

Abstract
Purpose
Individuals with sex chromosome aneuploidies (SCAs) are born with an atypical number of X and/or Y chromosomes, and present with a range of medical, developmental, educational, behavioral, and psychological concerns. Rates of SCA diagnoses in infants and children are increasing, and there is a need for specialized interdisciplinary care to address associated risks. The eXtraordinarY Kids Clinic was established to provide comprehensive and experienced care for children and adolescents with SCA, with an interdisciplinary team composed of developmental–behavioral pediatrics, endocrinology, genetic counseling, child psychology, pediatric neuropsychology, speech–language pathology, occupational therapy, nursing, and social work. The clinic model includes an interdisciplinary approach to care, where assessment results by each discipline are integrated to develop unified diagnostic impressions and treatment plans individualized for each patient. Additional objectives of the eXtraordinarY Kids Clinic program include prenatal genetic counseling, research, education, family support, and advocacy.
Methods
Satisfaction surveys were distributed to 496 patients, and responses were received from 168 unique patients.
Results
Satisfaction with the overall clinic visit was ranked as “very satisfied” in 85%, and as “satisfied” in another 9.8%. Results further demonstrate specific benefits from the clinic experience, the importance of a knowledgeable clinic coordinator, and support the need for similar clinics across the country. Three case examples of the interdisciplinary approach to assessment and treatment are included.
Keywords: XXY, Klinefelter syndrome, XYY, XXYY, trisomy X, XXX, Turner syndrome, XXXY, XXXXY, tetrasomy X, pentasomy X, prenatal diagnosis
Introduction
Sex chromosome aneuploidies (SCAs) are a common but under-recognized group of genetic conditions occurring in approximately 1:400 individuals characterized by an atypical number of X and/or Y chromosomes. While the typical male karyotype is 46,XY and female is 46,XX, the most common SCAs include males with 47,XXY (Klinefelter syndrome [KS]) and 47,XYY, and females with 47,XXX (trisomy X syndrome), and Turner syndrome (TS) (45,X and variations). Less common variations occur with the addition of more than one extra sex chromosome, including 48,XXYY, 48,XXXY, and 49,XXXXY in males and 48,XXXX (tetrasomy X) and 49,XXXXX (pentasomy X) in females.
Individuals with SCA may present with a range of medical, neurodevelopmental, educational, behavioral, and psychological concerns. There is significant variability among individuals with SCA in the presence and degree of involvement of these concerns. Table 1 lists the neurodevelopmental and psychological conditions that have been reported to be associated with SCA conditions involving extra X and/or Y chromosomes.1–10 In general, the neurodevelopmental and cognitive impairments of the trisomy conditions (XXY/KS, XYY, and trisomy X) are of decreased severity in comparison to the tetrasomy and pentasomy conditions (XXYY, XXXY, XXXXY, XXXX, XXXXX); however, there is still significant variability among individuals within each diagnostic category.11–13 Each SCA variation is also associated with a set of medical and physical features, some of which are core features that require close medical follow-up and treatment for almost all individuals with the condition (such as hypogonadism in KS and TS). Other medical conditions are known to occur at higher rates in SCA in comparison to the general population, and thus must be monitored and considered during medical evaluation and follow-up. Medical and physical features associated with SCAs are listed in Table 2.
Table 1.
Neurodevelopmental and psychological risks in SCA conditions
| Developmental delay Speech–language delay Motor skills delay Cognitive impairments/intellectual disability Learning disabilities Language-based learning disability/dyslexia Speech and language disorders Receptive–expressive language disorder Apraxia/dyspraxia of speech Motor skills disorders Motor coordination problems Hypotonia Graphomotor deficits |
Executive functioning deficits Slow processing speed Attention deficit hyperactivity disorder (ADHD) Adaptive functioning deficits Sensory processing differences Social difficulties Social cognitive deficits Social–emotional immaturity Autism spectrum disorders Emotional/psychological disorders: Anxiety disorder Depression Mood disorders/bipolar disorder Psychotic features/schizophrenia |
Abbreviation: SCA, sex chromosome aneuploidy.
Table 2.
Medical and physical features of SCA conditions
| 47,XXY | 48,XXYY
|
47,XYY | 47,XXX | 48,XXXX
|
|
|---|---|---|---|---|---|
| 48,XXXY
|
49,XXXXX
|
||||
| 49,XXXXY | |||||
| Strongly associated with: | |||||
| Endocrinologic | |||||
| Hypergonadotropic hypogonadism (testosterone deficiency/microorchidism) | X | X | |||
| Infertility/fertility problems | X | X | |||
| Increased risk for: | |||||
| Neurologic | |||||
| Seizure disorder | X | X | X | X | X |
| Hypotonia | X | X | X | X | X |
| Coordination/motor skills disorders | X | X | X | X | X |
| Tremor | X | X | X | X | X |
| Ear, Nose and Throat | |||||
| Cleft palate | X | X | X | X | X |
| Velopharyngeal insufficiency | X | X | X | X | X |
| Obstructive sleep apnea | X | X | X | X | X |
| Cardiopulmonary | |||||
| Congenital heart malformation | X | X | X | X | X |
| Asthma/reactive airway disease | X | X | X | X | X |
| Gastrointestinal | |||||
| Constipation | X | X | X | X | X |
| Functional abdominal pain | X | X | |||
| Eosinophilic esophagitis | X | X | X | X | X |
| Musculoskeletal | |||||
| Flat feet/pes planus | X | X | X | X | X |
| Joint hypermobility | X | X | X | X | X |
| Radioulnar synostosis/congenital elbow dislocation | X | X | X | X | X |
| Congenital hip dysplasia | X | X | |||
| Clubfoot | X | X | X | X | |
| Scoliosis | X | X | X | X | X |
| Endocrinologic | |||||
| Hypothyroidism | X | X | X | X | X |
| Diabetes mellitus/type II | X | X | X | X | X |
| Gynecomastia | X | X | |||
| Premature ovarian failure | X | X | |||
| Ovarian/uterine malformation | X | X | |||
| Rheumatologic/autoimmune | |||||
| Systemic lupus erythematosus | X | X | |||
| Rheumatoid arthritis | X | X | |||
| Hematologic/oncologic | |||||
| Deep vein thromboses/blood clots | X | X | |||
| Venous ulcers/varicosities | X | X | |||
| Breast cancer | X | X | |||
| Non-Hodgkin’s lymphoma | X | ||||
| Teratoma/germ cell tumor (testicular or extragonadal) | X | X | |||
| Other medical | |||||
| Strabismus | X | X | X | X | X |
| Environmental allergies | X | X | X | X | X |
| Kidney aplasia/dysplasia | X | X | X | ||
| Cryptorchidism (undescended testicles) | X | X | |||
| Dental | |||||
| Atypical dental development | X | X | X | X | X |
| Thin enamel/dental caries | X | X | X | X | X |
| Taurodontism | X | X | X | X | X |
Given the long list of associated conditions in both neurodevelopmental and medical domains, comprehensive care for these patients often requires evaluation by many different providers. Since there can be considerable interplay between neurodevelopmental and medical features, an understanding of the role one may play on the other is critical for care. While there are some published treatment recommendations for specific SCA conditions (mostly XXY/KS and TS)14–16 or specific aspects of care (testosterone replacement therapy in XXY/KS),17 evidence-based treatment recommendations are lacking. Currently, children and adolescents with SCA often receive care by genetics clinics where families are provided with information about the SCA condition after diagnosis, and by pediatric endocrinologists who often have experience with hormone treatment in XXY and TS. Beyond these specialties, there remains little expertise with SCA among other medical providers, psychologists, developmental pediatricians, and other specialists/therapists involved in the care of children with SCA. As a result, parents/caregivers often do not have access to experienced providers who can incorporate all different aspects of SCA into understanding how the child’s disorder affects their health, behavior, learning, and overall daily functioning. This requires caregivers to “piece together” evaluations, as well as to educate the professionals working with children about SCA.
In response to the need for comprehensive and coordinated care for children and adolescents with SCA, the eXtraordinarY Kids Clinic was established at Children’s Hospital Colorado and the University of Colorado School of Medicine to provide specialized, interdisciplinary assessment and treatment in this population. Figure 1 illustrates the components of the eXtraordinarY Kids Clinic program. The clinic is composed of a primary team of professionals from different disciplines including:
developmental–behavioral pediatrics (clinic director)
pediatric endocrinology
genetic counseling (clinic coordinator)
child psychology
pediatric neuropsychology
speech–language therapy
occupational therapy
nursing
social work.
Figure 1.

Diagram of eXtraordinarY Kids Clinic model.
Abbreviations: SCA, sex chromosome aneuploidy; ADHD, attention deficit hyperactivity disorder; CVS, chorionic villus sampling; DBPediatrics, Developmental-Behavioral Pediatrics.
The specific evaluations and team members involved are individualized for each patient based on an intake phone call and review of records. In response to the varied needs in this population, our clinical services have also evolved to include prenatal genetic counseling, developmental monitoring of infants diagnosed in the prenatal period or in infancy, comprehensive evaluations for school-age and adolescent patients, interdisciplinary endocrinology and developmental–behavioral pediatrics follow-up, and psychopharmacologic medication management. If needed, additional consultative services are available in the areas of physical therapy, clinical genetics, child neurology, psychiatry, otolaryngology, orthopedics, adolescent gynecology, and reproductive urology. Clinic team members collaborate to provide integrated diagnostic impressions and treatment recommendations specific to the child’s assessment and SCA diagnosis. Families receive feedback from the clinic director and pertinent team members at the conclusion of the visit, as well as a comprehensive written report with findings and recommendations. Figure 1 shows the components of the eXtraordinarY Kids Clinic program.
Additional objectives of the eXtraordinarY Kids Clinic program include research, education, family support, and advocacy. The large clinic population allows for clinical and translational research related to developmental trajectories, neurodevelopmental disorders, genotype–phenotype studies, clinical trials, and maintenance of a DNA biobank. The location of the clinic at a teaching hospital and its affiliation with training programs at the University of Colorado School of Medicine offers medical students and residents the opportunity to rotate through the clinic to learn about SCA conditions, as well as trainees in genetic counseling, psychology, and speech and occupational therapy. The program provides support and educational opportunities for families by sponsoring local social and educational events, and also collaborates with national advocacy organizations (AXYS and The XXYY Project) to host or provide speakers at conferences for families and professionals.
Since the inception of the eXtraordinarY Kids Clinic in the fall of 2007, more than 400 new patients have been seen. Many patients continue to be seen for ongoing medical care and follow-up evaluation, with over 1000 patient visits to date. Approximately 50% of patients have been from Colorado, while the other half have traveled from around the USA and internationally. Among unique patients, SCA diagnoses have included 43% with XXY/KS, 17% with XXX, 11% with XYY, 18% with tetrasomy and pentasomy conditions, and 11% with other variations or rearrangements. A small number of girls with TS have been followed through the clinic (5% of visits). A specialized program for TS is being developed within the eXtraordinarY Kids Clinic for 2015 that will address the more complex medical needs in TS while maintaining an interdisciplinary approach incorporating neurodevelopmental and psychological care.
In 2011, the clinic launched a satisfaction survey as a quality assurance and improvement measure. Goals of this measure were to assess satisfaction with clinical services provided by team members, perceived benefits of the evaluations, and satisfaction with clinical processes. Survey results are summarized and provide support for the eXtraordinarY Kids Clinic as an optimal model of care for children and adolescents with SCAs.
Materials and methods
Procedure
All families seen in clinic were sent a survey 4–8 weeks after their clinical visit. The first 363 surveys were mailed and included a cover letter and postage-paid return envelope. Later, a web-based version of the survey was launched utilizing REDCap, a secure web application designed to support data capture for research. Electronic surveys were disseminated to an additional 133 families via email, providing the cover letter and a web link to the survey. The cover letter specified that the responses were to be anonymous, and that electronic responses would remain de-identified. Families without an email address were mailed a paper copy and a return envelope.
Survey
The survey asked respondents to rate their overall satisfaction with the clinic experience (1= very satisfied, 2= somewhat satisfied, 3= somewhat disappointed, 4= very disappointed), and the quality of care provided by each team member (1= poor, 2= fair, 3= good, 4= very good, 5= excellent). Next, respondents were asked to select the benefits they received from their visit from a list of 13 possible benefits. They were asked to describe using free text the parts of the clinic experience that they found most helpful and suggestions for improvements. Finally, respondents ranked satisfaction with respect to various aspects of clinical operations (1= very disappointed to 5= very satisfied).
Results
A total of 168/496 surveys were completed (33.7% response rate), of which 85/363 surveys were paper-based (23.42%) and 83/133 were web-based (62.4%). One survey was discarded due to comments indicating they were seen in a different department, and another because no responses were completed. Thus, a total of 166 surveys were analyzed. Patient diagnoses were provided in 98.8% (164/166) of surveys and included: XXY (n=74), XXX (n=29), XXYY (n=26), XYY (n=22), XXXY (n=7), TS (n=5), and “other” (n=1). The proportion of respondents from each SCA condition was similar to the proportion of new patients with that condition, thus a specific SCA group was not disproportionately represented.
Eighty-five percent (139/163) of respondents ranked the overall satisfaction with the clinic visit as “very satisfied”, and another 9.8% as “satisfied” (16/163), as shown in Figure 2. There were not statistically significant differences in overall satisfaction scores between SCA subtypes (ANOVA, F(5,158) 0.66, P=0.65). Respondents also evaluated each specialist, and all team members received high satisfaction scores with mean scores ranging from 4.63 to 4.95 (4= very good, 5= excellent). The highest ranking specialties were genetic counseling/clinic coordination (4.95) and developmental–behavioral pediatrics (4.90).
Figure 2.
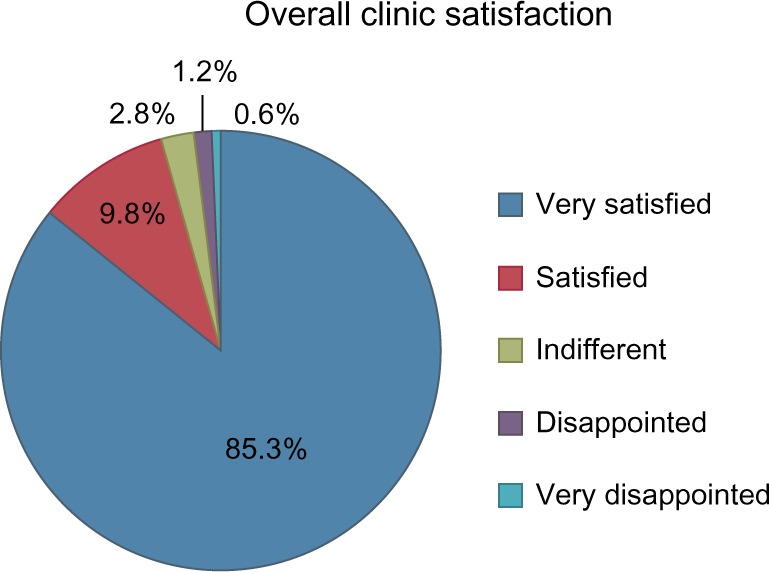
Overall satisfaction scores after a clinic visit (n=164).
Selected examples of positive free text write-in comments include:
“Our satisfaction comes from the approach of the clinic. It is complete. Coming from another state where no one knows about XXYY, it was wonderful to discuss the varied aspects of his condition with so many professionals in one facility. The sense of caring/compassion was wonderful!”
“We talked with the genetic counselor and doctor over the phone after our prenatal diagnosis, which was so helpful since the counselors at home had never met anyone with XXY and scared us with information they presented. We came for developmental testing at 8 months old, and they were very reassuring, had specific recommendations, and he is doing great now. What a relief to have a team who knows about XXY.”
“The Clinic’s team approach ensured that everyone knew in advance about my daughter’s issues and consulted with each other for final recommendations. We got a lot of action items to address specific issues. However, the best advice we received was to remember to focus on her strengths. Being reminded of her strengths and encouraged to celebrate them was refreshing.”
“Every provider genuinely cared about my son. From when I received the prenatal XXY diagnosis until now (3 years), no one has been able to confidently answer my questions about his development. Every provider was very thorough, we did not feel rushed, and there was adequate time to ask questions.”
“The report serves as important feedback to local providers who typically have no experience with XXX, and forces them to incorporate an understanding of XXX into their therapies. It’s otherwise easy for others to disregard the impact of the chromosomal condition, so having the reports from the eXtraordinarY Kids Clinic adds an important layer to the child’s care.”
“The medical and speech evaluations led to identification of a medical condition (VPI) which could be corrected by surgery, and has helped my son’s speech and self-esteem. I am disappointed it was not identified by other doctors earlier.”
“We received a lot of information about treatment plans, what to expect, and other supports. Knowing there is a team approach to helping our son is a great relief. Also, it was the first time that our son was really included in conversations about his own care. We left feeling much more encouraged about his future.”
Constructive feedback comments included:
“I was really anxious to have the 1st appointment after the diagnosis by genetics, and 2 months seemed like a long wait.”
“It is unfortunate that it takes so long to get an appointment, I waited 7 months.”
“The week was very intensive and exhausting for my son – a bit overwhelming.”
“The appointments could have been spaced closer together so that we could have spent fewer days there.”
“We were seen in late October, but didn’t get some of the reports until after the holidays. When dealing with an IEP and much of the school year had passed, it was frustrating.”
“His school didn’t listen to all the recommendations – partly because of funding but also because they said the evaluation was from out-of-state and there are different requirements here.”
“If a child presents with something unexpected, let the parent know before the final feedback so that they have time to react and get the most from the visit.”
When asked to select benefits of the clinic evaluation from a provided list, the most commonly reported benefits of clinic evaluations from patients seen by a team of three or more providers (n=82) included:
“Helped to better understand the child’s diagnosis” (78%)
“Helped to better understand my child’s strengths and weaknesses” (76.8%)
“Improved our understanding of the services and supports needed at school” (64.6%)
“Led to changes in my child’s support services, curriculum, therapies, and/or IEP at school” (62.2%)
“Helped me feel more encouraged for my child’s future” (57.3%).
Lastly, respondents provided their satisfaction with experiences with various aspects of clinical operations (from 1 for very disappointed to 5 for very satisfied). The highest rated clinical operations included: directions and maps to the clinic (4.78), information regarding medications (4.64), and thoroughness of reports (4.62). Other processes surveyed scored >4.4 and included process of blood draws, length of appointments, intake/scheduling process, and educational materials provided. The lowest scores were in timeliness of getting a report (4.17), and timeliness in getting an appointment (3.2).
Discussion
The eXtraordinarY Kids Clinic is a unique program at Children’s Hospital Colorado that we present as a model of care for children and adolescents with SCA, supported by survey results demonstrating a high level of satisfaction with the clinic experience. Team clinics involving multiple relevant professionals and subspecialists have been established for many other pediatric medical and genetic conditions, including neurofibromatosis and Williams syndrome.18,19 Clinics specifically designed for TS currently exist at many centers across the USA, and children with SCA may also be served within clinics with expertise focused on genetic, endocrinologic, or psychological features of the SCA conditions. To our knowledge, the eXtraordinarY Kids Clinic is the first and most comprehensive clinic specializing in other SCA conditions to be established at an academic medical center, and also the first to report results of patient surveys of the clinic experience and parental report of impact to the child’s health and development.
An important emphasis of the clinic model is on the interdisciplinary approach to patient care as opposed to a multidisciplinary approach. In multidisciplinary clinic models, patients and families receive care from multiple providers within the context of a single clinic visit; however, each discipline generally approaches the patient from its own perspective and develops its own diagnoses and treatment plans without explicit collaboration with the other disciplines involved. However, the approach of interdisciplinary care is to integrate results and considerations from different disciplines to develop unified diagnostic impressions and treatment plans individualized for each patient. Thus, given the long list of medical, neurodevelopmental, and psychological risks to consider in the SCA conditions (Tables 1 and 2), and considering the significant variability of features among individuals, the interdisciplinary approach allows children with SCA and their parents to understand how different features of the syndrome may be contributing to their health, behavior, learning, and overall daily functioning.
The three most commonly reported benefits of a clinic visit were items indicating an overall improved understanding of the child, his or her diagnosis, and the supports needed to help them (Table 3). Results suggest that most parents do not feel that they have a solid understanding of how the SCA condition affects their child prior to the clinic visit and that this improved with information provided by the interdisciplinary team. Efforts need to be made to educate healthcare providers, psychologists, therapists, and educators nationally about SCA conditions so that families receive better information after receiving a diagnosis. The next two most common benefits (Table 3) indicate that the evaluation led to direct changes in school supports, therapies, or medications in 57%–62% of cases, indicating that the clinic is effective in leading to positive changes and interventions for the child. It is important to note that percentages in Table 3 reflect responses of the entire group; however, some listed items were not applicable to all patients (ie, adolescent patients would not qualify for early intervention services or infants would not need special education/individualized education plan (IEP) supports if they are not yet in school). Thus, lower percentages need to be interpreted cautiously considering the broad spectrum of ages and differing needs among patients.
Table 3.
Parent reported benefits following team evaluation*
| Perceived benefits following team evaluation | Percentage (%) (n=82) |
|---|---|
| Helped me to better understand my child’s diagnosis | 78.0 |
| Helped to better understand my child’s strengths and weaknesses | 76.8 |
| Improved our understanding of the services and supports needed at school | 64.6 |
| Led to changes in my child’s support services, curriculum, therapies, and/or IEP at school | 62.2 |
| Led to recommendations for medications or medication changes that were helpful for my child | 57.3 |
| Helped me feel more encouraged for my child’s future | 56.0 |
| Helped to identify useful behavioral strategies to help my child | 40.2 |
| Led to receiving additional services or therapies outside of school, such as speech therapy, occupational therapy, psychological or behavioral supports | 37.8 |
| Helped to identify new medical problems or medical evaluations needed for my child | 32.9 |
| Helped to identify new developmental problems I was unaware of before | 26.8 |
| Helped to identify community resources and/or programs to help my child | 13.4 |
| Helped my child qualify for early intervention services | 13.3 |
| Helped my child qualify for state or community disability services | 4.9 |
| Other | 4.9 |
Note:
Team evaluation was defined as an evaluation including three or more providers on the team.
Abbreviation: IEP, individualized education plan.
Survey results demonstrated high satisfaction for all team members; however, the highest satisfaction scores were reported for the genetic counselor, who also serves as the clinic coordinator. The clinic coordinator is the first and single point of contact for families contacting the clinic. A phone intake interview is conducted, and pertinent medical and educational records are collected and reviewed to determine which specialists the patient needs to address the clinical concerns. Having a master’s level genetic counselor in the role of coordinator has been critical to success and efficiency due to training in taking medical histories, understanding of the complexities of genetics and SCA conditions, and skill in providing counseling to families. Further, families have a trusted and knowledgeable contact person who is sensitive to the personal nature of many concerns specific to SCA such as behavioral difficulties, pubertal changes, and confidentiality. Frequent genetic counseling is provided for families with a prenatal diagnosis either in clinic or over the phone for nonlocal families.
The clinic generates revenue from billing of clinical services by each professional. General infrastructure is provided by the hospital and incorporated within clinical operations of the Child Development Unit and Section of Neurodevelopmental-Behavioral Pediatrics, including the use of clinical and meeting space, scheduling support, front desk staff, computer medical records systems, nursing and social work support, laboratory services, radiology, and other general services offered within a large hospital system. Currently, additional administrative time for directorship or salaries of the clinic coordinator are not funded by the hospital or academic department, and require fundraising through grants and individual donations to the clinic.
The clinic also serves as a base for many clinical and translational research projects, as there is comprehensive data collected on a high volume of patients with a variety of SCA conditions. At the conclusion of clinical care, patients are offered participation in an Institutional Review Board-approved patient registry and DNA biobank, in which medical and psychological data are included in a large research database for descriptive and longitudinal studies. The optional collection of DNA provides a source for genotype–phenotype studies, as there is currently little known about specific genes on the X and Y chromosomes contributing to the SCA phenotype. Additional research projects include a clinical trial evaluating the psychological and motor effects of testosterone replacement therapy in adolescents with XXY, and further descriptive and comparative studies of motor skills, language, executive functioning, social development/autism spectrum disorders, psychopharmacology, and endocrine function in all SCA conditions. Results of these studies further guide clinical care and interventions for individuals with SCA.
While the overall survey responses reflect a high level of satisfaction, it is important to point out that survey response was only 33%. It is possible that some of the parents who did not respond had more neutral clinic experiences or found fewer benefits after the clinic experience. Further, the response rate may have been lower since surveys were sent after both new and follow-up visits, and families who had completed the survey after their first visit may not have responded subsequently. Results of the survey also highlight areas that need improvement. Specifically, the lowest satisfaction scores were related to time spent on the waiting list, and timeliness in receiving reports after completion of the visit. Strategies are being implemented to improve timeliness of compiling components of the team reports, and results of subsequent surveys will be analyzed for improvement. The issue of long wait times is more difficult to manage, as the demand for appointments has increased due to the unique clinic model, currently exceeding the capacity of providers in the clinic. This highlights the need for SCA clinics in geographic locations across the USA, which would allow for less expense for travel and missed work for families traveling to the clinic. Visiting a clinic closer to home would also allow for better overall care, as local providers are more familiar with medical and mental health providers, therapy options, community resources, and the education system in their area. Feedback from families both through this survey and in clinic has identified some difficulties implementing the recommended care plan in their communities outside of Colorado. Subsequent studies are needed to directly evaluate this clinic model in comparison to usual care and other models of care on outcomes such as developmental trajectories, academic and psychological outcomes, health status, and quality of life. Improved clinical services are important as SCA diagnoses are expected to increase due to the ability of new noninvasive prenatal testing (NiPT) methods to identify cases of SCA,20,21 and because the indications for genetic testing in evaluation of children with neurodevelopmental problems continue to broaden.
Case examples
The following case serves as an example of the interdisciplinary approach to assessment, formulation of diagnostic impressions and recommendations, and follow-up. Two additional case descriptions are included in the accompanying Supplementary materials, including that of an 8-year-old girl with XXX presenting with academic and social–emotional concerns, and an 18-month-old boy with a prenatal diagnosis of XXYY being monitored for risk of developmental delays.
Case 1
“Jack” was a 14-year-old boy with a prenatal diagnosis of XXY/KS. He had mild speech delays and received speech therapy from 2 to 4 years of age. Previous evaluation at 9 years of age due to academic concerns showed average IQ and no specific learning disabilities; however, he was diagnosed with attention deficit hyperactivity disorder (ADHD) – inattentive subtype. Symptoms had improved with medication treatment, but he does not always remember to take it. He has some accommodations to address ADHD symptoms in place at school. He presented to the clinic with concerns of increased conflict and frustration outbursts with his parents when doing homework, although without their help he would have difficulty completing many of his projects and assignments. There were occasional behavioral outbursts outside the home. He was slightly immature socially, but had successful friendships. He recently started puberty and his parents requested evaluation to determine if he needed testosterone treatment. They told Jack about his diagnosis of XXY when he was 11 years old, although they had not discussed it much with him and wanted him to learn more about his diagnosis.
Jack was evaluated in the eXtraordinarY Kids Clinic and he underwent evaluation by developmental pediatrics, neuropsychology, genetic counseling, and pediatric endocrinology experts. Cognitive and academic testing was consistent with previous testing and in the average range. Despite being on ADHD medication, he had moderate distractibility and attentional difficulties throughout testing, and significant executive functioning deficits in planning, organization, initiation, and working memory were identified. On medical evaluation he was healthy overall, and medication review showed treatment with a low dose of methylphenidate since 10 years of age. Physical examination showed pubertal development at Tanner stage 3, and laboratory results showed slightly elevated follicle stimulating hormone (FSH) (6.4 mIU/mL) and luteinizing hormone (LH) (5.25 mIU/mL) for his Tanner stage, and total testosterone in the average range (296 ng/dL).
Team recommendations included maximizing supports and treatments for executive functioning difficulties and ADHD symptoms. An increase in medication dose was recommended since he had grown considerably since starting the medication, as well as parental administration of medication to improve compliance. He and his parents were educated on how executive functioning deficits manifest and his 504 plan was reviewed with recommendations provided for more extensive executive functioning supports at school. Tutoring 2–3 days per week was recommended to help with organization and completion of assignments and homework, with the goal of decreasing the intensity of parental support needed for schoolwork, and subsequently minimizing conflicts with parents at home. It was felt that testosterone replacement therapy was not currently indicated; however, gonadotropins (LH and FSH) were starting to indicate testicular failure, and follow-up in 6 months was recommended. Options of testosterone replacement therapy formulations including daily topical gel versus monthly or bimonthly injections were introduced to Jack and his parents for consideration of future treatment. We discussed how ADHD symptoms may affect compliance with a daily treatment and, thus, if topical gel were selected, consistent involvement of his parents would be important. The genetic counselor met with Jack and his parents individually to further discuss his diagnosis and help him understand current and future implications of having XXY/KS, using research-supported counseling approaches.22
On follow-up 6 months later, he was 15 years of age and was finishing his first year of high school. The combination of increased medication dose, additional school supports, and tutoring had led to improved attention span in class and during homework, more consistent completion of assignments, and subsequent improvement in grades from C’s to mostly B’s. Conflicts with parents still occurred but were less frequent and the home environment was less tense overall. Puberty had progressed slightly, although he remained at Tanner stage 3, and laboratory results showed more significant elevation in FSH (10.2 mIU/mL) and LH (9.0 mIU/mL) and slightly lower testosterone levels (205 ng/dL). Testosterone replacement therapy was recommended, and he elected to start with injections to minimize the need for daily gel application over the upcoming summer vacation.
Conclusion
SCAs are among the most common chromosomal abnormalities and can be associated with a variety of developmental, psychological, and medical features. Consideration of the interplay of psychological and medical factors by experienced professionals is optimal in the assessment and care of the “whole child”. Survey results of patient and family experiences support the eXtraordinarY Kids Clinic as a unique and effective model of interdisciplinary care for children and adolescents with SCA. Additional clinics are needed in other geographic locations to optimally support the large number of patients across the country currently diagnosed with SCA, and to serve the increasing numbers of newly diagnosed cases as genetic testing indications and technologies broaden.
Supplementary materials
Case 2
“Katie” was an 8-year-old girl with trisomy X syndrome who was diagnosed at 2.5 years of age by genetic testing ordered for speech delays and hypotonia. She presented with parent concerns related to difficulty in school and behavioral problems. She had mild early delays in speech and motor skills, and received early intervention developmental therapies at home until age 3 when she was felt to be caught up. She attended a small private kindergarten and started public school in first grade where academic and behavioral difficulties began, initially attributed to the school transition and longer school days. At the time of evaluation, she was starting third grade and did not have current special education services. Emotional regulation was reported to be difficult and outbursts at home were frequent, intense, and easily triggered. While her teachers described her as generally compliant, she displayed difficulty remaining on task, required frequent redirection, had frequent meltdowns in class, and was particularly resistant to reading and writing tasks. She had no chronic medical problems, but sleep history included snoring and frequent nighttime awakening, with morning fatigue, poor physical endurance, and daily naps in the car after school.
Katie was evaluated in the eXtraordinarY Kids Clinic and underwent evaluation by developmental pediatrics, child psychology, neuropsychology, speech therapy, and occupational therapy (OT). She presented across all evaluations as cooperative and socially appropriate, with fatigue and mild attentional difficulties evident during the afternoon sessions. On the Wechsler Intelligence Scale for Children-Fourth Edition (WISC-IV) cognitive assessment, her general ability index was calculated at a standard score of 85 (16th percentile), with verbal comprehension in the borderline range and perceptual reasoning in the average range. Processing speed was very slow at less than the fifth percentile.
Emotional functioning was generally found to be consistent with immaturity and poor coping skills, but she also demonstrated strengths with respect to creativity and humor. Academic testing revealed reading, spelling, and writing skills well below what would be expected given intellectual abilities consistent with a language-based learning disability. Neuropsychological findings revealed no significant memory or executive functioning deficits. She showed mild attentional difficulties during difficult tasks and distractibility to environmental sounds that was more evident in the afternoon; however, she did not meet criteria for a diagnosis of ADHD. Speech/language testing revealed significant deficits in expressive language, which was estimated to be at a 5-year age equivalent, contributing to frustration and outbursts. OT evaluation showed normal gross motor skills, but fine motor and graphomotor skills were found to be an area of significant deficit and likely to impact her writing as well as adaptive functioning skills. Sensory processing concerns including auditory sensitivities were also identified and felt to contribute to behavioral outbursts and attentional difficulties. On medical evaluation, Katie was tall for her age (85th percentile), and otherwise a healthy, typical appearing female. Sleep study was ordered due to concerns for possible sleep apnea contributing to afternoon fatigue, irritability, and attention span.
Team recommendations led to initiation of special educational services in school with an IEP that included specialized instruction for her learning disability in reading and writing, as well as speech therapy and OT to address expressive language and graphomotor concerns. Additional school accommodations included extra time on tests due to slow processing speed, and seating/testing in quiet environments. A course of private speech therapy was initiated since difficulties with self-expression were felt to contribute to frequent behavioral outbursts, with goals and strategies coordinated with her school therapist. A 3-month course of OT helped her and her parents learn new strategies for helping with self-regulation and frustration, as well as addressing daily living skills that were affected by fine motor delays such as dressing and eating with utensils independently.
Sleep study showed mixed obstructive and central apnea, and she was referred to the sleep medicine clinic for treatment.
On follow-up 9 months later, her parents described a very positive school year overall, with rare behavioral outbursts in the school setting and markedly decreased frequency and intensity of outbursts at home. She had made marked progress in reading/writing and expressive language abilities, which led to improved confidence and self-expression, and ongoing school services and therapies were planned. Treatment of sleep apnea led to resolution of nighttime awakening and morning fatigue, and afternoon fatigue and attention span showed moderate improvement as well.
Case 3
“Chris” was an 18-month-old boy with a prenatal diagnosis of XXYY who was brought in for assessment due to his risk for developmental delays. He was described as a “good baby” since birth. Developmental screening through his pediatrician had been reassuring for meeting milestones within acceptable ranges, including sitting up at 7 months, walking independently at 14 months, and using three to five single word approximations at 14 months. However, his parents noted that his development had been much slower than his older sister, and they wanted further assessment due to the risk for developmental delays in XXYY and recommendations to maximize his progress. Medical history included five ear infections and recurrent wheezing with respiratory infections, including one hospitalization for bronchiolitis.
Chris was evaluated in the eXtraordinarY Kids Clinic and he underwent evaluation by developmental pediatrics, child psychology, speech–language, and OT. Results of developmental assessment using the Bayley-3 showed cognitive skills at the 38th percentile, motor skills at the tenth percentile, and language skills at the fifth percentile, with both receptive and expressive delays and early evidence of developmental apraxia of speech. During evaluation, concerns related to social development were noted by evaluators (ie, decreased frequency of eye contact and social referencing, repetitive play with cars) and, on questioning, his parents confirmed that they had also noticed these findings. Due to the increased risk for autism spectrum disorders (ASD) in XXYY, formal autism assessment was completed but Chris did not meet full criteria for a diagnosis of ASD. On medical evaluation, an elbow X-ray was obtained because his left elbow was prominent with decreased range of supination/pronation. He also had flat feet with moderate ankle pronation that affected balance and coordination.
Team recommendations included initiation of early intervention services to support speech and motor development. Speech–language recommendations included goals to target receptive–expressive language development, specific speech therapy techniques developed for children with apraxia of speech, as well as an emphasis on improving eye contact and social communication. While he did not currently meet criteria for ASD, it was recommended that therapies continue to include goals targeting social development and play skills, with repeat ASD evaluation considered at an older age if monitoring identified further concerns of autism. Results of the elbow X-ray showed radioulnar synostosis, and he was referred to orthopedics for discussion of this finding as well as fitting of orthotics for flat feet. Audiology testing obtained due to speech delays and recurrent ear infections was normal.
On follow-up at 28 months of age, developmental assessment showed steady cognitive development, and improved gross motor skills and coordination as a result of physical therapy and orthotics. He had made progress in speech therapy with acquisition of over 50 single words and word approximations, yet he was not yet putting together two-word phrases and there was further evidence of apraxia affecting expressive language for which more intensive and specialized clinic-based therapy was recommended. No treatment for the radioulnar synostosis was recommended unless there was pain or severe range of motion abnormalities affecting daily living skills after skeletal maturity had been achieved. His parents also expressed appreciation of integrated care by providers, since they had not encountered other physicians or therapists who could answer their specific questions about XXYY.
Acknowledgments
We thank caregivers who responded to surveys and our dedicated assistants (C D’Epagnier, V Patel, T Tanda, L Cordeiro, and C Deans) for compiling results. The AAP CATCH program funded a project to develop the clinic concept, and the Weckbaugh Foundation funded initial clinical operations. AXYS (formerly KS&A) and The XXYY Project provide ongoing referrals and collaborations. Work was also supported by NIH/NCATS Colorado CTSI Grant UL1TR001082 and NIH/NINDS K23NS070337. Contents are the authors’ sole responsibility and do not necessarily represent official NIH views.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Ross JL, Roeltgen DP, Kushner H, et al. Behavioral and social phenotypes in boys with 47,XYY syndrome or 47,XXY Klinefelter syndrome. Pediatrics. 2012;129(4):769–778. doi: 10.1542/peds.2011-0719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruining H, Swaab H, Kas M, van Engeland H. Psychiatric characteristics in a self-selected sample of boys with Klinefelter syndrome. Pediatrics. 2009;123(5):e865–e870. doi: 10.1542/peds.2008-1954. [DOI] [PubMed] [Google Scholar]
- 3.Leggett V, Jacobs P, Nation K, Scerif G, Bishop DV. Neurocognitive outcomes of individuals with a sex chromosome trisomy: XXX, XYY, or XXY: a systematic review. Dev Med Child Neurol. 2010;52(2):119–129. doi: 10.1111/j.1469-8749.2009.03545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Otter M, Schrander-Stumpel CT, Curfs LM. Triple X syndrome: a review of the literature. Eur J Hum Genet. 2010;18(3):265–271. doi: 10.1038/ejhg.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tartaglia NR, Ayari N, Hutaff-Lee C, Boada R. Attention-deficit hyperactivity disorder symptoms in children and adolescents with sex chromosome aneuploidy: XXY, XXX, XYY, and XXYY. J Dev Behav Pediatr. 2012;33(4):309–318. doi: 10.1097/DBP.0b013e31824501c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bishop DV, Jacobs PA, Lachlan K, et al. Autism, language and communication in children with sex chromosome trisomies. Arch Dis Child. 2010;96(10):954–959. doi: 10.1136/adc.2009.179747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Salbenblatt J, Meyers DC, Bender B, Linden MG, Robinson A. Gross and fine motor development in 47,XXY and 47,XYY males. Pediatrics. 1987;80:240–244. [PubMed] [Google Scholar]
- 8.van Rijn S, Bierman M, Bruining H, Swaab H. Vulnerability for autism traits in boys and men with an extra X chromosome (47,XXY): the mediating role of cognitive flexibility. J Psychiatr Res. 2012;46(10):1300–1306. doi: 10.1016/j.jpsychires.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Pennington BF, Bender B, Puck M, Salbenblatt J, Robinson A. Learning disabilities in children with sex chromosome anomalies. Child Dev. 1982;53(5):1182–1192. [PubMed] [Google Scholar]
- 10.Geerts M, Steyaert J, Fryns JP. The XYY syndrome: a follow-up study on 38 boys. Genet Couns. 2003;14(3):267–279. [PubMed] [Google Scholar]
- 11.Tartaglia N, Ayari N, Howell S, D’Epagnier C, Zeitler P. 48,XXYY, 48,XXXY and 49,XXXXY syndromes: not just variants of Klinefelter syndrome. Acta Paediatr. 2011;100(6):851–860. doi: 10.1111/j.1651-2227.2011.02235.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Linden MG, Bender BG, Robinson A. Sex chromosome tetrasomy and pentasomy. Pediatrics. 1995;96(4 pt 1):672–682. [PubMed] [Google Scholar]
- 13.Gropman AL, Rogol A, Fennoy I, et al. Clinical variability and novel neurodevelopmental findings in 49, XXXXY syndrome. Am J Med Genet A. 2010;152A(6):1523–1530. doi: 10.1002/ajmg.a.33307. [DOI] [PubMed] [Google Scholar]
- 14.Visootsak J, Aylstock M, Graham JM., Jr Klinefelter syndrome and its variations: an update and review for the primary pediatrician. Clin Pediatr (Phila) 2001;40(12):639–651. doi: 10.1177/000992280104001201. [DOI] [PubMed] [Google Scholar]
- 15.Bojesen A, Gravholt CH. Klinefelter syndrome in clinical practice. Nat Clin Pract Urol. 2007;4(4):192–204. doi: 10.1038/ncpuro0775. [DOI] [PubMed] [Google Scholar]
- 16.Bondy CA. Care of girls and women with turner syndrome: a guideline of the turner syndrome study group. J Clin Endocrinol Metab. 2007;92(1):10–25. doi: 10.1210/jc.2006-1374. [DOI] [PubMed] [Google Scholar]
- 17.Rogol AD, Tartaglia N. Considerations for androgen therapy in children and adolescents with Klinefelter syndrome (47, XXY) Pediatr Endocrinol Rev. 2010;8(Suppl 1):145–150. [PubMed] [Google Scholar]
- 18.Obringer AC, Zackai EH, Meadows AT. The neurofibromatosis clinic of the children’s hospital of Philadelphia. Neurofibromatosis. 1988;1(3):179–181. [PubMed] [Google Scholar]
- 19.Waxler JL, Levine K, Pober BR. Williams syndrome: a multidisciplinary approach to care. Pediatr Ann. 2009;38(8):456–463. doi: 10.3928/00904481-20090723-10. [DOI] [PubMed] [Google Scholar]
- 20.Zimmermann B, Hill M, Gemelos G, et al. Noninvasive prenatal aneuploidy testing of chromosomes 13, 18, 21, X, and Y, using targeted sequencing of polymorphic loci. Prenat Diagn. 2012;32(13):1233–1241. doi: 10.1002/pd.3993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mazloom AR, Džakula Ž, Oeth P, et al. Noninvasive prenatal detection of sex chromosomal aneuploidies by sequencing circulating cell-free DNA from maternal plasma. Prenat Diagn. 2013;33(6):591–597. doi: 10.1002/pd.4127. [DOI] [PubMed] [Google Scholar]
- 22.Dennis A, Howell S, Cordeiro L, Tartaglia N. “How should I tell my child?” Disclosing the diagnosis of sex chromosome aneuploidies. J Genet Couns. 2015;24(1):88–103. doi: 10.1007/s10897-014-9741-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aksglaede L, Wikstrom AM, Rajpert-De Meyts E, Dunkel L, Skakkebaek NE, Juul A. Natural history of seminiferous tubule degeneration in Klinefelter syndrome. Hum Reprod Update. 2006;12(1):39–48. doi: 10.1093/humupd/dmi039. [DOI] [PubMed] [Google Scholar]
- 24.Boisen E, Rasmussen L. Tremor in XYY and XXY Men. Acta Neurol Scand. 1978;58(1):66–73. doi: 10.1111/j.1600-0404.1978.tb02861.x. [DOI] [PubMed] [Google Scholar]
- 25.Bojesen A, Juul S, Birkebaek N, Gravholt CH. Increased mortality in Klinefelter syndrome. J Clin Endocrinol Metab. 2004;89(8):3830–3834. doi: 10.1210/jc.2004-0777. [DOI] [PubMed] [Google Scholar]
- 26.Bojesen A, Kristensen K, Birkebaek NH, et al. The metabolic syndrome is frequent in Klinefelter’s syndrome and is associated with abdominal obesity and hypogonadism. Diabetes Care. 2006;29(7):1591–1598. doi: 10.2337/dc06-0145. [DOI] [PubMed] [Google Scholar]
- 27.Campbell WA, Price WH. Venous thromboembolic disease in Klinefelter’s syndrome. Clin Genet. 1981;19(4):275–280. doi: 10.1111/j.1399-0004.1981.tb00709.x. [DOI] [PubMed] [Google Scholar]
- 28.Hutaff-Lee C, Cordeiro L, Tartaglia N. Cognitive and medical features of chromosomal aneuploidy. Handb Clin Neurol. 2014;111:273–279. doi: 10.1016/B978-0-444-52891-9.00030-0. [DOI] [PubMed] [Google Scholar]
- 29.Joseph M. Endodontic treatment in three taurodontic teeth associated with 48,XXXY Klinefelter syndrome: a review and case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2008;105:670–677. doi: 10.1016/j.tripleo.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 30.Lanfranco P, Kamichke A, Zitzmann M, Nieschlag E. Klinefelter’s syndrome. Lancet. 2004;364:273–283. doi: 10.1016/S0140-6736(04)16678-6. [DOI] [PubMed] [Google Scholar]
- 31.Lin HJ, Ndiforchu F, Patell S. Exstrophy of the cloaca in a 47,XXX child: review of genitourinary malformations in triple-X patients. Am J Med Genet. 1993;45(6):761–763. doi: 10.1002/ajmg.1320450619. [DOI] [PubMed] [Google Scholar]
- 32.Nielsen J, Pelsen B, Sorensen K. Follow-up of 30 Klinefelter males treated with testosterone. Clin Genet. 1988;33(4):262–269. doi: 10.1111/j.1399-0004.1988.tb03447.x. [DOI] [PubMed] [Google Scholar]
- 33.Ratcliffe SG. Longitudinal growth studies on children with sex chromosome abnormalities. Prog Clin Biol Res. 1985;200:301–309. [PubMed] [Google Scholar]
- 34.Simpson JL, de la Cruz F, Swerdloff RS, et al. Klinefelter syndrome: expanding the phenotype and identifying new research directions. Genet Med. 2003;5(6):460–468. doi: 10.1097/01.gim.0000095626.54201.d0. [DOI] [PubMed] [Google Scholar]
- 35.Tartaglia N, Davis S, Hench A, et al. A new look at XXYY syndrome: medical and psychological features. Am J Med Genet A. 2008;146A(12):1509–1522. doi: 10.1002/ajmg.a.32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tartaglia NR, Howell S, Sutherland A, Wilson R, Wilson L. A review of trisomy X (47,XXX) Orphanet J Rare Dis. 2010;5:8. doi: 10.1186/1750-1172-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Villanueva AL, Rebar RW. Triple-X syndrome and premature ovarian failure. Obstet Gynecol. 1983;62(3 Suppl):70s–73s. [PubMed] [Google Scholar]
- 38.Bardsley MZ, Kowal K, Levy C, et al. 47,XYY syndrome: clinical phenotype and timing of ascertainment. J Pediatr. 2013;163(4):1085–1094. doi: 10.1016/j.jpeds.2013.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]