaction, the curve reaches a plateau that leads us to believe that real discrimination is not occurring among higher dilutions. Detection of viral RNA was observed from up to 12.5 copies/reaction, the smallest concentration that gave positive results in a few number of replicas (Table 2). This result corroborates the findings by Mantel et al.,17 where the authors were able to detect yellow fever virus from up to 12 copies/reaction. However, our group adopted a detection limit (DL) of 25 copies/reaction to give a margin of safety and reliability in the reports generated. In our study, this concentration did not provide a precise quantification; instead, there was wide variation among replicas despite of turning possible the virus detection. As indicated in Table 2, we assumed a quantification limit (QL) of 102 copies/reaction, considering the accurate quantification of all assayed replicas (8/8). As inferred by Burd,21 the detection limit is usually below the range of linearity of an assay and could not be assumed as higher than the quantification limit.
The experiments performed to evaluate the Real-Time qPCR precision demonstrated optimal repeatability and reproducibility. For both panel of samples analyzed, we observed a homogeneity in the Ct values with small variations relative to the number of viral copies per reaction, despite changing the operators or the days for conducting the assays (Fig. 3). It is important to emphasize that the 2 operators conducted the tests since their initial stage, including sample processing for recovering viral RNA, a factor that could introduce errors concerning distinct operators. It should be emphasized the inclusion of clinical samples presenting low viral load, very close to the established detection limit, in order to assess the precision of the quantification method. Even so, the test was sensitive enough with slight discrepancy between results. Following the ANVISA requirements, the acceptance criteria for coefficient of variation between replicas should not bypass 20%.14 This was respected, as we found the highest variation as 3.35% (for the reconstituted serological panel), a value lower than the one preconized by ANVISA.
According to Sloan,20 FDA recommends 3 replicates each for 2 distinct concentrations in experiments performed on the same day and on different days. Therefore, the test developed in the present study not only conforms the quality standards recognized by the National Health Surveillance Agency from Brazil, as also shows adequacy to the FDA requirements. Overall, the parameters herein analyzed gave support for the application of YFV RT-qPCR, with reliable reports, in clinical samples from vaccinated individuals, especially when related to adverse reactions.
Our standardization results suggest the use of human RNAse P gene as an internal amplification control gene, for Real-Time PCR assays in multiplexed reactions, together with the NS5 viral region. We observed a small variation for the RNase P Cts between samples, despite great viral load differences presented by each sample. For all tested samples, Ct values for the human gene were consistent, being included in the acceptable range (around Ct = 30). Through this analysis, there were no false negative results regarding the detection/quantification of yellow fever virus in human clinical samples, supporting the accuracy of the results. Due to the impossibility of using human constitutive RNAse P gene as an internal reaction control in free-serum samples or hemolyzed clinical samples, an exogenous internal positive control (EXO IPC, Applied Biosystems), was used in separate reactions (singleplex). From the analysis of 17DD-YFV propagated in serum-free medium bioreactor vases, it was observed that Ct values for the exogenous control were homogenous (close to Ct = 30), suggesting that it can be used as viral RNA extraction control, before the qPCR reaction.
Bae et al.16 argued against using PCR based assays to replace the conventional plaque lysis method, due to the detection of genetic material and not the infectious particle. Yet, our findings indicate that by calculating a conversion factor, the Real-Time qPCR could be used as effective as the plaque assay, to monitor viral propagation kinetics. After statistically analyzing our results, a linear correlation was observed between the quantification data obtained by PFU/mL and copies/mL. The correlation factor indicated the possibility of using the molecular approach to monitor yellow fever virus kinetics in vitro and in vivo and eventually, may be adopted as the gold standard in laboratory analyzes.
The technique of real time-qPCR is efficient for determining yellow fever virus load in vivo and in vitro, becoming a promising tool to assure the quality control for vaccine production or viremia evaluation after vaccination, and also for yellow fever diagnosis in clinical samples. A reliable correlation was found between PFU/mL and copies/mL, but more tests are still needed to be conducted with a higher number of samples, in order to confirm the applicability of this correlation factor between both assays.
Material and Methods
Samples
The reconstituted serological panel was generated by artificially contaminating non-infected serum using a Negative Quality Control Serum from NIBSC (Product number 11/B606–03) with 3 concentrations of purified 17DD yellow fever virus: high viral load (104 copies/reaction), average viral load (103 copies/reaction) and low viral load (102 copies/reaction). The samples were assayed in 8 replicates each.
A pool of clinical samples constituted by human sera from 17DD yellow fever virus vaccinated individuals representing 3 different viral concentrations was also tested: high load (103 copies/reaction), average load (102 copies/reaction) and low viral load (50 copies/reaction). The samples were assayed in 10 replicates each.
Aliquots from cultivated Dengue virus, Japanese encephalitis virus, Mumps, Measles viruses and 17DD yellow fever virus (YFV) were kindly provided by the staff from LATEV (Biomanguinhos/Fiocruz).
The study was approved by the Ethics Committee of the Evandro Chagas Clinical Research Institute from Fiocruz and by the Brazilian Regulatory Agency – ANVISA. This study is part of a clinical study on dose-responses to yellow fever vaccine, approved with an International Register: ISRCTN38082350.
RNA extraction and cDNA synthesis
Total RNA was extracted from 140 µL aliquots of different samples containing 17DD yellow fever virus: human sera from vaccinated patients, human serum from healthy individual spiked with purified 17DD virus, and supernatant from propagated 17DD virus in cell culture or bioreactor. The protocol was carried out using QIAamp® Viral RNA Mini kit, Qiagen (Catalog number 52906) according to the manufacturer's instructions and the RNA was eluted with 60 µL of elution (AE) buffer. Human serum of healthy individual was used as negative control. Reverse transcription reaction was performed with random primers in 20 µL of extracted RNA added to 20 µL of High-Capacity cDNA Reverse Transcription mix, Applied Biosystems (Catalog number 4368814). The obtained cDNA was stored at − 80° C until use.
TaqMan quantitative Real-Time PCR (RT-qPCR)
The RT-qPCR assays were performed in the ABI Prism 7500 (Applied Biosystems, Foster City, CA), directed to the NS5 region of the yellow fever virus, as described by Mantel et al.16 The following primers and probe were used: Forward - 5′ GCA CGG ATG TAA CAG ACT GAA GA, Reverse - 5′ CCA GGC CGA ACC TGT CAT and Probe: 5′ FAM-CGA CTG TGT GGT CCG GCC CT C -TAMRA. The reactions were set up with 0.3 µM of each primer, 0.15 µM TaqMan fluorogenic probe, 1X TaqMan® Universal PCR Master Mix, Applied Biosystems (Catalog number 4304437), and 5 µL cDNA template in a final volume of 25 uL. The thermal conditions were 50°C, 2 min; 95°C, 10 min and 40 cycles of 95°C, 15 s and 60°C, 1 min.
For generating the standard curves, the 83 bp fragment from the NS5 viral region obtained by PCR was cloned into TOPO TA Cloning Vector, Invitrogen (Catalog number K4575–40), according to the manufacturer's instructions. Serial dilutions from 107 to 102 plasmid copies per reaction were used to generate the calibration curves for the RT-qPCR assays.
Each assay was designed in order to include negative (human serum of healthy individual) and non-template controls (NTC), positive control (spiked virus in human serum of healthy individual with viral load of 103 copies/reaction). All samples were tested in at least 3 replicates, according to the design of each experiment.
Standardization of RT-qPCR for yellow fever virus
Each assay was designed in order to include negative and positive controls, the standard curve and reference controls, in multiplex or singleplex tests, together with the test samples. All controls and samples were assayed in at least 3 replicates. The experiment models followed the ANVISA requirements (RE 899), the Brazilian agency that regulates the parameters to be evaluated in bioanalytical assays.14
The specificity of the Yellow Fever Virus (YFV) RT-qPCR was evaluated against a panel of other flaviviruses, including dengue virus (DENV1, DENV2 and DENV3) and Japanese encephalitis virus (JEV). In parallel, the analytical specificity was inferred by serially diluting the recombinant plasmid containing the NS5 region of the 17DD YFV in healthy human serum or in reconstituted sera pool containing dengue virus (DENV1, DENV2 and DENV3) with 105 copies/reaction, mumps virus (MuV) with 103 copies/reaction and measles virus (MV) with 103 copies/reaction.
The analytical sensitivity was established using 8 replicates of serially diluted plasmid DNA standard samples, starting from 107 copies up to 1.5 copies/reaction in 2 independent experiments. For simultaneous evaluation of both, the detection and quantification limits, dilutions beginning with 107 up to 1.5 plasmid copies/reaction were performed in 8 replicas each.
To analyze the precision of the method, 2 sample panels were used as mentioned: reconstituted serological panel and clinical samples from vaccinated individuals. The tests were repeated by the same operator on different days and by different operators on the same day. The serological reconstituted panel was processed by 2 different operators and repeated in 4 independent trials: distinct operators conducted 2 assays on the same day and 2 other tests were performed on different days. For the panel constituted by clinical samples, 3 independent tests were conducted: 2 of them performed on the same day with different operators and only one operator held the third test on a different day.
Reference controls
To ensure the quality of the test, excluding the possibility of false negative due to the presence of eventual inhibitors or the quality and integrity of cDNA samples, the TaqMan® RNase P Control Reagents kit, Applied Biosystems (Catalog number 4316844) was used as an endogenous internal control for the analysis of human clinical samples. Alternatively, for the assays using in vitro free serum samples, the TaqMan® Exogenous Internal Positive Control Reagents, EXO IPC, Applied Biosystems (Catalog number 4308323) was employed. In this case, samples were spiked with EXO IPC before processing, and the reaction was conducted according to the manufacturer's specifications. In both situations, TaqMan probes were 5′ labeled with VIC fluorophore and 3′MGB. For testing clinical samples, the reference control was included in multiplexed assays (targeted to the genomic NS5 viral region and human RNase P gene). Separate reactions in the same plate (singleplex) were established for the analysis of in vitro free serum samples spiked with the exogenous positive control (EXO IPC).
Virus titration using VERO cells
Virus titration was performed on confluent Vero cells (ATCC, CCL 81) using a multiplicity of infection (MOI) of 0.02 in 6-well plates. After an incubation of 1 h at 37°C, the inoculum was removed by aspiration and the cells were overlaid with 3% carboxymethylcellulose in Earle´s medium. On day 7 after infection, virus plaques were fixed with 10% formaldehyde and visualized using 0.02% crystal violet.
Statistical analysis
Statistical analysis was performed with the GraphPad Prism® version 5.01 using Pearson's correlation test. All samples were tested at least in 3 replicates and p-values ≤0.05 were considered statistically significant.
Acknowledgments
C. Britto is research fellow of CNPq. The authors thank the Program for Technological Development in Tools for Health (PDTIS-Fiocruz) and Bio-Manguinhos, for the facilities on the Real Time PCR and DNA sequencing platforms. The authors would like to acknowledge Marcelo Ribeiro-Alves for his input on the statistical analyses; Reinaldo de Menezes Martins who contributed to the central coordination of the study and regulatory affairs support.
Disclosure of Potential Conflicts of Interest
Alice G. Fernandes-Monteiro, Gisela F. Trindade, Anna M. Y. Yamamura, Ana Cláudia M. Duarte, and Sheila Maria B. Lima work permanently for Bio-Manguinhos/Fiocruz, a government-owned, not for profit, a Brazilian vaccine producer, including the yellow fever vaccine assessed in the study. Constança Britto, Otacilio C. Moreira, and Vanessa S. de Paula work permanently for the Oswaldo Cruz Institute (IOC), a government-owned, not for profit, a Brazilian institute of research from Fiocruz.
Funding
This work was supported by PROEP/IOC-Fiocruz/CNPq (National Council for Scientific and Technological Development) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ E-26/110.594/2012 and CNE E-26/102.775/2012).
References
- 1.Monath TP. Pathobiology of Flaviviruses “Togaviridae and the Flaviviridae”. Schlesinger S, Schlesinger MJ, eds. New York: Plenum Press; 1986:375-440. [Google Scholar]
- 2.Chambers TJ, Hahn CS, Galler R, Rice CM. Flavivirus genome organisation, expression and replication. Annu Rev Microbiol 1990; 44:649-88; PMID:2174669; http://dx.doi.org/ 10.1146/annurev.mi.44.100190.003245 [DOI] [PubMed] [Google Scholar]
- 3.Lindenbach BD, Rice CM. Flaviviridae: The viruses and their replication. Philadelphia: Lippincott Williams & Wilkins; 2001. 991-1042 (Knipe DM, Howley PM, editors. Fields Virology; vol. 1) [Google Scholar]
- 4.Brinton MA. The molecular biology of West Nile Virus: a new invader of the western hemisphere. Annu Rev Microbiol 2002; 56:371-402; PMID:12142476; http://dx.doi.org/ 10.1146/annurev.micro.56.012302.160654 [DOI] [PubMed] [Google Scholar]
- 5.Burke DS, Monath TP. Flaviviruses. Philadelphia: Lippincott, Williams & Wilkins; 2001. 1043–1125 (Knipe DM, Howley PM, editors. Fields Virology; vol. 1) [Google Scholar]
- 6.Martins RM, Maia MD, Farias RH, Camacho LA, Freire MS, Galler R, Yamamura AM, Almeida LF, Lima SM, Nogueira RM, Sá GR, et al.. 17DD yellow fever vaccine: a double blind, randomized clinical trial of immunogenicity and safety on a dose-response study. Hum Vaccin Immunother 2013:30-9; PMID:23364472; http://dx.doi.org/ 10.4161/hv.22982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deubel V, Huerre M, Cathomas G, Drouet MT, Wuscher N, Le Guenno B, Widmer AF. Molecular detection and characterization of yellow fever virus in blood and liver specimens of a non-vaccinated fatal human case. J Med Virol 1997; 53:212-7; PMID:9365884; http://dx.doi.org/ 10.1002/(SICI)1096-9071(199711)53:3%3c212::AID-JMV5%3e3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- 8.De Brito T, Siqueira SA, Santos RT, Nassar ES, Coimbra TL, Alves VA. Human fatal yellow fever. Immunohistochemical detection of viral antigens in the liver, kidney and heart. Pathol Res Pract 1992; 188:177-81; PMID:1594489; http://dx.doi.org/ 10.1016/S0344-0338(11)81176-3 [DOI] [PubMed] [Google Scholar]
- 9.Monath TP, Ballinger ME, Miller BR, Salaun JJ. Detection of yellow fever viral RNA by nucleic acid hybridization and viral antigen by immunocytochemistry in fixed human liver. Am J Trop Med Hyg 1989; 40:663-8; PMID:2500857 [DOI] [PubMed] [Google Scholar]
- 10.Monath TP, Nystrom RR. Detection of yellow fever virus in serum by enzyme immunoassay. Am J Trop Med Hyg 1984; 33:151-7; PMID:6364854 [DOI] [PubMed] [Google Scholar]
- 11.Porterfield JS. A plaque technique for the titration of yellow fever virus and antisera. Trans R Soc Trop Med Hyg 1959; 53:458-65; PMID:14434289; http://dx.doi.org/ 10.1016/0035-9203(59)90021-5 [DOI] [PubMed] [Google Scholar]
- 12.Mackay IM, Arden KE, Nitsche A. Real-Time PCR in virology. Nucleic Acids Res 2002; 30:1292-305; PMID:11884626; http://dx.doi.org/ 10.1093/nar/30.6.1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manojkumar R, Mrudula V. Applications of Real-Time reverse transcription polymerase chain reaction in clinical virology laboratories for the diagnosis of human diseases. Am J Infectious Dis 2006; 2:204-9; http://dx.doi.org/ 10.3844/ajidsp.2006.204.209 [DOI] [Google Scholar]
- 14.ANVISA , 2003. http://www.anvisa.gov,br/legis/resol/2003/re/898_03re.htm (ANVISA – Agência Nacional de Vigilância Sanitária, 2003 Accessed in July24, 2012) [Google Scholar]
- 15.Barbana V, Girerda Y, Aguirrea M, Guliaa S, Pétiarda F, Rioub P, Barrereb B, Langa J. High stability of yellow fever 17D-204 vaccine: A 12-year restrospective analysis of 11 large-scale production. Vaccine 2007; 25:2941-50; PMID:16914238; http://dx.doi.org/ 10.1016/j.vaccine.2006.06.082 [DOI] [PubMed] [Google Scholar]
- 16.Bae HG, Nitsche A, Teichmann A, Biel SS, Niedrig M. Detection of yellow fever virus: a comparison of quantitative Real-Time PCR and plaque assay. J Virol Methods 2003; 110:185-91; PMID:12798247; http://dx.doi.org/ 10.1016/S0166-0934(03)00129-0 [DOI] [PubMed] [Google Scholar]
- 17.Mantel N, Aguirre M, Gulia S, Girerd-Chambaz Y, Colombani S, Moste C, Barban V. Standardized quantitative RT-PCR assays for quantitation of yellow fever and chimeric yellow fever-dengue vaccines. J Virol Methods 2008; 151:40-6; PMID:18501437; http://dx.doi.org/ 10.1016/j.jviromet.2008.03.026 [DOI] [PubMed] [Google Scholar]
- 18.Gurukumar KR, Priyadarshini D, Patil JA, Bhagat A, Singh A, Shah PS, Cecilia D. Development of real time PCR for detection and quantitation of Dengue Viruses. Virol J 2009; 23:6-10; PMID:19166574; http://dx.doi.org/ 10.1186/1743-422X-6-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.da Cunha RV, Dias M, Nogueira RMR, Chagas N, Miagostovich MP, Schatzmayr H. Secondary dengue infection in school childrens in a dengue endemic área in the state of Rio de Janeiro, Brazil. Rev Inst Med Trop S Paulo 1995; 37:517-21; PMID:8731265; http://dx.doi.org/ 10.1590/S0036-46651995000600008 [DOI] [PubMed] [Google Scholar]
- 20.Sloan LM. Real-Time PCR in clinical microbiology: verification, validation, and contamination control. Clin Microbiol Newsletter 2007; 29; http://dx.doi.org/ 10.1016/j.clinmicnews.2007.05.002 [DOI] [Google Scholar]
- 21.Burd EM. Validation of laboratory-developed molecular assays for infectious diseases. Clin Microbiol Rev 2010; 23:550-76; PMID:20610823; http://dx.doi.org/ 10.1128/CMR.00074-09 [DOI] [PMC free article] [PubMed] [Google Scholar]