

Abstract
Hydrogen sulfide (H2S) intoxication produces a rapid cardio-circulatory failure leading to cardiac arrest. In non-lethal forms of sulfide exposure, the presence of a circulatory shock is associated with long-term neurological sequelae. Our aim was to clarify the mechanisms of H2S-induced circulatory failure.
In anesthetized paralyzed and mechanically ventilated rats, cardiac output, arterial pressure and ventricular pressures were determined while NaHS was infused to increase arterial concentration of soluble H2S (CgH2S) from undetectable to levels leading to circulatory failure.
Compared to control/saline infusion, blood pressure started to decrease significantly along with a modest drop in peripheral vascular resistance (-19 ± 5%, P<0.01), when CgH2S reached about 1 microM. As CgH2S exceeded 2-3 microM, parameters of ventricular contractility diminished with no further reduction in peripheral resistance. Whenever H2S exposure was maintained at a higher level (CgH2S over 7 microM), a clear inhibition of cardiac contractility was observed, leading to asystole within minutes, but with no evidence of peripheral vasoplegia.
The immediate and long-term neurological effects of specifically counteracting sulfide induced cardiac contractility depression following H2S exposure remain to be investigated.
Keywords: cardiac toxicity, H2S, vascular blood flow, vasodilation
Introduction
Hydrogen sulfide (H2S) intoxication produces a coma associated to a depression in breathing and a profound circulatory failure [1]. This gas remains a potential source of dreadful intoxication in oil and gas industry [2] and has more recently used as a method of suicide [3]. In subjects or animals surviving the initial exposure, various neurological after-effects can occur spanning from minor defects -such as a persistent anosmia- to major cognitive and motor dysfunctions [4]. These neurological lesions appears to be provoked by the presence of a circulatory shock induced brain ischemia rather than by the direct exotoxicity of H2S alone, as shown for instance by Baldelli et al [5].
The mechanisms of the circulatory shock produced during H2S intoxication remains however poorly understood. Indeed, an increasing number of in-vitro studies are supporting the view that H2S has vasodilatory properties, akin to the effects of nitric oxide (NO) [6-8]. In most of these studies, soluble H2S at a concentration of 10 microM or above have been shown to produce a potent vasodilation [6-8]. Since similar blood levels of sulfide are sufficient to produce a terminal apnea in-vivo [9, 10], these observations suggest that exogenous H2S-induced vasodilation is an effect of sulfide toxicity. On the other hand, H2S has also been shown to reduce cardiac contractility, at least in-vitro, by acting like a calcium channel blocker [11, 12]. Since H2S-induced circulatory failure seems to be the major determinant of the neurological outcome of patients surviving H2S exposure [5], establishing the mechanisms of H2S-induced circulatory failure, i.e. vasoplegia versus cardiogenic shock, will help us defining the strategy to be used in the symptomatic treatment of sulfide exposure and hopefully limit its potential devastating neurological sequelae of hydrogen sulfide, which appear to be greatly potentiated by the presence of low perfusion of the brain [4]. To clarify this outstanding issue, we determined the hemodynamic changes, i.e. cardiac output, peripheral arterial blood flow, intracardiac and arterial pressures and stroke volume along with the change in peripheral resistances and lactate production as a function of the concentrations of H2S in the blood during stepwise increase or constant rate of sulfide infusion in anesthetized mechanically ventilated rats.
In this manuscript, the term sulfide or H2S will be used interchangeably, as dissolved/soluble H2S when in solution in the blood or in cells is, at physiological pH, mainly (70-80%) in the form of the anion HS-, while the gaseous form of H2S, which is responsible for H2S partial pressure and thus its diffusion across membranes, is only 20-30% of the total pool of free sulfide [13, 14]. Of note, is that majority of H2S produced or infused is combined with proteins and metallo-compounds sulfide (see [9, 10] and [15] for review and references). To describe the toxicology of the members of this “family” of molecules, H2S is often used for convenience.
Material and Methods
Animal preparation
Adult male Sprague-Dawley rats weighing 490 ± 84 g (Charles River) were studied. All experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, 8th Edition (National Research Council (US) Institute for Laboratory Animal Research).
The study was approved by the Pennsylvania State University College of Medicine Institutional Animal Care and Use Committee. All rats were housed in cages on a temperature-controlled room (24 ± 1°C) with 12 hour light/dark cycle, and were provided with standard laboratory diet and water ad libitum.
Anesthesia was induced with inhalation of isoflurane 3.5% in O2, followed by the urethane (1.2 g/kg, i.p.). Rat were tracheostomized (14G surflo catheter, Terumo, Tokyo, Japan) and mechanically ventilated (frequency of breathing: 80 breaths/min, minute ventilation: ∼350 ml/min) to maintain PaO2 around 80 mmHg. To prevent any spontaneous breathing movement, a muscular relaxant was infused via tail vein (10 mg/kg/h, Rocuronium Bromide, Hospira, Inc. Lake Forest, IL). A catheter (PE-50 tubing) was inserted into the right femoral artery for continuous monitoring of systemic arterial blood pressure. Additional catheters were inserted into the right femoral vein for NaHS infusion and left carotid artery for arterial blood sampling. Adequate ventilation was monitored by periodic arterial blood gas measurements. Arterial blood (0.1 ml) was sampled and the gases partial pressures in O2 (PaO2), CO2 (PaCO2), and blood lactate concentration were measured using i-STAT1 blood gas analyzer (Abaxis, Union City, CA).
The left femoral artery was isolated and a transonic flow probe (MA1PRB, Transonic Systems Inc. Ithaca, NY) was placed around the artery.
Our initial purpose was to determine the change in cardiac output and peripheral resistance. Therefore, in a first group of rats (see protocol section for further information), a catheter (20G surflo catheter, TERUMO, Tokyo, Japan) was inserted into the right ventricle through the right jugular vein for continuous monitoring of right ventricular pressure. Cardiac output was determined as follows: The 1st and 2nd rib on the left side were partially resected and left upper lobe of the lung was retracted to expose the pulmonary artery trunk. The small incision of the pericardium was performed and the pulmonary artery was isolated from surrounding tissues. A transonic flow probe (MA2PSB) was placed around the isolated pulmonary trunk just above the right ventricle.
To further quantify the effects of sulfide on the left ventricle function, a separate group of rats was equipped with a catheter (PE-50 tubing) inserted into left ventricle through the right carotid artery. This allowed the continuous monitoring of left ventricular pressure while measuring cardiac output via the determination of aortic blood. A median sternotomy and a small incision of the pericardium were performed to expose the ascending aorta. A transonic flow probe (MA2PSB) was placed around the ascending aorta just above the coronary arteries. Great care was taken to prevent compression of vessels or obstruction of circulation during the procedure.
At the end of the experiment, rats were euthanized by injection of a lethal dose of barbiturate (200 mg/kg) into the right heart through the jugular catheter, followed by aortic dissection.
Measurements
The arterial and ventricular catheters were connected to pressure transducer (BD DTXPIus Transducer, Becton Dickinson, Franklin Lakes, NJ; amplifier TA-100, CWE, Ardmore, PA) to monitor blood pressure.
The blood flow probes were connected to the transit-time flowmeter (TS420, Transonic Systems Inc., Ithaca, NY).
Expiratory flow was measured using a pneumotachograph (1100 Series, Hans Rudolph, Inc. Shawnee, KS), as previously described [9]. Mixed-diluted expired O2 (FchO2), CO2 (FchCO2) and H2S fractions (FchH2S) were measured continuously using O2 (Oxystar-100, CWE Inc. Ardmore, PA), CO2 (model 17630, VacuMed, Ventura, CA) and H2S (InterScan RM series; range: 0-200 ppm, InterScan Corporation, Simi Valley, CA) analyzers.
All signals were digitized at 400 Hz using an analog-to-digital data acquisition system (Power Lab 16/35, AD Instruments, Inc. Colorado Springs, CO) and were visualized on-line. All data were stored for further analysis by LabChart7 (AD Instruments, Inc. Colorado Springs, CO).
Data Analysis
Blood flow was determined by positive integration of the flow signals every 3 seconds then expressed in ml/min. Total peripheral resistance and hindlimb peripheral resistance were computed by dividing mean arterial pressure by mean cardiac output or mean femoral artery blood flow respectively. For the evaluation of left ventricular function, left ventricular systolic pressure (LVSP) and heart rate (HR) were determined by peak detection, and dP/dt max was analyzed by derivative of the left ventricular pressure signal.
Breathing frequency (f) and tidal volume (VT) was determined using peak detection and integration of the expiratory flow signal, respectively, and minute ventilation (V̇E) was computed as f × VT in body temperature and pressure, saturated (BTPS) conditions.
Mixed expired fraction of H2S (FEH2S) were computed as previously described [9]. The partial pressure of expired H2S (PEH2S) was then calculated as FEH2S × barometric pressure (PB, 760 mmHg). Then alveolar partial pressure of H2S (PAH2S) assimilated to the arterial partial pressure of H2S (PaH2S) was computed [9]. Finally the concentration of gaseous H2S in the blood (CgH2S) was then calculated as: CgH2S = 0.00012 × PaH2S, with 0.00012 being the coefficient of solubility of H2S (0.09 mole/l/760 mmHg at 37°C in saline). Assuming that H2S is under the form of H2S gas and its sulfhydryl anion HS- at a ratio of 1/3 and 2/3 in the arterial blood [16], the concentration of dissolved H2S could be estimated as three times CgH2S.
Experimental protocols: intravenous NaHS infusion
H2S solution was prepared using sodium hydrosulfide hydrate (NaHS, Sigma, St. Louis, MO). The molecular weight of the “hydrated” form of NaHS was 74.08 g/mol as the crystals used for preparing the solution contained 75% NaHS - 25% H20. NaHS was diluted in saline at a concentration of 0.8 mg/ml (10.8 mM), and prepared immediately prior each experiment and kept in airtight syringes. The level of exposure to H2S was determined based on the relationship we have previously established between the blood concentrations, rate of exogenously administered H2S, and the corresponding clinical symptoms, i.e., H2S-induced apnea using continuous infusion of NaHS solution [9, 10].
First, a group of 14 rats was divided into 2 sets, randomly selected, one received NaHS infusion (n = 7) while the other received saline (n = 7). After 10 minute of stable recording, intravenous NaHS infusion was started at the rate of 0.05 ml/min (0.5 micromoles/min) using a syringe pump (Fusion 100, Chemyx Inc., Stafford, TX). Infusion rate was increased by 0.05 ml/min every 2 minutes until 0.6 ml/min (6.5 micromoles/min). Based on our previous experiments, the rate of 0.6 ml/min will increase CgH2S by more than 2 microM and produce a breathing stimulation [9, 10]. To limit the number of animals that were sacrificed and after having observed that the cardiovascular effects were extremely short lasting (few minutes at the best after the cessation of NaHS infusion all circulatory parameters returned to normal), two of seven rats in both group received a second series of saline or NaHS infusion. Animals were then given a recovery period long enough so that blood pressure and flow returned to normal before repeating the same infusion. All parameters were monitored continuously and averaged over the last 30 seconds of each step of infusion. Arterial blood was sampled before and after the periods of NaHS infusion. Blood gas analysis was performed immediately after sampling. Corresponding volume of saline was infused at the same rate of NaHS infusion in the control group.
In an additional group of 6 rats, left ventricular pressure and aortic flow were determined while NaHS was infused at a constant rate just above the level found to decrease cardiac output (0.7 ml/min see result section), in keeping with the data obtained in the protocol described above.
Finally, at the end of the experiment, NaHS was infused at a higher rate of 2.0 ml/min in 5 rats while the changes in cardiac function versus peripheral resistance were determined.
Statistical Analysis
All results are presented as mean ± SD. All variables of interest were compared between the vehicle control group and the group receiving NaHS at the corresponding rate of infusion using two-way ANOVA for repeated measurements. Post-hoc comparisons were performed using a Bonferroni correction. P < 0.05 was regarded as significant for all these comparisons. Data were compared before and during each rate of infusion using ANOVA for repeated measurements. For left ventricular function determined during constant infusion, the values were compared between baseline and 2 minutes into NaHS infusion, using paired t test. All statistical analyses were conducted using GraphPad Prism 6 (Graphpad Software, La Jolla, CA).
Results
1- Effect of incremental infusion of saline vs NaHS (from 0 to 6 micromoles/min) on circulation
Figure 1 shows an example of the effects of saline versus NaHS infusion on mean arterial blood pressure (Mean ABP), mean right ventricular pressure (Mean RV pressure), mean cardiac output, total peripheral vascular resistance, and estimated CgH2S (displayed in linear and log scale) in 2 different rats. Averaged data are shown on figures 2-4. At the end of the protocol, the total volume of fluid infused to each animal of both group was the same (7.8 ml, ∼15 ml/kg).
Figure 1.
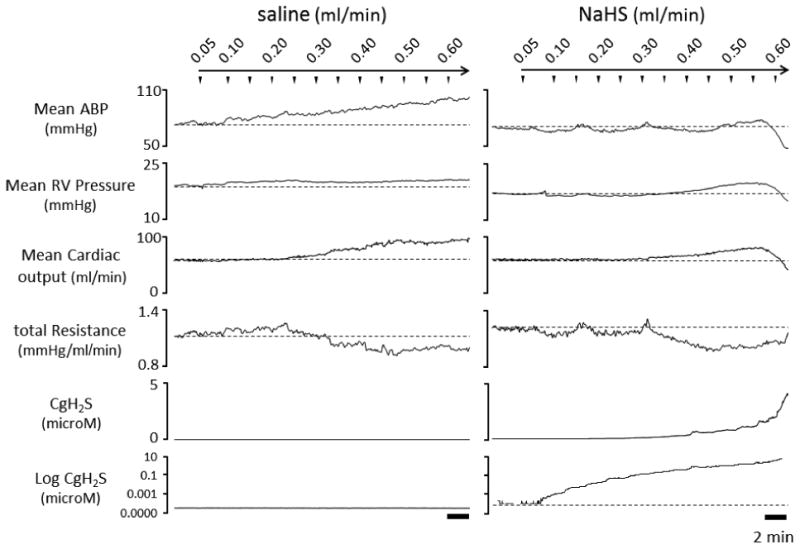
Example of the effects of saline (left panel) and sulfide infusion (right panel) on mean arterial blood pressure (Mean ABP), mean right ventricular pressure (Mean RV pressure), mean cardiac output, total peripheral vascular resistance (total Resistance), and estimated CgH2S (displayed in normal and log scale) in 2 different rats for rate up to 0.6 ml/min. Mean ABP and cardiac output displayed a progressive and continuous rise during saline infusion, with a modest reduction in total resistance. During sulfide infusion (10.8 micromoles/ml), the normal increase in mean ABP was virtually abolished which was associated with the reduction of total resistance, which was clearly visible above 0.4 ml/min. A very rapid drop in mean ABP, mean RV pressure, and mean cardiac output was observed after 0.55 ml/min (5.9 micromoles/min) of NaHS infusion. CgH2S increased exponentially during NaHS infusion.
Figure 2.
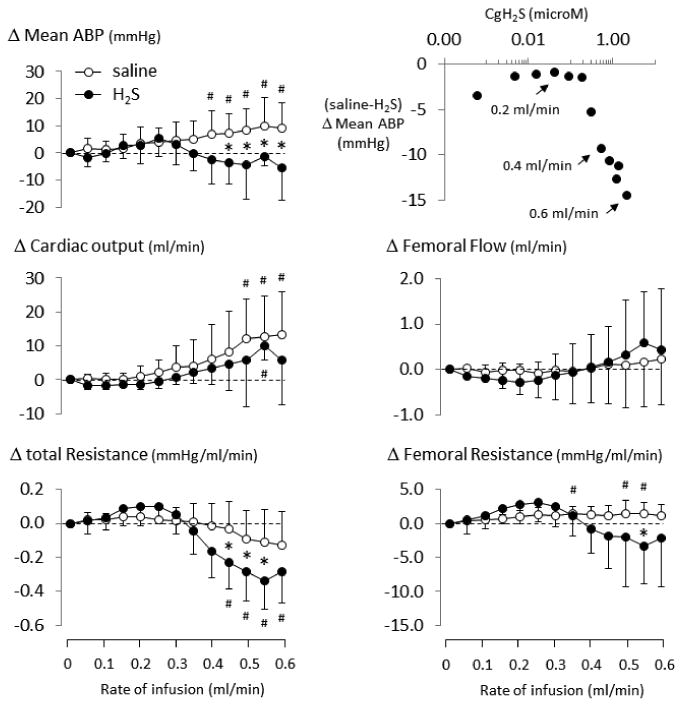
Averaged blood pressure, blood flow, and vascular resistance in the groups of rats receiving saline (open circle) vs NaHS (closed circle) infusion as a function of rate of infusion (for rate up to 0.6 ml/min). Mean arterial blood pressure (ABP) and cardiac output are increasing throughout the period of saline infusion, without major changes in femoral blood flow. This resulted in a modest reduction of total peripheral resistance. NaHS infusion induced similar changes in all parameters between 0-0.35 ml/min (3.8 micromoles/min), above these levels, mean ABP, femoral resistance, and total resistance departed from saline infusion showing a significantly lower blood pressure associate to a decrease in peripheral resistance. At 0.6 ml/min (6.5 micromoles/min) there was a trend for a decrease in cardiac output in the NaHS group. The iso-rate (iso-volume) difference in the variation in mean ABP between saline and H2S group is illustrated as a function of CgH2S (top right). Compared to saline infusion, NaHS infusion clearly reduced blood pressure when CgH2S increased. Values are shown in mean ± SD. *significantly different from saline/control at P<0.05. #significantly different from baseline at P<0.05.
Figure 4.
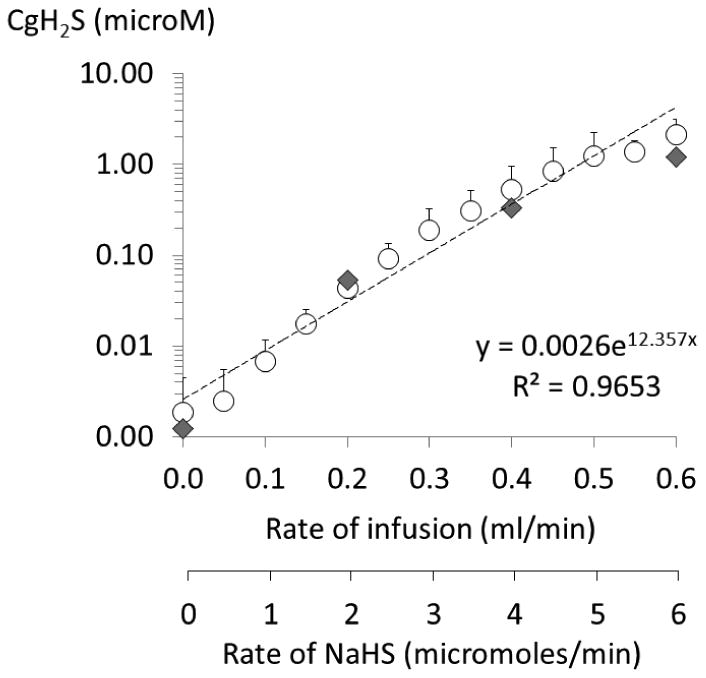
CgH2S as a function of the rate of NaHS infusion, for rate up to 0.6 ml/min). CgH2S showed exponential-like increase during NaHS infusion in keeping with the rate of infusion. The data from the previous study (Klingerman et al. 2013, gray diamond) fits with the relationship in this study. Values are shown as mean ± SD.
1-a) Effects of saline infusion (control)
Hemodynamic changes
As illustrated on figure 1-3, mean ABP, mean RV pressure, and cardiac output increased progressively during vehicle saline infusion in keeping to the rate of sulfide infusion. Mean ABP, which baseline levels averaged 79 ± 12 mmHg, rose throughout the period of infusion becoming significantly higher than the pre-infusion values at 0.40 ml/min (P<0.01). Mean ABP increased by 9 ± 10 mmHg at 0.6 ml/min of infusion (P<0.01) as shown on figure 2. This increase in mean ABP was clearly related to a rise in cardiac output, which (baseline 55 ± 14 ml/min) rose by 23 ± 18% at 0.6 ml/min of infusion (P<0.01). Baseline heart rate was 402 ± 125 bpm and did not change significantly throughout the period of infusion (fig. 3), as a result, the rise in cardiac output was exclusively related an increase in stroke volume (fig. 3): stroke volume averaged 0.147 ± 0.047 ml at baseline, and increased significantly at 0.50 ml/min infusion (P<0.01) to reached +22 ± 21% at 0.6 ml/min infusion (P<0.01). This increase in cardiac output related to volume that was infused was associated to a modest reduction in peripheral resistance; total peripheral vascular resistance (fig. 2) decreased by -7 ± 12% at 0.6 ml/min (not significant). Femoral flow did not change significantly throughout the infusion period (fig. 2).
Figure 3.
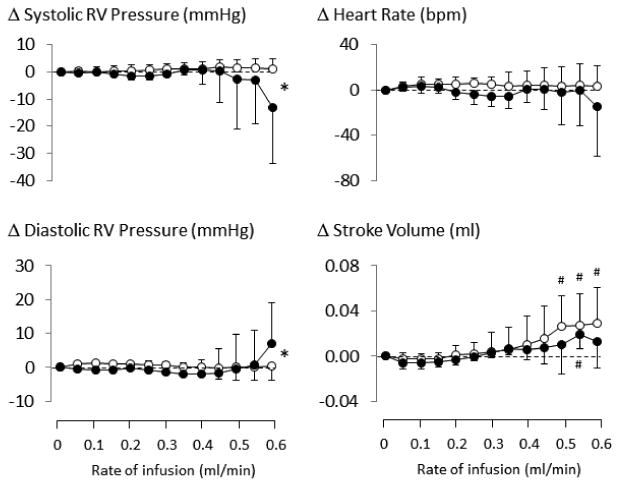
Averaged cardiac parameters (stroke volume, heart rate, and right ventricular (RV) pressure) in the groups of rats receiving saline (open circle) vs NaHS (closed circle) infusion as a function of rate of infusion, for rate up to 0.6 ml/min. During saline infusion, stroke volume increased according to infusion rate, but systolic RV pressure, diastolic RV pressure, and heart rate did not change. The only significant changes produced by NaHS infusion were observed at 0.6 ml/min (6.5 micromoles/min) wherein systolic RV pressure decreased and diastolic RV pressure significantly increased, associated to a non-significant decrease in stroke volume. Values are shown in mean ± SD. *significantly different from saline/control at P<0.05. #significantly different from baseline at P<0.05.
Blood gas and lactate
Baseline PaO2 and PaCO2 were 79 ± 16 mmHg and 34 ± 6 mmHg, respectively, and were not affected by the saline infusion. At the end of period of infusion PaO2 and PaCO2 averaged 75 ± 11 mmHg and 36 ± 7 mmHg, respectively. Blood lactate at the end of saline infusion was 1.69 ± 0.29 mM.
1-b) Effects of NaHS infusion
CgH2S during NaHS infusion
As illustrated in figure 1 and figure 4, CgH2S increased in keeping with the rate of NaHS infusion following an exponential pattern.
Hemodynamic changes
Baseline levels of mean ABP (83 ± 18 mmHg), stroke volume (0.124 ± 0.034 ml), cardiac output (53 ± 12 ml/min), and femoral flow (3.5 ± 1.7 ml/min) were not different from the saline group.
When compared to saline infusion, the effects of H2S on circulation can be described in keeping with the rate of infusion as follows:
-
Up to 0.35 ml/min (∼3.8 micromoles/min)
The changes in mean ABP, blood flow, pressure in the heart and peripheral resistance were indistinguishable from the effects of saline infusion. Mean ABP increased by 5 ± 7 mmHg from baseline i.e. within the same range as saline infusion (fig. 2 and 3). CgH2S reached a value, which averaged 0.31 microM.
-
From 0.35 – 0.55 ml/min (3.8-5.9 micromoles/min)
Mean ABP during NaHS infusion diverged from saline infusion and decreased significantly along with a reduction in peripheral resistance. This change in mean ABP was significantly different from the saline group above 0.45 ml/min infusion (4.9 micromoles/min, P<0.05). Cardiac output continuously increased by 20 ± 10% of the baseline value at 0.55 ml/min (5.9 micromoles/min, P<0.05 from baseline). Femoral flow also increased by 14 ± 25% from baseline at 0.55 ml/min (although this difference was not significant). Total vascular resistance showed significant decrease from baseline at 0.45 ml/min (P<0.05), and continued to decrease reaching -19 ± 5% at 0.55 ml/min (P<0.01). All the changes in vascular resistance were significantly different from saline group between 0.45-0.55 ml/min of NaHS infusion (P<0.01). This progressive reduction in mean ABP and peripheral resistance was associated with an increase in stroke volume (19 ± 16% at 0.55 ml/min, P<0.05). These effects were observed at CgH2S between 0.31-1.36 microM.
-
From 0.55-0.6 ml/min (5.9- 6.4micromoles/min)
Figure 5 displays an example of the effects of infusion during the transition from 0.55 to 0.6 ml/min on arterial pressure, cardiac output, femoral flow, and CgH2S in one rat. On average, blood pressure continued to decrease (fig. 2), but this time along with a decrease in systolic RV pressure, stroke volume (fig. 3) and cardiac output as soon as the rate of infusion was above 0.55 ml/min. These effects were observed for CgH2S, which averaged 1.36 microM. The most striking change was an increase in diastolic right ventricular pressure and drop in systolic RV pressure (fig. 1 and 3), which was significant at 0.6 ml/min, reflecting a reduction in cardiac performance. Infusion was stopped at this rate and both blood pressure and cardiac output were able to recover very rapidly i.e. as soon as the infusion was interrupted.
Figure 5.
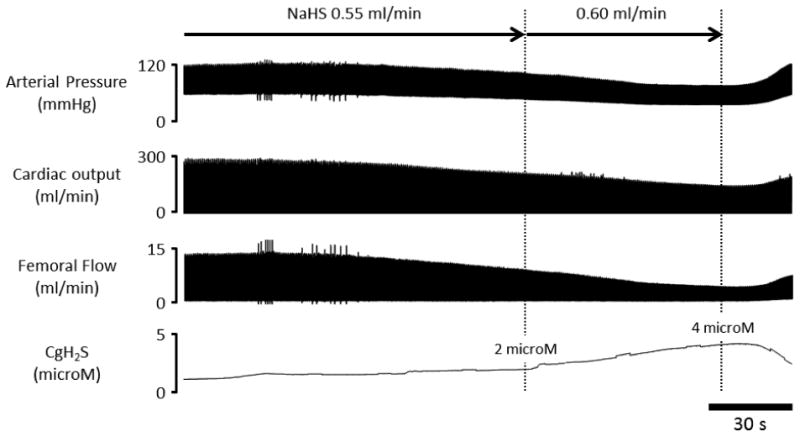
Example of the effects of NaHS infusion at 0.55-0.60 ml/min (5.9-6.5 micromoles/min) on arterial pressure, cardiac output, femoral flow, and CgH2S in one rat. Both blood pressure and blood flow started to drop when CgH2S increased above 2 microM.
Blood gas and lactate
Baseline PaO2 and PaCO2 were 87 ± 11 mmHg and 35 ± 6 mmHg respectively. After the period of NaHS infusion, PaO2 and PaCO2 were 82 ± 8 mmHg and 36 ± 8 mmHg, respectively, and therefore were not affected by NaHS infusion, while blood lactate averaged 2.59 ± 0.69 mM (P<0.01, vs saline).
2- Effect of H2S infusion on left ventricular function a rate of 0.7 ml/min (7.5 micromoles/min)
We found that left ventricular and blood pressure dropped along with cardiac output at a rate of 0.7 ml/min (fig. 6 and 7). Cardiac output dropped by half (152 ± 47 to 75 ± 33 ml/min/kg) 88-141 seconds (median 108 seconds) after the onset of sulfide infusion. At the same time, left ventricular systolic pressure (LVSP) decreased from 94 ± 19 to 62 ± 11 mmHg (P<0.01), and heart rate decreased from 433 ± 78 bpm to 311 ± 100 bpm (P<0.01); dP/dt max also dropped by about half from 3260 ± 946 to 1626 ± 432 mmHg/s (P<0.01). This severe reduction in cardiac contractility occurred when H2S concentration in the arterial blood reached 7.6 ± 0.6 microM.
Figure 6.
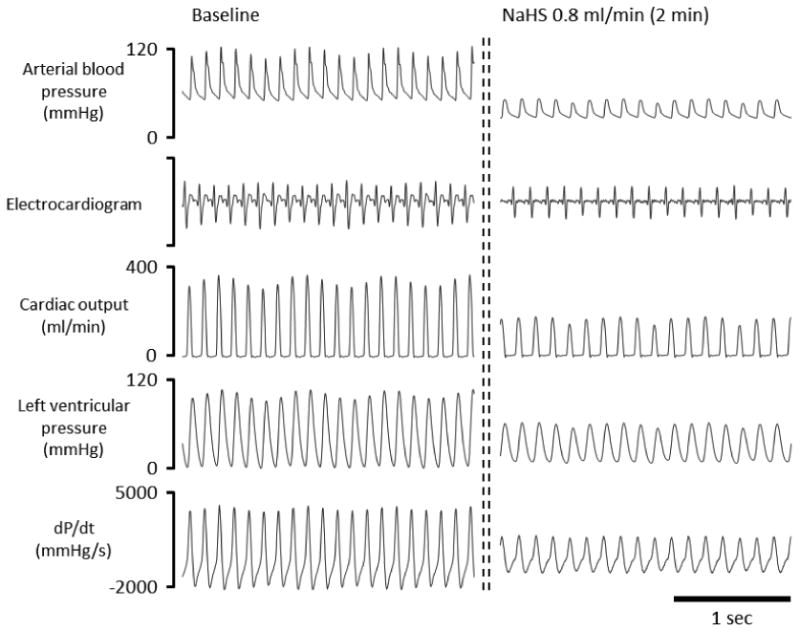
Example of effects of NaHS infusion at a rate of 0.8 ml/min (8.6 micromoles/min) on the arterial blood pressure, electrocardiogram, cardiac output, and left ventricular pressure in one rat. Two to three minutes into NaHS infusion, arterial blood pressure, cardiac output, and left ventricular pressure largely decreased compared to baseline.
Figure 7.
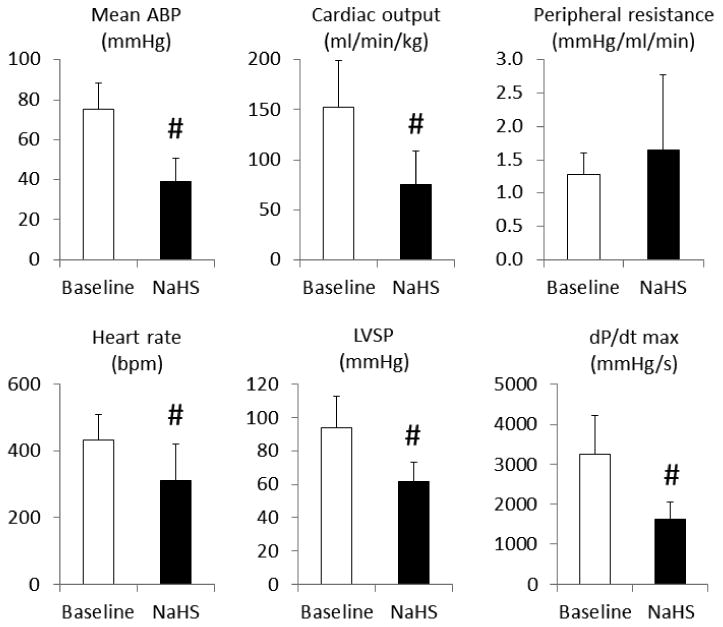
Mean ± SD values of left ventricular functions: mean arterial blood pressure (ABP), cardiac output, peripheral resistance, left ventricular systolic pressure (LVSP), dP/dt max, and heart rate at baseline (before infusion) and at 2 minutes into an infusion of 0.6-0.8 ml/min NaHS in 6 rats. All the left ventricular-related parameters significantly dropped during NaHS infusion, without any reduction in peripheral resistance. #significantly different from baseline, P<0.01.
CgH2S and circulatory effects of H2S
Figure 8 displays all individual data obtained from all NaHS infusion tests, showing the change in cardiac output and blood pressure as a function of gaseous H2S concentration from infusions ranging from 0 to 0.7 ml/min. Clearly, the decrease in blood pressure was associated with a drop in cardiac output as soon as CgH2S increased above 2-3 microM.
Figure 8.
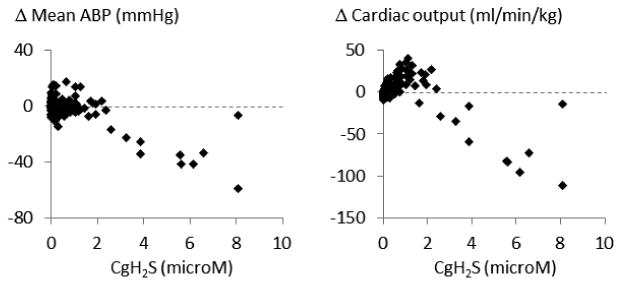
Individual data points obtained from all NaHS infusion tests. Note that when CgH2S reached concentrations about 2-3 microM, a progressive in drop blood pressure associated to a reduction in cardiac output was observed leading to sever reduction in cardiac output a soon as CgH2S was above 3 microM (see discussion section for more details).
Finally, In 5 rats, the infusion of NaHS was resumed at the end of the experiment and maintained at about twice the levels required to affect cardiac function. Both arterial pressure and cardiac output dropped dramatically (fig. 9A). In all cases, effective cardiac contractions stopped being discernible on the arterial pressure or cardiac output signals after 3 minutes, without any reduction in peripheral vascular resistance (fig. 9B). In other words, at the time required for cardiac output to drop by half when a rate of sulfide infusion of 7-8 micromoles/min was used, 15-16 micromoles/min led to a complete cessation of cardiac contractions.
Figure 9.
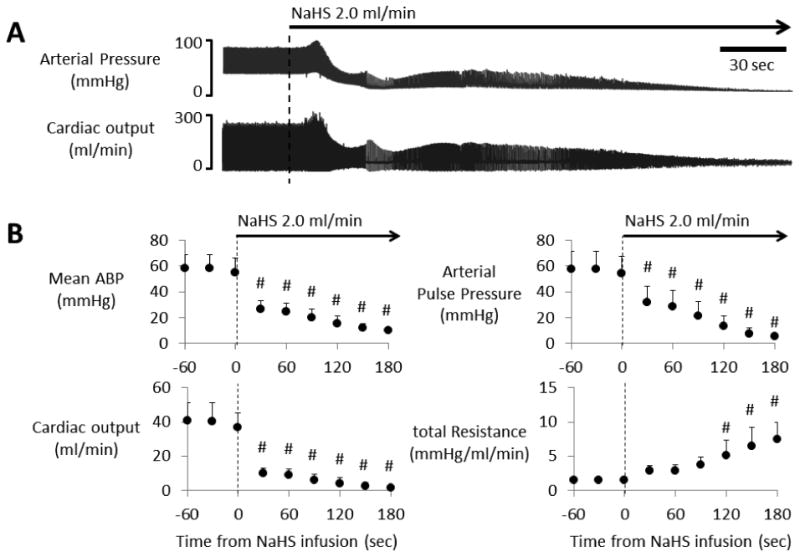
A) Example of the effects of NaHS infusion at 2.0 ml/min (21.6 micromoles/min) on arterial pressure and cardiac output in one rat. Both blood pressure and cardiac output rapidly dropped during continuous infusion of high-dose of NaHS. Pulse pressure was undetectable within 3 minutes. B) Averaged arterial blood pressure (ABP), arterial pulse pressure, cardiac output, and total peripheral resistance in 5 rats receiving NaHS infusion at 2.0 ml/min. During NaHS infusion, arterial pressure, pulse pressure, and cardiac output significantly dropped within the first minute of infusion, while estimated total peripheral resistance increased until cardiac asystole occurred. Values are shown in mean ± SD. #significantly different from the value at -60 seconds, P<0.05.
Discussion
Intravenous infusion of NaHS produces a small (less than 20%), but significant, reduction in peripheral vascular resistance in anesthetized ventilated rats at concentrations of gaseous H2S ranging between 0.5-1.0 microM. Below these levels, no circulatory changes were observed, whereas, as soon as CgH2S increased above 2-3 microM, which corresponds to levels not even associated with symptoms of toxicity [9, 10], cardiac contractility decreased very rapidly, leading to a dramatic reduction in cardiac output but with no additional reduction in peripheral vascular resistance.
H2S and peripheral vascular resistance
We found that H2S decreased the peripheral vascular resistance by an average of 19%, leading to a relative reduction in ABP (in keeping to the effect of saline infusion) only in a narrow and low ranges of H2S infusion and concentrations (fig. 1-3). Cardiac output increases along with the rate of infusion in both saline and NaHS infusion. These results are qualitatively consistent with earlier findings showing that H2S can produce a vascular relaxation in various in-vitro vascular preparations [8]. As discussed in the following paragraphs, it should be pointed out that the concentrations required to produce such an effect, in-vitro, were one to two orders of magnitude higher than the concentrations found to be effective in our in-vivo experiments. Previous data obtained in small animals using H2S intravenous injection have resulted in a drop of blood pressure [6, 7]. The mechanism, which has been put forward to account for the effects of H2S, relate to the activation of K-ATP channels in smooth muscle [17]. In addition, H2S induced vaso-relaxation, although not directly mediated by cyclic guanosine 5′-monophosphate (cGMP), appears to be potentiated by the NO pathway [18, 19]. Our present findings depart from most in-vitro studies, in the sense that the effects we observed remain very modest and are only produced in a very small range of diffusible H2S concentrations in contact with the vessels. Certainly, various factors could have blunted or limited the vascular of H2S in-vivo, one of them being the stimulation of the chemoreceptors, which occurs in the same ranges of CgH2S as those producing a reduction in peripheral resistance [9]. Such stimulation could have activated the sympathetic system triggering in turn a classical vasoconstriction, which can oppose any direct vascular relaxation effects (chemoreceptor induced sympathetic stimulation [20, 21]). Intravenous bolus injection of an exogenous H2S solution has long been shown to increase breathing by stimulation of carotid body chemoreceptor [22, 23]. In our study, the rats were mechanically ventilated and were given a muscular relaxant to prevent the confounding effects of spontaneous breathing on circulation. As a consequence, the determination of expired H2S and blood flow were not influenced by the changes in breathing pattern. Still H2S-induced direct stimulation of chemoreceptors, cardiac receptors, or even lung receptors might have affected peripheral blood flow regulation [21, 24]. Incidentally, the ratio between systolic RV pressure and pulmonary blood flow, a marker of right ventricular afterload, was not different between the two groups. This is consistent with the study using human pulmonary artery which could only be dilated with a concentration 200 times higher than the concentrations we used in-vivo (500 microM of H2S [25]). Sun et al. [26] also found differences in the response to NaHS between rat aortic and pulmonary arterial rings, the latter being less responsive to NaHS than aorta. They also found that pulmonary artery rings could produce a vasoconstriction after 2 minutes of administration of very large concentrations of soluble NaHS (50-200 microM).
Toxicity of H2S for the heart, depression of cardiac contractility
H2S at levels, lower that those we previously found to be able to depress breathing [9], decreases cardiac contractility (see also fig. 8). As soon as level of soluble H2S in the blood increases to higher concentrations and reaches about 7 microM, cardiac output was severely depressed leading to asystole within a few minutes.
H2S can affect cardiac contractility via a direct inhibition of ATP production [27] or an inhibition of L-type calcium channels [11, 12], producing in turn a negative inotropic effect. In addition, H2S appears to potentiate the effect of NO [18, 28, 29]. NO is thought to be one of the main mechanisms of cardiac depression in various conditions wherein a cardiogenic shock can develops, e.g. septic shock [30, 31]. The “blocking” effect of H2S on calcium channels appears to be the major factor leading to the reduction in the contractility of the cardiomyocytes [11, 12]. At least 2 different mechanisms could be put forward to account for this effect. The first one relates the sulfhydration of cysteine residues on calcium channels [11]. This mechanism has important potential implications since sulfhydration could be antagonized through the creation of reducing environment of the heart to “release” H2S. Second, calcium channel activity seems to be able to be modulated by the red-ox environment of the cells [32], the latter could be greatly affected by the presence of H2S. Of note, is that all the circulatory parameters returned to their baseline values very rapidly following the cessation of H2S exposure, suggesting that the mechanisms of depression in cardiac contractility is rapidly reversible, and thus compatible with the very rapid oxidation of H2S [10, 33] as well as the trapping of free H2S by the metallo-compounds [34] present in the heart (myoglobin). Maintaining H2S exposure or leading to levels of more than 5-10 microM, will lead to major cardiac depression.
Circulatory effects as a function of H2S levels
In our previous studies [9], we found that breathing was stimulated in sedated rats by H2S at CgH2S of about 1 microM, while a terminal apnea occurred around 8 microM, a level that would be also associated with a severe reduction in cardiac contractility. In in-vitro experiments, H2S is usually diluted in saline, or phosphate buffered solution, and is therefore only present in soluble forms (H2S and HS-). In contrast, in the blood, a large part of H2S that, can be have measured for instance using monobromobimane technic [9, 35], is combined with plasma proteins or hemoglobin (and thus is not diffusible). We used the level of alveolar H2S to estimate the gaseous and thus soluble forms of H2S interacting with the heart or the vascular structure in-vivo. Assuming that H2S is under the form of H2S gas and HS- at a ratio of 1/3 in the arterial blood [13, 14], the total free concentration of H2S and HS- was therefore approximately 4 microM, when the cardiac function was affected. This effect seems therefore to develop very “early” during H2S intoxication. These data are consistent with results obtained in the perfused rat heart, wherein a reduction in cardiac function can be observed starting at only 1 microM of NaHS [36]. Incidentally, such low microM levels of H2S have also been shown to be able to already affect the cytochrome c oxidase activity [27, 37].
Model limitations
The modality of sulfide exposure used in this study, i.e. intravenous infusion of sulfide, is not faithful to a clinical scenario of H2S intoxication, wherein H2S is typically inhaled. When in solution of NaHS, sulfides are actually present under the form of H2S/HS-, i.e. in gaseous form (20-30%) and under the form of HS- (70-80%) [13, 14, 16]. These 2 forms also constitute the pool of soluble sulfide (and in the same proportion) in the blood and in the cells. Indeed, as mentioned in the introduction, whether “H2S” is produced endogenously, inhaled, infused or formed from a slow releasing agent, it is never present in its gaseous form only: as soon as H2S reaches the blood or increases in the tissues, the majority of soluble H2S is under the form of HS-, while an even larger pool combined with proteins. All these pools are always in equilibrium in keeping with the level of H2S partial pressure [9]. The general term sulfide should always be preferable to the use of H2S, as H2S in its gaseous form is never present alone in a biological milieu (it would be present under a pure gaseous form in a very acidic solution).
Certainly the toxicity of sulfide towards the lungs and the upper-airways [38, 39] is expected to be blunted, at any given level of sulfide in the blood, if sulfides are infused rather than inhaled [40]. However, as soon as H2S reaches the blood, the original site of diffusion, whether the alveolar regions to or a direct venous infusion, is not going to affect the distribution of the various pools of sulfide in the blood and the concentration of H2S in the tissues (see above paragraph and ref for discussion [9, 10, 35]). It is however likely that when infused into the venous blood, concentrations of free H2S reaching the right heart would be higher than those entering the left heart (exposed to a lower concentration after a passage through the lungs), while the opposite is expected to occur during H2S inhalation, i.e. when sulfide reached directly the left heart from the pulmonary circulation. Finally, recent studies have been using promising slow H2S releasing agent such as GYY4137 [41] to study the physiological effects of sulfide on vessels. The interest of using these compounds in investigating sulfide toxicity is yet to be established. Indeed, a slow H2S releasing compound [41] is not faithful to a real life scenario created by an exposure to toxic levels of H2S, in keeping with the very fast kinetics of H2S toxicity (coma, respiratory arrest or circulatory failure), which can be produced within seconds during inhalation of H2S or infusion of solution of sulfide obtained from NaHS [40, 42]. Perhaps more importantly, whenever GYY4137 “releases” H2S, the latter will, for most of it, be immediately transformed into HS- along with the creation of a pool of sulfides combined to proteins in various ways, including in sulfhydration form [15, 43], just like any sources of exogenous or endogenous H2S. For all intends and purposes, the reduction in left cardiac contractility during H2S infusion that we are reporting in the present study appears to be an early sign of sulfide toxicity and thus represents a relevant mechanism responsible of the circulatory failure that occur very early during sulfide inhalation. The limits of using an sedated animal has been discussed in details in one of our previous paper [44].
Conclusion
Continuous intravenous infusion of a sulfide solution prepared from NaHS produces a modest reduction in peripheral vascular resistance in rats, which occurs when CgH2S reaches 0.5 to 1 microM (2-4 microM of total soluble sulfide, i.e. comprising H2S and HS-) in the arterial blood, corresponding to levels of sulfide we previously found capable of stimulating breathing. However, the range of exogenous H2S able to produce this effect is relatively narrow and as soon as gaseous H2S concentration reaches 2-3 microM, i.e. still below levels of sulfide producing an apnea, a significant depression in cardiac contractility occurs leading to a fall in cardiac output and blood pressure as sulfide concentrations increases. This finding supports the view that H2S-induced circulatory failure is predominantly related to a cardiogenic shock, with little involvement of the peripheral vascular network.
Acknowledgments
This work has been supported in part by the CounterACT Program, National Institutes of Health Office of the Director (NIH OD), and the National Institute of Neurological Disorders and Stroke (NINDS), Grant Number 1R21NS080788-01.
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest.
References
- 1.Reiffenstein RJ, Hulbert WC, Roth SH. Toxicology of hydrogen sulfide. Annu Rev Pharmacol Toxicol. 1992;32:109–134. doi: 10.1146/annurev.pa.32.040192.000545. [DOI] [PubMed] [Google Scholar]
- 2.Arnold IM, Dufresne RM, Alleyne BC, Stuart PJ. Health implication of occupational exposures to hydrogen sulfide. J Occup Med. 1985;27:373–376. doi: 10.1097/00043764-198505000-00018. [DOI] [PubMed] [Google Scholar]
- 3.Bott E, Dodd M. Suicide by hydrogen sulfide inhalation. Am J Forensic Med Pathol. 2013;34:23–25. doi: 10.1097/PAF.0b013e31827ab5ad. [DOI] [PubMed] [Google Scholar]
- 4.Tvedt B, Skyberg K, Aaserud O, Hobbesland A, Mathiesen T. Brain damage caused by hydrogen sulfide: a follow-up study of six patients. Am J Ind Med. 1991;20:91–101. doi: 10.1002/ajim.4700200109. [DOI] [PubMed] [Google Scholar]
- 5.Baldelli RJ, Green FH, Auer RN. Sulfide toxicity: mechanical ventilation and hypotension determine survival rate and brain necrosis. J Appl Physiol. 1993;75:1348–1353. doi: 10.1152/jappl.1993.75.3.1348. 1985. [DOI] [PubMed] [Google Scholar]
- 6.Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, Snyder SH, Wang R. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322:587–590. doi: 10.1126/science.1162667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;237:527–531. doi: 10.1006/bbrc.1997.6878. [DOI] [PubMed] [Google Scholar]
- 9.Klingerman CM, Trushin N, Prokopczyk B, Haouzi P. H2S concentrations in the arterial blood during H2S administration in relation to its toxicity and effects on breathing. Am J Physiol Regul Integr Comp Physiol. 2013;305:R630–638. doi: 10.1152/ajpregu.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haouzi P, Sonobe T, Torsell-Tubbs N, Prokopczyk B, Chenuel B, Klingerman CM. In Vivo Interactions Between Cobalt or Ferric Compounds and the Pools of Sulphide in the Blood During and After H2S Poisoning. Toxicol Sci. 2014;141:493–504. doi: 10.1093/toxsci/kfu140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang R, Sun Y, Tsai H, Tang C, Jin H, Du J. Hydrogen sulfide inhibits L-type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PLoS One. 2012;7:e37073. doi: 10.1371/journal.pone.0037073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun YG, Cao YX, Wang WW, Ma SF, Yao T, Zhu YC. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. 2008;79:632–641. doi: 10.1093/cvr/cvn140. [DOI] [PubMed] [Google Scholar]
- 13.Olson KR. Is hydrogen sulfide a circulating “gasotransmitter” in vertebrate blood? Biochim Biophys Acta. 2009;1787:856–863. doi: 10.1016/j.bbabio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 14.Olson KR. A practical look at the chemistry and biology of hydrogen sulfide. Antioxid Redox Signal. 2012;17:32–44. doi: 10.1089/ars.2011.4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- 16.Millero FJ. The Thermodynamics and Kinetics of the Hydrogen-Sulfide System in Natural-Waters. Mar Chem. 1986;18:121–147. [Google Scholar]
- 17.Tang G, Wu L, Liang W, Wang R. Direct Stimulation of K(ATP) channels by exogenous and endogenous hydrogen sulfide in vascular smooth muscle cells. Mol Pharmacol. 2005;68:1757–1764. doi: 10.1124/mol.105.017467. [DOI] [PubMed] [Google Scholar]
- 18.Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Modis K, Panopoulos P, Asimakopoulou A, Gero D, Sharina I, Martin E, Szabo C. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, Cantalupo A, Dhayade S, Karalis KP, Wang R, Feil R, Cirino G. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS One. 2012;7:e53319. doi: 10.1371/journal.pone.0053319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Timmers HJ, Rongen GA, Karemaker JM, Wieling W, Marres HA, Lenders JW. The role of carotid chemoreceptors in the sympathetic activation by adenosine in humans. Clin Sci (Lond) 2004;106:75–82. doi: 10.1042/CS20030174. [DOI] [PubMed] [Google Scholar]
- 21.Marshall JM. Peripheral chemoreceptors and cardiovascular regulation. Physiol Rev. 1994;74:543–594. doi: 10.1152/physrev.1994.74.3.543. [DOI] [PubMed] [Google Scholar]
- 22.Winder CV, Winder HO. The seat of action of sulfide on pulmonary ventilation. Am J Physiol. 1933;105:337–352. [Google Scholar]
- 23.Haouzi P. Ventilatory and metabolic effects of exogenous hydrogen sulfide. Respir Physiol Neurobiol. 2012;184:170–177. doi: 10.1016/j.resp.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Abboud FM, Thames MD. Interaction of Cardiovascular Reflexes in Circulatory Control. Compr Physiol Supplement. 2011;8:675–753. [Google Scholar]
- 25.Ariyaratnam P, Loubani M, Morice AH. Hydrogen sulphide vasodilates human pulmonary arteries: a possible role in pulmonary hypertension? Microvasc Res. 2013;90:135–137. doi: 10.1016/j.mvr.2013.09.002. [DOI] [PubMed] [Google Scholar]
- 26.Sun Y, Tang CS, Jin HF, Du JB. The vasorelaxing effect of hydrogen sulfide on isolated rat aortic rings versus pulmonary artery rings. Acta Pharmacol Sin. 2011;32:456–464. doi: 10.1038/aps.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper CE, Brown GC. The inhibition of mitochondrial cytochrome oxidase by the gases carbon monoxide, nitric oxide, hydrogen cyanide and hydrogen sulfide: chemical mechanism and physiological significance. J Bioenerg Biomembr. 2008;40:533–539. doi: 10.1007/s10863-008-9166-6. [DOI] [PubMed] [Google Scholar]
- 28.Eberhardt M, Dux M, Namer B, Miljkovic J, Cordasic N, Will C, Kichko TI, de la Roche J, Fischer M, Suarez SA, Bikiel D, Dorsch K, Leffler A, Babes A, Lampert A, Lennerz JK, Jacobi J, Marti MA, Doctorovich F, Hogestatt ED, Zygmunt PM, Ivanovic-Burmazovic I, Messlinger K, Reeh P, Filipovic MR. H2S and NO cooperatively regulate vascular tone by activating a neuroendocrine HNO-TRPA1-CGRP signalling pathway. Nat Commun. 2014;5:4381. doi: 10.1038/ncomms5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Altaany Z, Yang G, Wang R. Crosstalk between hydrogen sulfide and nitric oxide in endothelial cells. J Cell Mol Med. 2013;17:879–888. doi: 10.1111/jcmm.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paciullo CA, McMahon Horner D, Hatton KW, Flynn JD. Methylene blue for the treatment of septic shock. Pharmacotherapy. 2010;30:702–715. doi: 10.1592/phco.30.7.702. [DOI] [PubMed] [Google Scholar]
- 31.Preiser JC, Lejeune P, Roman A, Carlier E, De Backer D, Leeman M, Kahn RJ, Vincent JL. Methylene blue administration in septic shock: a clinical trial. Crit Care Med. 1995;23:259–264. doi: 10.1097/00003246-199502000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Zima AV, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 33.Bouillaud F, Blachier F. Mitochondria and sulfide: a very old story of poisoning, feeding, and signaling? Antioxid Redox Signal. 2011;15:379–391. doi: 10.1089/ars.2010.3678. [DOI] [PubMed] [Google Scholar]
- 34.Haouzi P, Klingerman CM. Fate of intracellular H2S/HS(-) and metallo-proteins. Respir Physiol Neurobiol. 2013;188:229–230. doi: 10.1016/j.resp.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wintner EA, Deckwerth TL, Langston W, Bengtsson A, Leviten D, Hill P, Insko MA, Dumpit R, VandenEkart E, Toombs CF, Szabo C. A monobromobimane-based assay to measure the pharmacokinetic profile of reactive sulphide species in blood. Br J Pharmacol. 2010;160:941–957. doi: 10.1111/j.1476-5381.2010.00704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng B, Yang J, Qi Y, Zhao J, Pang Y, Du J, Tang C. H2S generated by heart in rat and its effects on cardiac function. Biochem Biophys Res Commun. 2004;313:362–368. doi: 10.1016/j.bbrc.2003.11.130. [DOI] [PubMed] [Google Scholar]
- 37.Khan AA, Schuler MM, Prior MG, Yong S, Coppock RW, Florence LZ, Lillie LE. Effects of hydrogen sulfide exposure on lung mitochondrial respiratory chain enzymes in rats. Toxicol Appl Pharmacol. 1990;103:482–490. doi: 10.1016/0041-008x(90)90321-k. [DOI] [PubMed] [Google Scholar]
- 38.Dorman DC, Moulin FJ, McManus BE, Mahle KC, James RA, Struve MF. Cytochrome oxidase inhibition induced by acute hydrogen sulfide inhalation: correlation with tissue sulfide concentrations in the rat brain, liver, lung, and nasal epithelium. Toxicol Sci. 2002;65:18–25. doi: 10.1093/toxsci/65.1.18. [DOI] [PubMed] [Google Scholar]
- 39.Brenneman KA, Meleason DF, Sar M, Marshall MW, James RA, Gross EA, Martin JT, Dorman DC. Olfactory mucosal necrosis in male CD rats following acute inhalation exposure to hydrogen sulfide: reversibility and the possible role of regional metabolism. Toxicol Pathol. 2002;30:200–208. doi: 10.1080/019262302753559533. [DOI] [PubMed] [Google Scholar]
- 40.Lopez A, Prior MG, Reiffenstein RJ, Goodwin LR. Peracute toxic effects of inhaled hydrogen sulfide and injected sodium hydrosulfide on the lungs of rats. Fundam Appl Toxicol. 1989;12:367–373. doi: 10.1016/0272-0590(89)90053-5. [DOI] [PubMed] [Google Scholar]
- 41.Li L, Whiteman M, Guan YY, Neo KL, Cheng Y, Lee SW, Zhao Y, Baskar R, Tan CH, Moore PK. Characterization of a novel, water-soluble hydrogen sulfide-releasing molecule (GYY4137): new insights into the biology of hydrogen sulfide. Circulation. 2008;117:2351–2360. doi: 10.1161/CIRCULATIONAHA.107.753467. [DOI] [PubMed] [Google Scholar]
- 42.Evans CL. The toxicity of hydrogen sulphide and other sulphides. Q J Exp Physiol Cogn Med Sci. 1967;52:231–248. doi: 10.1113/expphysiol.1967.sp001909. [DOI] [PubMed] [Google Scholar]
- 43.Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, Barrow RK, Yang G, Wang R, Snyder SH. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haouzi P, Chenuel B, Sonobe T. High-dose hydroxocobalamin administered after H2S exposure counteracts sulfide-poisoning-induced cardiac depression in sheep. Clin Toxicol (Phila) 2015;53:28–36. doi: 10.3109/15563650.2014.990976. [DOI] [PMC free article] [PubMed] [Google Scholar]