

Abstract
Patient: Male, 60
Final Diagnosis: IgG4 related disease
Symptoms: Cough • hemoptysis
Medication: —
Clinical Procedure: None
Specialty: Pulmonology
Objective:
Rare disease
Background:
Immunoglobulin (Ig) G4-related disease, previously referred to as IgG4-related sclerosing disease or hyper-IgG4 disease, may occur in the lung, involving alveolar parenchyma, airways, and pleura. Various pulmonary manifestations of IgG4-related disease have been reported, but to the best of our knowledge a cavitating lung disease has not been reported previously.
Case Report:
We describe a 60-year-old man who presented with hemoptysis and cavitating lung disease with clinical, laboratory, and histopathologic findings compatible with IgG4-related disease. Other potential causes of cavitation were excluded. Treatment was initiated with oral prednisone and subsequently mycophenolate mofetil was added. Follow-up 1 year later shows stable pulmonary function with complete resolution of the cavitary lesions.
Conclusions:
We present a case of cavitating lung disease as a previously unreported manifestation of IgG4-related disease. Our patient had an excellent response to immunosuppression. An increased awareness of IgG4-related disease and its myriad of manifestations is very important for pulmonologists.
MeSH Keywords: Hemoptysis, Immunoglobulin G, Cough
Background
Immunoglobulin (Ig) G4-related disease, previously referred to as IgG4-related sclerosing disease or hyper-IgG4 disease, may occur in the lung, involving alveolar parenchyma, airways, mediastinum, and pleura. Various pulmonary manifestations of IgG4-related disease have been reported but a cavitating lung disease has not been documented in this setting. Herein, we describe a 60-year-old man who presented with hemoptysis and cavitating lung disease with clinical, laboratory, and histopathologic findings compatible with IgG4-related disease. To the best of our knowledge, this is the first reported case of IgG4-related disease manifesting as a cavitating lung lesion.
Case Report
A 60-year-old white male presented for a second opinion regarding a 2-year history of cough and streaky hemoptysis. He was an active smoker with history of smoking for 40 pack-years. Before presenting to our center, he had undergone a diagnostic evaluation elsewhere that included a chest CT (which revealed a thick-walled cavitating lesion in the left upper lobe) and a bronchoscopy with nondiagnostic transbronchial biopsy results. He was treated with a course of broad-spectrum antibiotics with no improvement, followed by a thoracotomy with resection of the cavitary lesion. The biopsy specimen apparently revealed an inflammatory lesion with no evidence of malignancy. Review of all microbial cultures, including mycobacterial culture of the lung tissue, was negative.
His cough was associated with whitish phlegm, and had minimal nocturnal symptoms. He never had a frank hemoptysis, and noted blood-streaked sputum on many occasions. He complained of shortness of breath, which was grade 1 on the mMRC scale. He denied night sweats, weight loss, or loss of appetite. His medical history was significant for COPD, dyslipidemia, reflux esophagitis, and obstructive sleep apnea. His cough and hemoptysis continued even after complete resection of the cavitary lesion.
On examination, he was afebrile with a SpO2 of 97% on room air at rest. Bilateral nontender parotid gland enlargement was noted. The rest of the physical exam and review of systems was without abnormality. Laboratory studies revealed anemia with a hemoglobin of 11.5 gm/dl, erythrocyte sedimentation rate of 67 mm/hour, and an elevated C-reactive protein of 17.3 mg/L. Anti-nuclear antibody was positive at 3.6 U (normal, <1 U). Tests for anti-neutrophil cytoplasmic antibody (ANCA), rheumatoid factor, and anti-cyclic citrullinated peptide (anti-CCP) antibodies were negative.
Pulmonary function test (PFT) revealed moderate obstruction with a normal diffusing capacity. Chest radiography demonstrated multiple cavitary lesions in the left perihilar region (Figure 1). A CT chest (Figure 2) confirmed 3 thick-walled new cavitary lesions; 1 in the left upper and 2 in the left lower lobes. The main diagnostic considerations included chronic infections (especially fungi and mycobacteria), vasculitis (especially granulomatosis with polyangiitis [GPA]), and neoplasms. His outside surgical lung biopsy specimens were obtained to review and revealed a cavitary lesion with necrosis; it was surrounded by histiocytes, dense lymphoplasmacytic infiltrate, and fibrosis (Figure 3A). Focal neutrophilic infiltration was present. Obliterative phlebitis was not identified. There were no microorganisms noted on hematoxylin eosin stain or on special stains, including Gomori Methenamine Silver and Ziehl Neelson for fungi and mycobacteria, respectively. In view of the dense lymphoplasmacytic infiltrate, immunostaining was requested.
Figure 1.
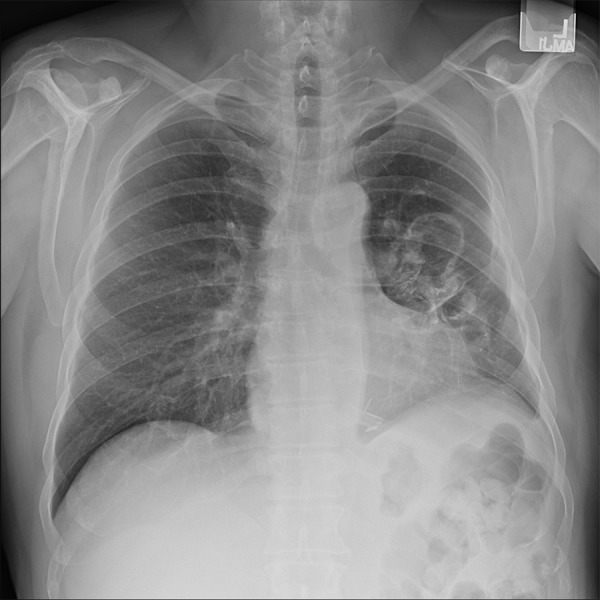
Chest x-ray showing multiple relatively thick-walled cystic air spaces in the left perihilar region.
Figure 2.

CT chest at initial presentation showing multiple thick-walled cavitary lesions in the left upper and lower lobes.
Figure 3.
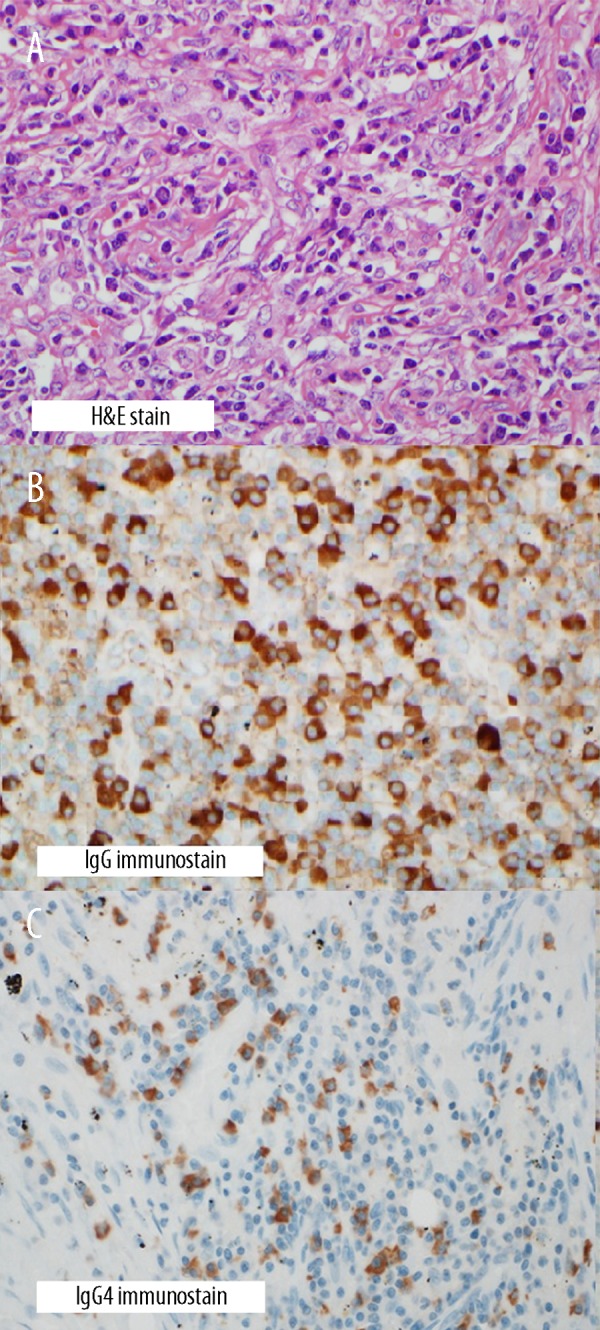
(A) Hematoxylin and eosin (HE) stain showing histiocytes, dense lymphoplasmacytic infiltrate, and fibrosis (400× original magnification). (B) IgG immunostain showing increased number of IgG+ plasma cells (400× original magnification). (C) IgG4 immunostain showing about 60 IgG4+ plasma cells per high power field (400× original magnification).
This (Figure 3B, 3C) showed approximately 60 IgG4-positive (+) cells per high-power field and an IgG4+/IgG+ plasma cell ratio of 23%. No classic features of GPA such as necrotizing vasculitis, parenchymal necrosis with surrounding granuloma, heavy neutrophilic infiltrates, or evidence of hemorrhage were observed. Total serum immunoglobulin (Ig)-G assay was done, and was found to be elevated at 1820 mg/dl (normal, 767–1590 mg/dl) with a mildly elevated IgG4 at 171 mg/dl (normal, 2.4–121 mg/dl).
Although the presence of neutrophilic aggregates has been noted in pulmonary IgG4-related lung disease (IgG4-RLD), a cavitary lesion has not been documented as a feature of IgG4-RLD. We believe that this case could be a possible cavitary IgG4-RLD in light of his other manifestations, including the parotid gland enlargement elevated serum IgG4 level, and the lack of any viable alternative diagnoses. While the histopathologic diagnosis was challenging due to the presence of cavitary lesion, the remaining histopathologic findings with increased IgG4+ plasma cells were acceptable for IgG4-RLD. There was no evidence of extrapulmonary IgG4-related disease other than parotid gland enlargement. Treatment was initiated with oral prednisone 60 mg/day, and subsequently mycophenolate mofetil (MMF) was added with slow tapering of prednisone over the following 2 months. Follow-up 1 year later while on 2 gm/day of MMF and a tapering dose of prednisone at 5 mg per day showed stable pulmonary function. Follow-up CT chest (Figure 4) 1 year later revealed complete resolution of the cavitary lesions, with residual scarring. Parotid gland swelling also resolved. Serum IgG4 levels normalized 6 weeks after initiation of prednisone.
Figure 4.

Follow-up CT chest 1 year later showing complete resolution of previously noted cavitary lesions in the left upper and lower lobes, with residual scarring.
Discussion
IgG4 accounts for only 4–6% of the total serum IgG in normal subjects, and is the least abundant of the IgG subclasses [1]. Serum IgG4 level can be elevated in a number of allergic and auto-immune disorders like autoimmune pancreatitis, primary sclerosing cholangitis, ANCA vasculitis, rheumatoid arthritis, inflammatory bowel disease, cutaneous plasmacytosis, and autoimmune atrophic gastritis [2,3,4].
IgG4-RD is a systemic fibro-inflammatory disease associated with IgG4-positive plasma cell infiltration and elevated circulating levels of IgG4 [5,6]. Virtually any organ system can be involved in IgG4-RD [7]. The 3 major histopathological features associated with IgG4-RD are dense lymphoplasmacytic infiltrate, storiform fibrosis, and obliterative phlebitis [8]. However, the last 2 features are often absent in some sites, including lung [8], lymph nodes [9], minor salivary glands, and lacrimal glands [10]. Intrathoracic manifestations of IgG4-RD include parenchymal nodules or masses, interstitial lung disease, tracheobronchial stenosis, pleural nodules, pleural effusion, mediastinal lymphadenopathy, and fibrosing mediastinitis [11]. However, cavitary lung disease has not been previously reported in IgG4-RD.
The consensus statement requires an elevated number of IgG4-positive plasma cells within tissues along with characteristic histopathologic features for diagnosis [12]. In surgical lung biopsy specimens, >50 IgG4-positive plasma cells per high-power field and the IgG4+/IgG+ plasma cell ratio of >40% is reportedly highly specific [13,14].
There are no randomized treatment trials for IgG4-RD, and treatment decisions are based on data from retrospective studies. Although most patients respond favorably to glucocorticoids [15], the dosing and duration of treatment are largely undefined. A minority of patients are steroid-resistant. Azathioprine, MMF, and rituximab have been reported to be effective in these cases, as well as those requiring prolonged therapy [16,17].
Conclusions
We present a case of cavitating lung disease as a possible manifestation of IgG4-RD that has not been reported previously. Our patient had an excellent response to immunosuppression, with complete resolution of all lesions. An increased awareness of IgG4-RD and its myriad pulmonary manifestations is very important for the pulmonologist.
References:
- 1.Van der Zee JS, Aalberse RC. Immunochemical characteristics of IgG4 antibodies. N Engl Reg Allergy Proc. 1998;9:31–33. doi: 10.2500/108854188778984554. [DOI] [PubMed] [Google Scholar]
- 2.Kamisawa T, Egawa N, Nakajima H. Autoimmune pancreatitis is a systemic autoimmune disease. Am J Gastroenterol. 2003;98:2811–12. doi: 10.1111/j.1572-0241.2003.08758.x. [DOI] [PubMed] [Google Scholar]
- 3.Narula N, Vasudev M, Marshall JK. IgG-related sclerosing disease: a novel mimic of inflammatory bowel disease. Dig Dis Sci. 2010;55:3047–51. doi: 10.1007/s10620-010-1287-1. [DOI] [PubMed] [Google Scholar]
- 4.Ryu JH, Horie R, Sekiguchi H, et al. Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. Int J Rheumatol. 2012;2012:232960. doi: 10.1155/2012/232960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol. 2008;14(25):3948–55. doi: 10.3748/wjg.14.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zen Y, Nakanuma Y. IgG4-related disease: a cross-sectional study of 114 cases. Am J Surg Pathol. 2010;34(12):1812–19. doi: 10.1097/PAS.0b013e3181f7266b. [DOI] [PubMed] [Google Scholar]
- 7.Stone JH, Zen Y, Deshpande V. IgG4-related disease. N Engl J Med. 2012;366:539–51. doi: 10.1056/NEJMra1104650. [DOI] [PubMed] [Google Scholar]
- 8.Shrestha B, Sekiguchi H, Colby TV, et al. Distinctive pulmonary histopathology with increased IgG4-positive plasma cells in patients with auto-immune pancreatitis: report of 6 and 12 cases with similar histopathology. Am J Surg Pathol. 2009;33:1450–62. doi: 10.1097/PAS.0b013e3181ac43b6. [DOI] [PubMed] [Google Scholar]
- 9.Cheuk W, Yuen HK, Chu SY, et al. Lymphadenopathy of IgG4-related sclerosing disease. Am J Surg Pathol. 2008;32:671–81. doi: 10.1097/PAS.0b013e318157c068. [DOI] [PubMed] [Google Scholar]
- 10.Cheuk W, Yuen HK, Chan JK. Chronic sclerosing dacryoadenitis: part of the spectrum of IgG4-related Sclerosing disease? Am J Surg Pathol. 2007;31:643–45. doi: 10.1097/01.pas.0000213445.08902.11. [DOI] [PubMed] [Google Scholar]
- 11.Ryu JH, Sekiguchi H, Yi ES. Pulmonary manifestations of immunoglobulin G4-related sclerosing disease. Eur Respir J. 2012;39(1):180–86. doi: 10.1183/09031936.00025211. [DOI] [PubMed] [Google Scholar]
- 12.Deshpande V, Zen Y, Chan JK, et al. Consensus statement on the pathology of IgG4-related disease. Mod Pathol. 2012;25(9):1181–92. doi: 10.1038/modpathol.2012.72. [DOI] [PubMed] [Google Scholar]
- 13.Zen Y, Kitagawa S, Minato H, et al. IgG4-positive plasma cells in inflammatory pseudotumor (plasma cell granuloma) of the lung. Hum Pathol. 2005;36(7):710–17. doi: 10.1016/j.humpath.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 14.Cheuk W, Chan JK. IgG4-related sclerosing disease: a critical appraisal of an evolving clinicopathologic entity. Adv Anat Pathol. 2010;17:303–32. doi: 10.1097/PAP.0b013e3181ee63ce. [DOI] [PubMed] [Google Scholar]
- 15.Zen Y, Inoue D, Kitao A, et al. IgG4-related lung and pleural disease: a clinicopathologic study of 21 cases. Am J Surg Pathol. 2009;33(12):1886–934. doi: 10.1097/PAS.0b013e3181bd535b. [DOI] [PubMed] [Google Scholar]
- 16.Khosroshahi A, Bloch DB, Deshpande V, et al. Rituximab therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum. 2010;62(6):1755–62. doi: 10.1002/art.27435. [DOI] [PubMed] [Google Scholar]
- 17.Ghazale A, Chari ST, Zhang L, et al. Immunoglobulin G4-associated cholangitis: clinical profile and response to therapy. Gastroenterology. 2008;134:706–15. doi: 10.1053/j.gastro.2007.12.009. [DOI] [PubMed] [Google Scholar]