

Abstract
Objective
Clinical evidence suggests an association between oxidative stress and vascular disease, and in vitro studies have demonstrated that reactive oxygen species (ROS) can have prothrombotic effects on vascular and blood cells. It remains unclear, however, whether elevated levels of ROS accelerate susceptibility to experimental thrombosis in vivo.
Approach and Results
Using a murine model with genetic deficiency in superoxide dismutase-1 (SOD1), we measured susceptibility to carotid artery thrombosis in response to photochemical injury. We found that SOD1-deficient (Sod1−/−) mice formed stable arterial occlusions significantly faster than wild-type (Sod1+/+) mice (P < 0.05). Sod1−/− mice also developed significantly larger venous thrombi than Sod1+/+ mice after inferior vena cava ligation (P < 0.05). Activation of protein C by thrombin in lung was diminished in Sod1−/− mice (P < 0.05 vs. Sod1+/+ mice), and generation of activated protein C in response to infusion of thrombin in vivo was decreased in Sod1−/− mice (P < 0.05 vs. Sod1+/+ mice). SOD1 deficiency had no effect on expression of thrombomodulin, endothelial protein C receptor, or tissue factor in lung or levels of protein C in plasma. Exposure of human thrombomodulin to superoxide in vitro caused oxidation of multiple methionine residues, including critical methionine 388, and a 40% decrease in thrombomodulin-dependent activation of protein C (P < 0.05). SOD and catalase protected against superoxide-induced methionine oxidation and restored protein C activation in vitro (P < 0.05).
Conclusions
SOD prevents thrombomodulin methionine oxidation, promotes protein C activation, and protects against arterial and venous thrombosis in mice.
Keywords: protein C, methionine oxidation, superoxide, thrombomodulin, thrombosis
Introduction
Thrombotic complications, including myocardial infarction, stroke, pulmonary embolism, and deep vein thrombosis, are common causes of morbidity and mortality in patients with vascular disease. A high incidence of thrombotic events is also seen in cancer patients1 and in the aged population.2–4 Despite these well-established clinical observations, the mechanisms by which distinct pathologic states contribute to thrombosis remain poorly understood. One commonality among many of the prothrombotic conditions is an increase in vascular oxidative stress,5 which may generate excess reactive oxygen species (ROS). In vitro studies have suggested that ROS such as superoxide can have prothrombotic effects on vascular and blood cells, including enhanced platelet activation,6 increased expression or activity of tissue factor (TF),7 and dysregulation of anticoagulant pathways,8 all of which may predispose to arterial and/or venous thrombosis.
Findings from clinical studies have identified multiple sources for increased ROS in cardiovascular disease, including an increase in the expression and/or activity of prooxidant enzymes5 such as NADPH oxidase, nitric oxide synthase, and xanthine oxidase and a decrease in antioxidant enzymes such as superoxide dismutase (SOD), glutathione peroxidase, and catalase.9–14 It remains unclear, to what extent these oxidative mechanisms contribute to thrombosis in vivo.
The objective of this study was to determine the contribution of superoxide to the increased susceptibility to experimental thrombosis in vivo, using mice genetically deficient in superoxide dismutase-1 (SOD1). SOD1 is a copper- and zinc-containing enzyme that is the major cytosolic form of SOD, which catalyzes the conversion of superoxide to hydrogen peroxide. SOD1 is the predominant isoform of SOD expressed in the vasculature.15 Mice with homozygous deficiency of SOD1 (Sod1−/− mice) have increased superoxide in vascular tissue.16 Our new findings demonstrate that loss of SOD1 causes increased susceptibility to arterial and venous thrombosis in mice, and that SOD protects from superoxide-mediated oxidation of thrombomodulin and impairment of the protein C anticoagulant system.
Materials and Methods
Materials and Methods are available in the online-only data supplement.
Results
Vascular ROS are increased in mice with SOD1 deficiency
To confirm that mice deficient in SOD1 have increased vascular ROS, we measured tiron-quenchable lucigenin-enhanced chemiluminescence, a relatively selective indicator of superoxide,17 in the aorta. We detected a significant increase in fluorescence in aortic sections from Sod1−/− mice compared to Sod1+/+ mice (P = 0.035, Figure 1). This finding is consistent with a previous report of increased superoxide levels in the vascular wall.16
Figure 1.

Vascular ROS are increased in Sod1−/− mice. ROS levels were determined in aortic segments as tiron-quenchable lucigenin-enhanced chemiluminescence (relative light units (RLU)/sec/mg weight). Values are mean ± SEM (n = 5–6 mice per group).
Deficiency of SOD1 enhances susceptibility to both arterial and venous thrombosis
We next examined the effect of SOD1 deficiency on susceptibility to arterial thrombosis. Baseline prothrombin time (PT), partial thromboplastin time (PTT) and platelet count were similar in Sod1+/+ and Sod1−/− mice (Table 1). Following photochemical injury of the carotid artery, the time to first occlusion was nearly three times faster in Sod1−/− mice (8.5 ± 1.9 minutes) than in Sod1+/+ mice (24.5 ± 8.4 minutes; P = 0.010) (Figure 2A). Similarly, the time to stable occlusion was significantly faster in Sod1−/− mice than in Sod1+/+ mice (16.2 ± 3.3 vs. 34.0 ± 9.1 minutes, respectively; P = 0.032) (Figure 2B). An inferior vena cava (IVC) ligation method was used to assess susceptibility to stasis-induced venous thrombosis. Sod1−/− mice developed significantly heavier (14.8 ± 4.4 mg in Sod1−/− mice vs. 3.9 ± 2.3 mg in Sod1+/+ mice; P = 0.047) and longer (5.5 ± 1.5 mm in Sod1−/− mice vs. 1.6 ± 0.8 mm in Sod1+/+ mice; P = 0.044) IVC thrombi compared to Sod1+/+ mice (Figure 2C, D). These data demonstrate that deficiency of SOD1 increases susceptibility to both arterial and venous thrombosis.
Table 1.
Platelet count, hemostasis assays and plasma protein C levels in Sod1+/+ and Sod1−/− mice.
|
Sod1+/+ mice n = 4 |
Sod1−/− mice n = 4 |
P-value | |
|---|---|---|---|
| Platelet count (×103/μl) | 859 ± 48 | 945 ± 89 | 0.40 |
| Prothrombin time (minutes) | 11.8 ± 0.5 | 11.7 ± 0.3 | 0.93 |
| Partial thromboplastin time (minutes) | 42.5 ± 5.2 | 36.1 ± 1.8 | 0.28 |
| Protein C (ng/mL) | 21.0 ± 3.0 | 36.4 ± 8.4 | 0.13 |
Values represent mean ± SEM.
Figure 2.

Deficiency of SOD1 enhances susceptibility to arterial and venous thrombosis. The time to first (A) and stable (B) occlusion were measured in carotid arteries after photochemical injury (n = 11 to 15 mice per group). The weight (C) and length (D) of thrombi that developed in the IVC 48 hours following ligation was measured. Values are mean ± SEM (n=10 to 11 mice per group).
SOD1 deficiency does not enhance platelet activation in response to thrombin
In previous work, we have demonstrated that hydrogen peroxide (H2O2), which is generated by the action of SOD1 on superoxide, contributes to increased thrombotic susceptibility and thrombin-stimulated platelet activation.18 We therefore asked if deficiency of SOD1 alters platelet activation. However, in response to thrombin, we did not observe a difference in the surface expression of P-selectin or fibrinogen binding to Sod1−/− platelets as compared to Sod1+/+ platelets (Figure 3). These data suggest that deficiency of SOD1 in platelets does not influence alpha granule release or activation of platelet integrin α2bβ3.
Figure 3.
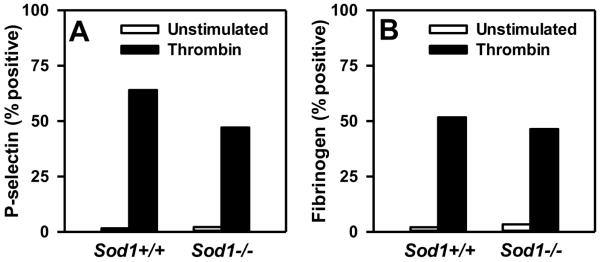
SOD1 deficiency does not enhance platelet activation in response to thrombin. P-selectin surface expression (A) and fibrinogen surface binding (B) were measured in unstimulated or thrombin-activated platelets using flow cytometric analysis. Values are mean ± SEM (n = 5 to 6 mice per group).
Deficiency of SOD1 does not influence tissue factor (TF) expression or activity
We next investigated the effect of SOD1 deficiency on the expression and activity of TF, a major trigger of coagulation and thrombosis.19 Because TF expression has been reported to be redox-sensitive,7 real-time qPCR was performed to quantitatively measure levels of TF mRNA in lung homogenates. We found, however, that the expression of TF mRNA was not altered in Sod1−/− mice (P = 0.3 vs. Sod1+/+ mice) (Supplemental Figure IA). Since de-encryption of TF leading to its activation is also redox-regulated, we measured TF activity in a factor Xa activation assay. Again, we did not detect any difference in TF activity between Sod1+/+ and Sod1−/− mice (P = 0.5) (Supplemental Figure IB). These results suggest that accelerated thrombosis in Sod1−/− mice is not likely due to oxidative upregulation of TF gene expression or activity.
SOD1 deficiency impairs generation of activated protein C (APC)
Thrombomodulin (TM), an endothelial transmembrane glycoprotein, can undergo methionine oxidation, limiting its anticoagulant activity to support thrombin-mediated generation of APC from protein C.8 Therefore, to determine if the activation of endogenous protein C is altered in SOD1-deficient mice, circulating plasma levels of APC were measured after intravenous injection of thrombin. Plasma levels of APC were 45% lower in Sod1−/− mice compared to Sod1+/+ mice (Figure 4A, P = 0.035). The decreased thrombin-induced generation of APC in Sod1−/− mice was not due to lower baseline levels of protein C, since plasma protein C levels tended to be higher, rather than lower, in Sod1−/− mice compared to Sod1+/+ mice (P = 0.07) (Table 1).
Figure 4.
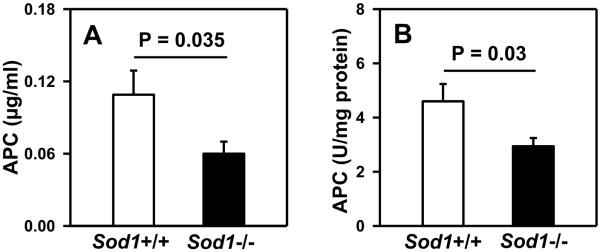
SOD1 deficiency impairs generation of APC from thrombin in vivo and in vitro. (A) Following thrombin infusion, levels of APC were measured in plasma by enzyme capture ELISA. (B) Activation of protein C was measured in lung lysates in presence of exogenous thrombin and protein human C. Values are mean ± SEM (n = 5 to 6 mice per group).
Next, we measured generation of exogenous APC by thrombin in lung homogenates from Sod1−/− and Sod1+/+ mice. The data revealed a 40% decrease in TM-dependent APC generation in Sod1−/− mice as compared to Sod1+/+ mice (Figure 4B, P = 0.030). These findings suggest that deficiency of SOD1 leads to impaired TM anticoagulant activity (decreased TM-dependent activation of protein C by thrombin).
Another endothelial transmembrane protein, the endothelial protein C receptor (EPCR), can act in concert with TM to promote protein C activation.20 Therefore, we further investigated whether SOD1 deficiency affects the expression of TM or EPCR. No differences in TM mRNA (measured by real-time qPCR) or TM protein (measured by Western blotting) were detected in the lungs of Sod1−/− mice compared with Sod1+/+ mice (P = 0.4 and P = 0.2, respectively) (Figure 5). Similarly, EPCR mRNA levels were comparable in the lungs of Sod1−/− and Sod1+/+ mice (P = 0.6) (Supplemental Figure II). These data suggest that the decreased generation of APC with SOD1 deficiency is not due to loss of expression of TM or EPCR.
Figure 5.
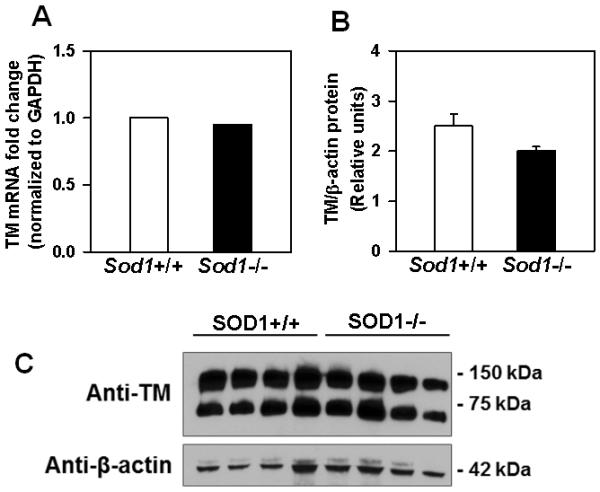
Deficiency of SOD1 does not influence levels of TM mRNA or protein in mice. (A) mRNA levels for TM in lung homogenates were determined by qRT-PCR. Levels were normalized to GAPDH, and data are displayed as fold-change relative to control (Sod1+/+ mice). Comparisons of normalized expression values (ΔCt) employed the conventional ΔΔCt fold change method. The Ct values for TM were 19.8±0.18 in Sod1+/+ vs. 19.6±0.13 in Sod1−/− mice. P = 0.4 vs. Sod1+/+ mice (n=5 to 6 mice per group). (B) Protein levels of TM and β actin were determined in lung lysates by Western blotting. Immunoreactive bands were quantified by densitometry. P = 0.2 vs. Sod1+/+ mice. Values are mean ± SEM (n = 4 mice per group). (C). A representative Western blot image showing detection of both dimer and monomer forms of TM.
Exposure of human TM to superoxide leads to oxidation of methionine 388 and loss of TM anticoagulant activity
To determine if SOD can protect TM from methionine oxidation, we exposed recombinant human thrombomodulin to hypoxanthine plus xanthine oxidase (X-XO) to generate superoxide in vitro. Exposure to superoxide inhibited TM anticoagulant activity, measured as TM-dependent APC generation, by 40% (P = 0.0002) (Figure 6A). The inhibitory effect of superoxide was partially prevented by the addition of SOD or catalase, and almost completely prevented by co-incubation with both SOD and catalase (P = 0.0003 vs. catalase alone) (Figure 6A).
Figure 6.
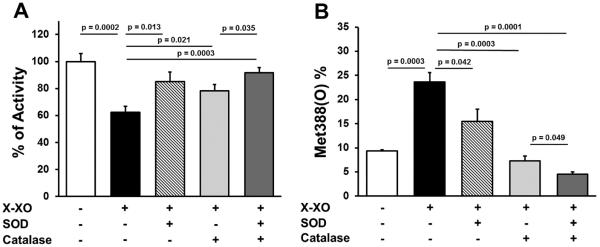
Superoxide-induced inhibition of TM anticoagulant activity and oxidation of TM Met388. Recombinant human TM (containing only the extracellular domain) was incubated with or without 5 mU/ml xanthine oxidase and 1 mM hypoxanthine (X-XO) in the presence or absence of 50 U/ml PEG-SOD and/or 250 U/ml PEG-catalase. (A) TM activity was measured in a protein C activation assay (n = 6). (B) The content of TM Met388(O) was determined by nano-LC-MS/MS (n = 4).
Human TM contains four methionine residues in its extracellular domain, and it has been demonstrated previously that oxidation of methionine 388 to methionine sulfoxide (Met388(O)) causes loss of TM anticoagulant activity.8, 21, 22 We therefore performed nano-LC-MS/MS to quantify the extent of TM methionine oxidation at Met388. We found that exposure to superoxide resulted in a 2.6-fold increase in Met388(O) (P = 0.0003) (Figure 6B). Oxidation of Met388 was partially prevented by SOD (P = 0.042) and almost completely prevented by catalase or co-incubation with SOD and catalase (P = 0.0001) (Figure 6B). The other three extracellular methionine residues in human TM (Met42, Met205, and Met 291) also underwent oxidation after exposure to superoxide (Supplemental Figure III), but these methionine residues are unlikely to affect TM anticoagulant activity because they are outside of the critical EGF-like domains of TM required for protein C activation (Supplemental Figure IIID).21
Taken together, these data suggest that superoxide induces loss of TM anticoagulant activity at least in part via increased oxidation of Met388. The protective effects of both SOD and catalase suggest that both superoxide and H2O2 contribute to methionine oxidation and inhibition of TM anticoagulant activity.
Discussion
Despite a large body of literature demonstrating activation of oxidant pathways and increased production of ROS in vascular diseases,23 relatively little is known about the role of ROS in driving thrombosis in vivo. Several studies have demonstrated increased levels of superoxide in blood vessels of humans and animals with atherosclerosis and other vascular conditions.24–26 Previous studies using SOD1-deficient mice have implicated superoxide in the mechanism of endothelial dysfunction27 and cerebral hypertrophy.28 One major finding of the current study is that genetic deficiency of SOD1 increases susceptibility to both arterial and venous thrombosis in mice. This finding suggests that superoxide, when present in excess, can be considered to be a prothrombotic mediator in vivo. Another key finding from our study is that deficiency of SOD1 is associated with an impaired protein C anticoagulant response to thrombin. These findings suggest that elevation of superoxide due to SOD1 deficiency may lead to thrombosis in part by impairing TM-dependent protein C activation.
TM is a cell surface glycoprotein expressed on the luminal surface of endothelial cells.29 When bound to thrombin, TM functions as a potent anticoagulant by converting circulating protein C to APC. APC then proteolytically cleaves activated factors V and VIII to prevent thrombin generation. Disruption of the TM/APC anticoagulant pathway in mice results in a prothrombotic phenotype. For example, Weiler and colleagues demonstrated that mice with TM deficiency have increased susceptibility to carotid artery thrombosis.30 Mice with endothelial-specific deficiency of TM exhibit severe thrombosis at an early age and die due to consumptive coagulopathy.31 Similarly, mice deficient in EPCR32 or partially deficient in PC33 also are prothrombotic. Our new findings that Sod1-deficient mice with decreased APC generation have increased susceptibility to arterial and venous thrombosis are consistent with these previous observations.
TM contains a critical redox sensitive methionine residue (Met388) that, when oxidized, limits its ability to activate protein C.8 Therefore, we hypothesized that increased vascular superoxide in Sod1−/− mice would promote the oxidative inactivation of TM and thereby decrease APC generation. Following thrombin infusion, we observed a significant decrease in the levels of plasma APC in Sod1−/− mice compared with Sod1+/+ mice. Similarly, exogenous generation of APC by thrombin also was decreased in lung tissue from Sod1−/− mice. Importantly, no differences in the expression of TM mRNA or protein, EPCR mRNA, or plasma levels of protein C were detected between Sod1+/+ and Sod1−/− mice, which is consistent with a post-translational oxidative modification of TM as the likely mechanism of decreased APC generation in Sod1−/− mice.
Using nano-LC-MS/MS to quantify methionine oxidation in human TM, we found that superoxide-induced inhibition of recombinant human TM anticoagulant activity in vitro was associated with significantly increased levels of TM Met388(O). Both oxidation of Met388 and loss of TM anticoagulant activity were partially prevented by incubation with SOD. Interestingly, co-incubation with SOD and catalase conferred an even greater degree of protection, with almost complete prevention of superoxide-induced inhibition of TM anticoagulant activity and Met388 oxidation, which suggests a role for both superoxide and H2O2 in the redox regulation of TM. An important limitation of these findings is that the oxidation status of TM was assessed in a cell-free system. Additional work is needed to determine the specific role of SOD in protecting against TM Met388 oxidation in vivo, particularly because SOD1 is an intracellular enzyme and TM is a cell-surface protein with extracellular oxidation sites. We speculate that deficiency of cytosolic SOD1 may cause the accumulation of not only superoxide (a charged anion that cannot cross biological membranes) but also other uncharged ROS that can diffuse across the plasma membrane to interact with surface proteins such as TM. 34, 35
Three additional TM methionine residues (Met42, Met205, and Met291) also underwent oxidation after exposure to superoxide. However, these oxidized methionine residues are less likely to affect TM anticoagulant activity because they are outside of the critical EGF-like domains of TM required for protein C activation and it has been demonstrated that TM Met388 is the key methionine residue involved in redox regulation of its anticoagulant activity.8, 21, 22 TM also has antifibrinolytic effects through its ability to enhance thrombin-mediated activation of thrombin activatable fibrinolysis inhibitor (TAFI).36 However, oxidation of Met388 on TM has no effect on TAFI activation despite producing a significant decrease in protein C activation.37
In a previous study, we reported that H2O2 contributes to a prothrombotic phenotype in aged mice.18 Unlike the effects of superoxide seen in Sod1−/− mice, however, the increased thrombotic susceptibility induced by H2O2 in aged mice was associated with platelet hyperactivation rather than an effect on protein C activation.18 In the current study, we did not observe enhanced activation of platelets from Sod1−/− mice, which suggests that platelet SOD1 does not directly protect from platelet hyperactivation. It remains possible, however, that indirect antioxidant effects of SOD1, such as protection from endothelial dysfunction27 and diminished nitric oxide bioavailability,38, 39 may help blunt platelet activation and protect from thrombosis in vivo. We also considered the possibility that the protective antithrombotic effect of endogenous SOD1 may be mediated through decreased TF activity, since both the expression and activity of TF are known to be redox regulated.7 However, we did not detect any changes in TF mRNA expression or TF activity in Sod1−/− mice.
Clinical studies demonstrate a clear link between increased oxidative stress and vascular complications in a number of disease settings. SOD1 gene variants have been associated with increased risk of cardiovascular death in patients with type 2 diabetes.40 Relevant to our studies in Sod1−/− mice, increased oxidative stress in patients with type 2 diabetes has been reported to be associated with decreased levels of APC,41 which may lead to a prothrombotic phenotype. Gene variants in other SOD enzymes also have been linked with cardiovascular events.42 For example, the SOD3 (the gene encoding extracellular SOD) polymorphism R213G is associated with an increased risk for ischemic heart disease.43 Moreover, expression of this variant has been linked to increased cardiovascular and cerebrovascular death in diabetic patients.44 Similarly, an A16V polymorphism in SOD2 is associated with increased cardiovascular events.45
Together with the data from Sod1−/− mice reported herein, these studies strongly suggest that oxidative stress leads to increased thrombotic susceptibility and that SOD is required for redox regulation of hemostatic pathways. It is likely that endogenous SOD protects against thrombosis by modulating multiple oxidation-sensitive pathways, including the superoxide-mediated oxidation of TM and impairment of APC generation.
Supplementary Material
Significance.
Using mice deficient in superoxide dismutase, this study provides in vivo evidence that superoxide modulates the thrombomodulin / protein C anticoagulant pathway and protects against thrombosis. These data have important clinical implications for vascular diseases characterized by increased oxidative stress and thrombotic susceptibility. Strategies to reduce the inhibitory effect of oxidative stress on thrombomodulin anticoagulant activity may represent an approach to prevent thrombosis.
Acknowledgements
We thank Kristina W. Thiel for assistance in manuscript editing.
Sources of Funding This study was supported in part by an American Heart Association grant 0860052Z and SDG14050002 to SD, American Heart Association 12PRE9430065 and MSTP T32 GM007337 to SXG, and National Institutes of Health grants HL063943 and NS024621 to SRL, a grant from the American Society of Hematology to SRL, and Puget Sound Blood Center departmental funding to XF.
Abbreviations
- APC
activated protein C
- EPCR
endothelial protein C receptor
- H2O2
hydrogen peroxide
- IVC
inferior vena cava
- ROS
reactive oxygen species
- TM
thrombomodulin
- TF
tissue factor
- X-XO
hypoxanthine plus xanthine oxidase
Footnotes
Disclosures None
References
- 1.Timp JF, Braekkan SK, Versteeg HH, Cannegieter SC. Epidemiology of cancer-associated venous thrombosis. Blood. 2013;122:1712–1722. doi: 10.1182/blood-2013-04-460121. [DOI] [PubMed] [Google Scholar]
- 2.Wolf PA, Lewis A. Conner Lecture. Contributions of epidemiology to the prevention of stroke. Circulation. 1993;88:2471–2478. doi: 10.1161/01.cir.88.5.2471. [DOI] [PubMed] [Google Scholar]
- 3.Kelly-Hayes M, Beiser A, Kase CS, Scaramucci A, D'Agostino RB, Wolf PA. The influence of gender and age on disability following ischemic stroke: the Framingham study. J Stroke Cerebrovasc Dis. 2003;12:119–126. doi: 10.1016/S1052-3057(03)00042-9. [DOI] [PubMed] [Google Scholar]
- 4.Silverstein MD, Heit JA, Mohr DN, Petterson TM, O'Fallon WM, Melton LJ., 3rd Trends in the incidence of deep vein thrombosis and pulmonary embolism: a 25-year population-based study. Arch Intern Med. 1998;158:585–593. doi: 10.1001/archinte.158.6.585. [DOI] [PubMed] [Google Scholar]
- 5.Munzel T, Gori T, Bruno RM, Taddei S. Is oxidative stress a therapeutic target in cardiovascular disease? Eur Heart J. 2010;31:2741–2748. doi: 10.1093/eurheartj/ehq396. [DOI] [PubMed] [Google Scholar]
- 6.Ferroni P, Vazzana N, Riondino S, Cuccurullo C, Guadagni F, Davi G. Platelet function in health and disease: from molecular mechanisms, redox considerations to novel therapeutic opportunities. Antioxid Redox Signal. 2012;17:1447–1485. doi: 10.1089/ars.2011.4324. [DOI] [PubMed] [Google Scholar]
- 7.Herkert O, Diebold I, Brandes RP, Hess J, Busse R, Gorlach A. NADPH oxidase mediates tissue factor-dependent surface procoagulant activity by thrombin in human vascular smooth muscle cells. Circulation. 2002;105:2030–2036. doi: 10.1161/01.cir.0000014611.28864.1e. [DOI] [PubMed] [Google Scholar]
- 8.Glaser CB, Morser J, Clarke JH, Blasko E, McLean K, Kuhn I, Chang RJ, Lin JH, Vilander L, Andrews WH, et al. Oxidation of a specific methionine in thrombomodulin by activated neutrophil products blocks cofactor activity. A potential rapid mechanism for modulation of coagulation. J Clin Invest. 1992;90:2565–2573. doi: 10.1172/JCI116151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freedman JE, Loscalzo J, Benoit SE, Valeri CR, Barnard MR, Michelson AD. Decreased platelet inhibition by nitric oxide in two brothers with a history of arterial thrombosis. J Clin Invest. 1996;97:979–987. doi: 10.1172/JCI118522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blankenberg S, Rupprecht HJ, Bickel C, Torzewski M, Hafner G, Tiret L, Smieja M, Cambien F, Meyer J, Lackner KJ. Glutathione peroxidase 1 activity and cardiovascular events in patients with coronary artery disease. N Eng J Med. 2003;349:1605–1613. doi: 10.1056/NEJMoa030535. [DOI] [PubMed] [Google Scholar]
- 11.Serdar Z, Aslan K, Dirican M, Sarandol E, Yesilbursa D, Serdar A. Lipid and protein oxidation and antioxidant status in patients with angiographically proven coronary artery disease. Clin Biochem. 2006;39:794–803. doi: 10.1016/j.clinbiochem.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 12.Kaur K, Bedi G, Kaur M, Vij A, Kaur I. Lipid peroxidation and the levels of antioxidant enzymes in coronary artery disease. Indian J Clin Biochem. 2008;23:33–37. doi: 10.1007/s12291-008-0008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Misra P, Reddy PC, Shukla D, Caldito GC, Yerra L, Aw TY. In-stent stenosis: potential role of increased oxidative stress and glutathione-linked detoxification mechanisms. Angiology. 2008;59:469–474. doi: 10.1177/0003319707309651. [DOI] [PubMed] [Google Scholar]
- 14.Gupta S, Sodhi S, Mahajan V. Correlation of antioxidants with lipid peroxidation and lipid profile in patients suffering from coronary artery disease. Expert Opin Ther Targets. 2009;13:889–894. doi: 10.1517/14728220903099668. [DOI] [PubMed] [Google Scholar]
- 15.Faraci FM, Didion SP. Vascular protection: superoxide dismutase isoforms in the vessel wall. Arterioscler Thromb Vasc Biol. 2004;24:1367–1373. doi: 10.1161/01.ATV.0000133604.20182.cf. [DOI] [PubMed] [Google Scholar]
- 16.Ramiro-Diaz JM, Nitta CH, Maston LD, Codianni S, Giermakowska W, Resta TC, Gonzalez Bosc LV. NFAT is required for spontaneous pulmonary hypertension in superoxide dismutase 1 knockout mice. Am J Physiol Lung Cell Mol Physiol. 2013;304:L613–625. doi: 10.1152/ajplung.00408.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Munzel T, Afanas'ev IB, Kleschyov AL, Harrison DG. Detection of superoxide in vascular tissue. Arterioscler Thromb Vasc Biol. 2002;22:1761–1768. doi: 10.1161/01.atv.0000034022.11764.ec. [DOI] [PubMed] [Google Scholar]
- 18.Dayal S, Wilson KM, Motto DG, Miller FJ, Jr., Chauhan AK, Lentz SR. Hydrogen peroxide promotes aging-related platelet hyperactivation and thrombosis. Circulation. 2013;127:1308–1316. doi: 10.1161/CIRCULATIONAHA.112.000966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mackman N. Role of tissue factor in hemostasis, thrombosis, and vascular development. Arterioscler Thromb Vasc Biol. 2004;24:1015–1022. doi: 10.1161/01.ATV.0000130465.23430.74. [DOI] [PubMed] [Google Scholar]
- 20.Stearns-Kurosawa DJ, Kurosawa S, Mollica JS, Ferrell GL, Esmon CT. The endothelial cell protein C receptor augments protein C activation by the thrombin-thrombomodulin complex. Proc Natl Acad Sci U S A. 1996;93:10212–10216. doi: 10.1073/pnas.93.19.10212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wood MJ, Becvar LA, Prieto JH, Melacini G, Komives EA. NMR structures reveal how oxidation inactivates thrombomodulin. Biochemistry. 2003;42:11932–11942. doi: 10.1021/bi034646q. [DOI] [PubMed] [Google Scholar]
- 22.Wood MJ, Helena Prieto J, Komives EA. Structural and functional consequences of methionine oxidation in thrombomodulin. Biochimica et Biophysica Acta. 2005;1703:141–147. doi: 10.1016/j.bbapap.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 23.Fukai T, Ushio-Fukai M. Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller FJ, Jr., Gutterman DD, Rios CD, Heistad DD, Davidson BL. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ Res. 1998;82:1298–1305. doi: 10.1161/01.res.82.12.1298. [DOI] [PubMed] [Google Scholar]
- 25.Dayal S, Arning E, Bottiglieri T, Boger RH, Sigmund CD, Faraci FM, Lentz SR. Cerebral vascular dysfunction mediated by superoxide in hyperhomocysteinemic mice. Stroke. 2004;35:1957–1962. doi: 10.1161/01.STR.0000131749.81508.18. [DOI] [PubMed] [Google Scholar]
- 26.Brown KA, Didion SP, Andresen JJ, Faraci FM. Effect of aging, MnSOD deficiency, and genetic background on endothelial function: evidence for MnSOD haploinsufficiency. Arterioscler Thromb Vasc Biol. 2007;27:1941–1946. doi: 10.1161/ATVBAHA.107.146852. [DOI] [PubMed] [Google Scholar]
- 27.Didion SP, Ryan MJ, Didion LA, Fegan PE, Sigmund CD, Faraci FM. Increased superoxide and vascular dysfunction in CuZnSOD-deficient mice. Circ Res. 2002;91:938–944. doi: 10.1161/01.res.0000043280.65241.04. [DOI] [PubMed] [Google Scholar]
- 28.Yoshida T, Maulik N, Engelman RM, Ho YS, Das DK. Targeted disruption of the mouse Sod I gene makes the hearts vulnerable to ischemic reperfusion injury. Circ Res. 2000;86:264–269. doi: 10.1161/01.res.86.3.264. [DOI] [PubMed] [Google Scholar]
- 29.Martin FA, Murphy RP, Cummins PM. Thrombomodulin and the vascular endothelium: insights into functional, regulatory, and therapeutic aspects. Am J Physiol Heart Circ Physiol. 2013;304:H1585–1597. doi: 10.1152/ajpheart.00096.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Weiler H, Lindner V, Kerlin B, Isermann BH, Hendrickson SB, Cooley BC, Meh DA, Mosesson MW, Shworak NW, Post MJ, Conway EM, Ulfman LH, von Andrian UH, Weitz JI. Characterization of a mouse model for thrombomodulin deficiency. Arterioscler Thromb Vasc Biol. 2001;21:1531–1537. doi: 10.1161/hq0901.094496. [DOI] [PubMed] [Google Scholar]
- 31.Isermann B, Hendrickson SB, Zogg M, Wing M, Cummiskey M, Kisanuki YY, Yanagisawa M, Weiler H. Endothelium-specific loss of murine thrombomodulin disrupts the protein C anticoagulant pathway and causes juvenile-onset thrombosis. J Clin Invest. 2001;108:537–546. doi: 10.1172/JCI13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu JM, Crawley JT, Ferrell G, Zhang F, Li W, Esmon NL, Esmon CT. Disruption of the endothelial cell protein C receptor gene in mice causes placental thrombosis and early embryonic lethality. J Biol Chem. 2002;277:43335–43343. doi: 10.1074/jbc.M207538200. [DOI] [PubMed] [Google Scholar]
- 33.Lay AJ, Liang Z, Rosen ED, Castellino FJ. Mice with a severe deficiency in protein C display prothrombotic and proinflammatory phenotypes and compromised maternal reproductive capabilities. J Clin Invest. 2005;115:1552–1561. doi: 10.1172/JCI24030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mueller CF, Laude K, McNally JS, Harrison DG. ATVB in focus: redox mechanisms in blood vessels. Arterioscler.Thromb.Vasc.Biol. 2005;25:274–278. doi: 10.1161/01.ATV.0000149143.04821.eb. [DOI] [PubMed] [Google Scholar]
- 35.Gu SX, Stevens JW, Lentz SR. Regulation of thrombosis and vascular function by protein methionine oxidation. Blood. 2015 doi: 10.1182/blood-2015-01-544676. Epub Apr 21st. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bouma BN, Mosnier LO. Thrombin activatable fibrinolysis inhibitor (TAFI)--how does thrombin regulate fibrinolysis? Ann Med. 2006;38:378–388. doi: 10.1080/07853890600852898. [DOI] [PubMed] [Google Scholar]
- 37.Wang W, Nagashima M, Schneider M, Morser J, Nesheim M. Elements of the primary structure of thrombomodulin required for efficient thrombin-activable fibrinolysis inhibitor activation. J Biol Chem. 2000;275:22942–22947. doi: 10.1074/jbc.M001760200. [DOI] [PubMed] [Google Scholar]
- 38.Mendelsohn ME, O'Neill S, George D, Loscalzo J. Inhibition of fibrinogen binding to human platelets by S-nitroso-N-acetylcysteine. J Biol Chem. 1990;265:19028–19034. [PubMed] [Google Scholar]
- 39.Murohara T, Parkinson SJ, Waldman SA, Lefer AM. Inhibition of nitric oxide biosynthesis promotes P-selectin expression in platelets. Role of protein kinase C. Arterioscler Thromb Vasc Biol. 1995;15:2068–2075. doi: 10.1161/01.atv.15.11.2068. [DOI] [PubMed] [Google Scholar]
- 40.Neves AL, Mohammedi K, Emery N, Roussel R, Fumeron F, Marre M, Velho G. Allelic variations in superoxide dismutase-1 (SOD1) gene and renal and cardiovascular morbidity and mortality in type 2 diabetic subjects. Mol Genet Metab. 2012;106:359–365. doi: 10.1016/j.ymgme.2012.04.023. [DOI] [PubMed] [Google Scholar]
- 41.De Cristofaro R, Rocca B, Vitacolonna E, Falco A, Marchesani P, Ciabattoni G, Landolfi R, Patrono C, Davi G. Lipid and protein oxidation contribute to a prothrombotic state in patients with type 2 diabetes mellitus. J Thromb Haemost. 2003;1:250–256. doi: 10.1046/j.1538-7836.2003.00072.x. [DOI] [PubMed] [Google Scholar]
- 42.Leopold JA, Loscalzo J. Oxidative risk for atherothrombotic cardiovascular disease. Free Radic Biol Med. 2009;47:1673–1706. doi: 10.1016/j.freeradbiomed.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Juul K, Tybjaerg-Hansen A, Marklund S, Heegaard NH, Steffensen R, Sillesen H, Jensen G, Nordestgaard BG. Genetically reduced antioxidative protection and increased ischemic heart disease risk: The Copenhagen City Heart Study. Circulation. 2004;109:59–65. doi: 10.1161/01.CIR.0000105720.28086.6C. [DOI] [PubMed] [Google Scholar]
- 44.Yamada H, Yamada Y, Adachi T, Fukatsu A, Sakuma M, Futenma A, Kakumu S. Protective role of extracellular superoxide dismutase in hemodialysis patients. Nephron. 2000;84:218–223. doi: 10.1159/000045580. [DOI] [PubMed] [Google Scholar]
- 45.Fujimoto H, Taguchi J, Imai Y, Ayabe S, Hashimoto H, Kobayashi H, Ogasawara K, Aizawa T, Yamakado M, Nagai R, Ohno M. Manganese superoxide dismutase polymorphism affects the oxidized low-density lipoprotein-induced apoptosis of macrophages and coronary artery disease. Eur Heart J. 2008;29:1267–1274. doi: 10.1093/eurheartj/ehm500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.