

Abstract
Salvinorin A (1), the main active ingredient of Salvia divinorum, is a potent and selective κ opioid receptor (KOPR) agonist. Based on the SAR, its C-2 position is one of the key binding sites and has very little space tolerance (3–4 carbons atoms) and limited to only lipophilic groups. In our attempt to prepare PET brain imaging agent for mapping KOPR, a series of C-2 halogenated analogs have been synthesized and screened for binding affinity at κ(KOPR), μ(MOPR), and δ (DOPR). These C-2 halogenated analogs with sequential changes of atomic radius and electron density serve as excellent molecular probes for further investigating the binding pocket at C-2, particularly on the effects of α verses β configuration at C-2 position. The results of KOPR binding and functional studies reveal β isomer in general binds better than α isomer with the exception of iodinated analogs and none of the C-2 halogenated analogs shows any improvement of KOPR binding affinity. Interestingly, functional assay has characterized that 6b is a partial agonist with Emax of 46% of the kappa receptor full agonist U50,488H at 250 nM (Ki). We have also observed that the affinity to the kappa receptor increases with atomic radius (I > Br > Cl > F) which is in good agreement with halogen bonding interactions reported in the literature.
Keywords: Salvinorin A, Kappa, Halogenation
Salvia divinorum, a Mexican psychoactive plant, has been used in traditional spiritual practices for centuries.1–4 Salvinorin A (1), a neoclerodane diterpenoid isolated from S. divinorum,5,6 is identified to be a potent and selective kappa (r) opioid receptor (KOR) agonist.7,8 Smoking of S. divinorum leaves fortified with salvinorin A extract has been the most efficient method for its hallucinogenic effects in human. Ingestion of S. divinorum in humans causes hallucinations or delusional episodes that mimic psychosis.9,10 It is increasingly used as a recreational drug in North America and Europe. Salvinorin A is the principal active ingredient responsible for the observed hallucinogen; however, its action in human brain remains unclear. Structurally, 1 represents the first non-alkaloid opioid receptor type-selective drug. The unique structural and biological features of 1 make it an attractive candidate for exploring the opioid pharmacology and a potential template for development of novel F-18 (PET) or I-123 (SPECT) substituted imaging agent for its sites of action in the brain.
Prior structure–activity relationship studies of 1 by our group and others have led to the synthesis of a significant number of C-2 analogs. Among them, methoxymethyl ether (2)11 and ethoxymethyl ether (3)12 display higher binding affinity and potency than 1 at KOPR. Compound 2 also exhibits longer duration of action than 1 in vivo.13 It has been demonstrated that the C-2 position of 1 is one of its key binding sites and highly sensitive to size and electronegativity of the substituent. In general, C-2 pocket has very little space tolerance (3–4 carbons atoms) and limited to only lipophilic groups. The synthesis of C-2 halogenated analogs would provide a series of interesting molecules for further investigation of the binding pocket at C-2, particularly with regard to the orientation of the C-2 α verses β isomers. In addition, F-18 and I-123 are usually the most popular radionuclides of choice for PET and SPECT studies, respectively, it would be necessary to evaluate the binding data with non-radioactive compounds. Since the synthesis of C-2 fluoro-and iodo-analogs of 1 and their corresponding binding data have not yet been reported and the halogen bonding in biomolecular systems are becoming popular for rational drug design,14 our study would be useful for providing such information particularly in view of the limited options of introducing halogens to the salvinorin A structure.
Salvinorin A (1) is isolated from S. divinorum and then converted to salvinorin B (2a) and 2-epi-salvinorin B (2b), as previously described.15 The reaction of 2a and 2b with Deoxo-Fluor reagent16 affords the C-2 fluoro analogs 3b and its C-2 epimer 3a (Scheme 1). Initial attempts to synthesize the C-2 chloro analogs 4a and 4b by treatment of 2a and 2b with TCT and DMF at room temperature 17 lead to the C-2 formate 4c,18 which may have formed due to presence of H2O in the solvent. However, an elevated temperature at 35 °C gives exclusively the chlorinated products.
Scheme 1.

Reagents and conditions: (a) Deoxo-Fluor reagent, CH2Cl2, −78 °C, 2 h, rt, 2 h, 3b (74%), 3a (80%); (b) TCT, DMF/CH2Cl2, 35 °C, 12 h, 4b (90%), 4a (81%); (c) TCT,DMF/CH2Cl2, rt, 2 h; yield varies according to reaction time.
Addition of NaBr in the synthesis of 4b provides a mixture of 4b and the bromo-analog 5b with a ratio of 3:2 (Scheme 2). Efforts to increase the yield of 5b by adding phase transfer catalyst 15-crown-5 or separate 5b from 4b with preparative HPLC are unsuccessful. Alternatively, PBr3 is exploited in brominating of 2a and an unexpected rearrangement product 5c is formed. To our knowledge, the opening of the lactone ring of 1 under basic conditions has been reported by several groups,19 however, acid catalyzed ring opening has not yet been addressed. Compound 5c is also obtained when 2a was treated with PPh3 and bromine in the presence of a catalytic amount of imidazole.20 Unfortunately, the previously reported procedure21 in converting 2a to 5b is not reproducible in our hands and the synthesis of 5a and 5b is eventually achieved by adopting a two-step-route as shown in Scheme 2.15
Scheme 2.

Reagents and conditions: (a) TCT, DMF/CH2Cl2, NaBr, overnight, 92%, 4b:5b = 3:2; (b) PBr3, C6H6, rt, 86%; (c) PPh3, Br2, imidazole, toluene, 48%; (d) (i) (CF3SO2)2O, pyridine, CH2Cl2, 0 °C; (ii) NaBr, DMF, rt, 75% in two-steps.
Reaction of 2a with I2, PPh3 and imidazole in toluene yields predominantly 6b with small amount of C-2 epimer 6a and deoxygenation product 6c.20 Surprisingly, when 2b is treated with the identical conditions, 6b is also the major product, which could be explained by a free-radical substitution mechanism. As shown in Scheme 3, when heated in toluene, the C-2 epimers 2a and 2b may proceed to form the common free-radical intermediate 6d with the loss of configuration on C-2. Since the axial attack of the iodo radical at C-2 is favored by both stereoelectronic effects and torsional effects,22,23 the C-2-β epimer 6b is preferentially formed as expected. The C-2-α epimer 6a is more favorable under SN2 conditions evidenced by treatment of 6b with lithium iodide in DMF leads to an exclusive conversion to 6a.
Scheme 3.

Reagents and conditions: (a) I2, PPh3, imidazole, Toluene, 80 °C, 1 h, 92–95%; (b) LiI, DMF, rt, overnight, 95%.
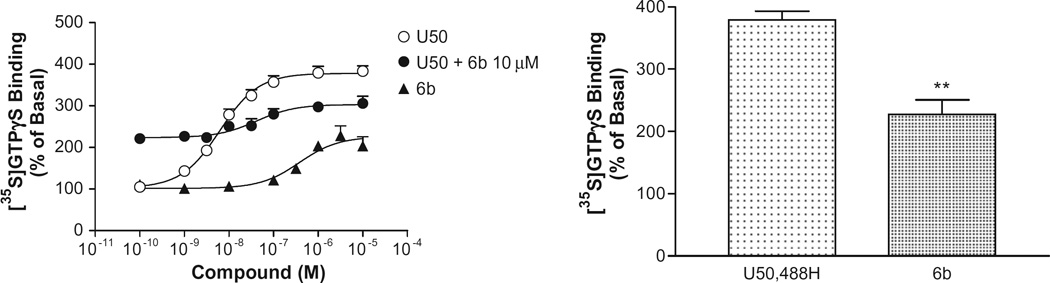
A total of 8 halogenated salvinorin A analogs is synthesized and evaluated for binding to the μ, δ, and κ-opioid receptor by competitive inhibition of [3H]diprenorphine (~0.3 nM) binding to membranes prepared from Chinese hamster ovary (CHO) cell lines stably transfected with rat μ, mouse δ, or human κ opioid receptor. The μ, δ and κ-opioid receptor agonist, etorphine, is used as the reference compound in the binding assays. At 3 μM, most of the compounds have exhibited >50% inhibition of [3H]diprenorphine binding to the kappa, but not to mu or delta, opioid receptor, indicating significant affinity for only the kappa opioid receptor. Ki values of these compounds for the kappa opioid receptor are determined. As shown in Table 1, β isomer in general binds better than α isomer with the exception of iodinated analogs and none of the C-2 halogenated analogs has shown any improvement of binding activity than the parent compound (1). It also appears that affinity to the kappa receptor increases with atomic radius (I > Br > Cl > F). Our results are in good agreement with halogen bonding interaction detected within small model complexes.14 Subsequent kappa receptor functional assay has been carried out to determine the potencies and efficacies of these compounds and U50,488H is used as the reference compound. [35S]GTPγS binding is used as the functional measure of activation of the kappaopioid receptor. DosëCresponse curves of these compounds are generated and EC50 and Emax values have been determined (Table 1). One noteworthy finding is that C-2 iodo analog 6b is a partial agonist with Emax of 46% of U50,488H.
Table 1.
Ki values and EC50 and Emax to kappa opioid receptor
| Sample | Ki (nM) (n =3) | EC50 (nM) (n = 3 and 4) | Emax(% of U50,488H) |
|---|---|---|---|
| 3a | >1000 | — | — |
| 4a | >1000 | — | — |
| 5a | 422± 44 | 1966 ± 94 | 87 |
| 6a | 198 ± 43 | 648 ± 152 | 92 |
| 3b | 197 ± 14 | 539 ± 50 | 77 |
| 4b | 167 ± 6.5 | 673 ± 111 | 112 |
| 5b | 42.3 ± 3.3 | 282 ± 8.5 | 90 |
| 6b | 245 ± 14 | 239 ± 23 | 46 |
| 6c | 51.6 ± 0.9 | 266 ± 40 | 124 |
| 4c | 24.3 ± 0.9 | 83.3 ± 10.9 | 103 |
| 5c | >1000 | — | |
| 1 | 7.5 ± 1.7 | 106 | |
| U50,488H | 1.2 ± 0.3 | 6.3 ± 0.7 | 100 |
In view of the fact that halogen bonding in biomolecular systems are becoming popular for rational drug design and the synthesis of C-2 fluoro- and iodo-analogs of 1 and their corresponding binding data have not yet been reported, our study would be useful for providing such information particularly with a limited options of introducing halogens to the salvinorin A structure. A total of 8 C-2-halogenated salvinorin A analogs have been synthesized and evaluated for the MOPR, DOPR, and KOPR affinities by competitive inhibition of [3H]diprenorphine (~0.3 nM) binding to membranes prepared from Chinese hamster ovary (CHO) cell lines stably transfected with rat MOPR, FLAG-mouse DOPR, and human KOPR. As summarized in Figure 1 and Table 1, C-2-b isomer in general binds better than a isomer with the exception of iodinated analogs and none of the C-2 halogenated analogs showed any improvement of binding affinity than the parent compound 1. It also appears that affinity to the kappa receptor increases with atomic radius (I > Br > Cl > F). These results agree with the characteristics reported within small model halogen bonded systems. Subsequent kappa receptor functional assay (Fig. 1) showed that 6b is a partial agonist with Emax of 46% of U-50,488 H at 250 nM (Ki). It is conceivable that with further improvement of affinity, 6b could be developed as a potential kappa antagonist.
Fig. 1.
Acknowledgements
We thank Dr. Yongxuan Su of University of California, San Diego for HR-MS measurements of all samples and National Institutes of Health for financial support (DA-019688).
References
- 1.Siebert DJ. J. Ethnopharmacol. 1994;43:53. doi: 10.1016/0378-8741(94)90116-3. [DOI] [PubMed] [Google Scholar]
- 2.Butelman ER, Prisinzano TE, Deng H, Rus S, Kreek MJ. J. Pharmacol. Exp. Ther. 2009;328:588. doi: 10.1124/jpet.108.145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harding WW, Tidgewell K, Schmidt M, Shah K, Dersch CM, Snyder J, Parrish D, Deschamps JR, Rothman RB, Prisinzano TE. Org. Lett. 2005;7:3017. doi: 10.1021/ol0510522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.González D, Riba J, Bouso JC, Gómez-Jarabo G, Barbanoj MJ. Drug Alcohol Depend. 2006;85:157. doi: 10.1016/j.drugalcdep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Ortega A, Blount JF, Manchand PS. J. Chem. Soc. Perkin Trans. 1. 1982:2505. [Google Scholar]
- 6.Valdes LJ, III, Butler WM, Hatfield GM, Paul AG, Koreeda MJ. Org. Chem. 1984;49:4716. [Google Scholar]
- 7.Roth BL, Baner K, Westkaemper R, Siebert DJ, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheffler DJ, Roth BL. Trends Pharmacol. Sci. 2003;24:107. doi: 10.1016/S0165-6147(03)00027-0. [DOI] [PubMed] [Google Scholar]
- 9.Roth BL, Baner K, Westkaemper R, Siebert D, Rice KC, Steinberg S, Ernsberger P, Rothman RB. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11934. doi: 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prisinzano TE, Rothman RB. Chem. Rev. 2008;108:1732. doi: 10.1021/cr0782269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee DYW, Karnati VVR, He M, Liu-Chen LY, Kondareti L, Ma Z, Wang Y, Chen Y, Beguin C, Carlezon WA, Cohen B. Bioorg. Med. Chem. Lett. 2005;15:3744. doi: 10.1016/j.bmcl.2005.05.048. [DOI] [PubMed] [Google Scholar]
- 12.Munro TA, Duncan KK, Xu W, Wang Y, Liu-Chen LY, Carlezon WA, Cohen BM, Beguin C. Bioorg. Med. Chem. 2008;16:1279. doi: 10.1016/j.bmc.2007.10.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Y, Chen Y, Xu W, Lee DYW, Ma Z, Rawls SM, Cowan A, Liu-Chen LY. J. Pharmacol. Exp. Ther. 2008;324:1073. doi: 10.1124/jpet.107.132142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Shi T, Wang Y, Yang H, Yan X, Luo X, Jiang H, Zhu WJ. Med. Chem. 2009;52:2854. doi: 10.1021/jm9000133. [DOI] [PubMed] [Google Scholar]
- 15.Beguin C, Richards MR, Li J, Wang Y, Xu W, Liu-Chen LY, Carlezon WA, Cohen BM. Bioorg. Med. Chem. Lett. 2006;16:4679. doi: 10.1016/j.bmcl.2006.05.093. [DOI] [PubMed] [Google Scholar]
- 16.Lal GS, Pez GP, Pesaresi RJ, Prozonic FM, Cheng HJ. Org. Chem. 1999;64:7048. [Google Scholar]
- 17.Luca LD, Giacomelli G, Porcheddu A. Org. Lett. 2002;4:553. doi: 10.1021/ol017168p. [DOI] [PubMed] [Google Scholar]
- 18.Munro TA, Rizzacasa MA, Roth BL, Toth BA, Yan FJ. Med. Chem. 2005;48:345. doi: 10.1021/jm049438q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.(a) Bikbulatov RV, Stewart J, Jin W, Yan F, Roth BL, Ferreira D, Zjawiony JK. Tetrahedron Lett. 2008;49:937. doi: 10.1016/j.tetlet.2007.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Beguin C, Duncan KK, Munro TA, Ho DM, Xu W, Liu-Chen LY, Carlezon WA, Cohen BM. Bioorg. Med. Chem. 2009;17:1370. doi: 10.1016/j.bmc.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skaanderup PR, Poulsen CS, Hyldtoft L, Jørgensen MR, Madsen R. Synthesis. 2002;12:1721. [Google Scholar]
- 21.Tidgewell K, Groer CE, Harding WW, Lozama A, Schmidt M, Marquam A, Hiemstra J, Partilla JS, Dersch CM, Rothman RB, Bohn LM, Prisinzano TE. J. Med. Chem. 2008;51:2421. doi: 10.1021/jm701162g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toru T, Okumara T, Ueno YJ. Org. Chem. 1990;55:1277. [Google Scholar]
- 23.Damm W, Giese B, Hartung J, Hasskerl T, Houk KN, Huter O, Zipse HJ. Am. Chem. Soc. 1992;114:4067. [Google Scholar]