

Abstract
Motivation: The 3D structure of the genome plays a critical role in regulating gene expression. Recent progress in mapping technologies for chromatin interactions has led to a rapid increase in this kind of interaction data. This trend will continue as research in this burgeoning field intensifies.
Results: We describe the 4DGenome database that stores chromatin interaction data compiled through comprehensive literature curation. The database currently covers both low- and high-throughput assays, including 3C, 4C-Seq, 5C, Hi-C, ChIA-PET and Capture-C. To complement the set of interactions detected by experimental assays, we also include interactions predicted by a recently developed computational method with demonstrated high accuracy. The database currently contains ∼8 million records, covering 102 cell/tissue types in five organisms. Records in the database are described using a standardized file format, facilitating data exchange. The vast major of the interactions were assigned a confidence score. Using the web interface, users can query and download database records via a number of annotation dimensions. Query results can be visualized along with other genomics datasets via links to the UCSC genome browser. We anticipate that 4DGenome will be a valuable resource for investigating the spatial structure-and-function relationship of genomes.
Availability and Implementation: 4Dgenome is freely accessible at http://4dgenome.int-med.uiowa.edu. The database and web interface are implemented in MySQL, Apache and JavaScript with all major browsers supported.
Contact: kai-tan@uiowa.edu
Supplementary Information: Supplementary Materials are available at Bioinformatics online.
1 Introduction
Determining the 3D structure of the genome and its impact on gene expression and other DNA transactions has been a long-standing question in cell biology. The pioneering method of chromosome conformation capture (3C) determines the relative frequency of direct physical contact between a pair of linearly separated chromatin segments (Dekker et al., 2002). By coupling 3C with short-read sequencing technology, chromosome conformation capture-on-chip (4C-Seq) (Splinter et al., 2011; Stadhouders et al., 2013) and chromosome conformation capture carbon copy (5C) (Dostie et al., 2006) can detect genome-wide interactions involving a single anchor region and interactions involving multiple genomic regions, respectively. The Hi-C protocol is a truly genome-wide technology that allows detection of all interactions in the genome. The ChIA-PET protocol (Fullwood et al., 2009) combines the principles of 3C and chromatin immunoprecipitation (ChIP) to identify chromatin interactions mediated by protein factors. Most recently, the Capture-C method (Hughes et al., 2014) combines oligonucleotide capture technology, 3C and short-read sequencing and enables researchers to interrogate cis interactions at hundreds of selected loci at high resolution in a single assay.
Application of these genome-wide assays in recent years has led to a dramatic increase in chromatin interaction data. This rapid increase in data has spurred the development of specialized databases to document chromatin interactions. However, existing databases only store interactions detected by either Hi-C (Li et al., 2014) or by 5C, ChIA-PET and Hi-C (Zhou et al., 2013). As a result, they miss a large number of interactions detected by other commonly used high-throughput methods, such as 4C and Capture-C. They also miss interactions detected by 3C, which tend to have higher quality and thus is very useful as the gold standard for a variety of purposes. Besides experimental assays, computational methods are (Corradin et al., 2014; Ernst and Kellis, 2010; He et al., 2014; Thurman et al., 2012) being continuously improved to predict chromatin interactions. In particular, the recently developed method, integrated method for predicting enhancer targets (IM-PET), has been demonstrated to have comparable accuracy as the 5C assay (He et al., 2014). Currently, there is a lack of a comprehensive depository for chromatin interactions identified by all major technologies. This dispersion of information hinders standardization, sharing and integration of the interaction data, which are critical for gaining insights into genome structure/function relationships.
Towards this goal, we have developed the 4DGenome database, a general repository for chromatin interactions. Records in 4DGenome are compiled through comprehensive literature curation of experimentally derived interactions. The database currently covers both low- and high-throughput assays, including 3C, 4C-Seq, 5C, Hi-C, ChIA-PET and Capture-C. The database also includes computational predictions made by the IM-PET algorithm (He et al., 2014) that integrates multiple types of genomic features to predict interactions between enhancers and promoters.
2 Materials and methods
2.1 Curation of chromatin interaction data
We conducted an extensive search of primary literature in the NCBI PubMed database, dating back to the original publication of 3C method in 2002 (Dekker et al., 2002). We used the following key words to conduct the literature search: ‘chromatin interaction’, ‘chromosome conformation capture’, ‘3C’, ‘4C’, ‘4C-Seq’, ‘5C’, ‘Hi-C’, ‘Capture-C’, ‘genome and 3D’, ‘enhancer-promoter interaction’ and ‘chromosome domain’.
We curated interactions with the following considerations in mind: (i) each record in the database should have a score representing the statistical confidence of the interaction. Because statistical analysis of chromatin interactions is a fast moving research area, we chose to report two types of confidence measures: those reported by study authors and those computed by a different method from the study but has shown improved accuracy. We reasoned that the authors of individual studies knew their data best and had done their best to report high quality interactions. On the other hand, as part of our ongoing development effort, as the field matures, we will periodically update the confidence scores using the best approach or using consensus scores from top approaches. We believe our strategy of reporting two confidence scores both maintains the historical context of the data and offers the flexibility of improving data annotation as the field moves forward with the development of more accurate statistical methods. To this end, we have used the Fit-Hi-C method (Ay et al., 2014) to compute a second confidence scores for all Hi-C interactions in the database. For interactions detected by 4C, 5C, Hi-C, ChIA-PET and Capture-C, since no methods other than those used by the study authors were shown to be better, we temporarily used author-reported confidence scores for both types of confidence scores. To help users better understand how the interactions were obtained from raw data, we have summarized the statistical method used in each study in Supplementary Table S1. (ii) If an interaction was detected using one of the short-read-sequencing-based methods, its contact frequency should be reported. (iii) We only included interaction data with a resolution (size of the interacting genomic loci) of 10 000 bp or higher, given the lengths of most functional DNA elements (e.g. promoter, enhancer, insulator, silencer) are shorter than 10 000 bp.
For interactions identified by 3C, there is no contact frequency or confidence measure. We thus just documented the interactions per se. For high-throughput assays (4C-Seq, 5C, ChIA-PET, Hi-C, Capture-C), we reported both contact frequency and confidence scores for the interactions. For computational predictions made by the IM-PET method, there is no measured contact frequency and we only reported the probability of the interaction as the confidence score.
2.2 File format for chromatin interaction records
We devised a standardized text file format for recording chromatin interactions. This file format is similar to formats used by many protein–protein interaction databases, such as the BioGrid (Chatr-Aryamontri et al., 2013) database. The standardized file format will facilitate future data exchange among databases. The data format is also recognizable by the widely used network visualization tool, Cytoscape (Saito et al., 2012), making it straightforward to visualize chromatin interactions using this powerful tool. Each record contains 15 tab-delimited columns (Supplementary Fig. S1): columns 1–6 describe the genomic coordinates of the interacting regions; columns 7–8 describe genes (if any) that are located in the interacting regions; columns 9–10 describe the organism and cell/tissue types from which the interaction was derived; column 11–14 describe the detection method, confidence scores, and contact frequency for the observed interaction; column 15 provides the PubMed ID of the publication that reported the interaction.
2.3 Database architecture and web interface
We have implemented the popular LAMP (Linux, Apache, MySQL and PHP) architecture (Supplementary Fig. S2), which uses 100% open source software to build the heavy-duty dynamic database backend. The user interface was implemented using JavaScript and Cascading Styling Sheets (CSS) languages and has been tested on all major internet browsers, including Firefox, Safari, Internet Explorer and Chrome. The current database server runs on a linux workstation with a 3.0G dual-core CPU and 3.6G RAM.
3 Results
The current release of 4DGenome contains 4 433 071 experimentally derived and 3 605 176 computationally predicted interactions across 102 cell/tissue types in five organisms. Figure 1A and 1B show the percentages of interactions by organism and detection method, respectively. In terms of data resolution, currently Hi-C data have the lowest resolution, followed by 5C, 4C-Seq, ChIA-PET and Capture-C (Fig. 1C). As sequencing becomes deeper, we anticipate that resolutions of the interaction data will improve (especially Hi-C data) although their theoretical limits are determined by the choice of the restriction enzyme used in the assays. In terms of genomic distance of interacting regions, current 4C-Seq and 5C data tend to capture interactions separated by longer genomic distance (Fig. 1D). This likely reflects the choices of genomic regions interrogated in the 5C and 4C-Seq studies instead of the inherent properties of these assays. Additional summary statistics regarding the database is presented in Supplementary Table S2.
Fig. 1.
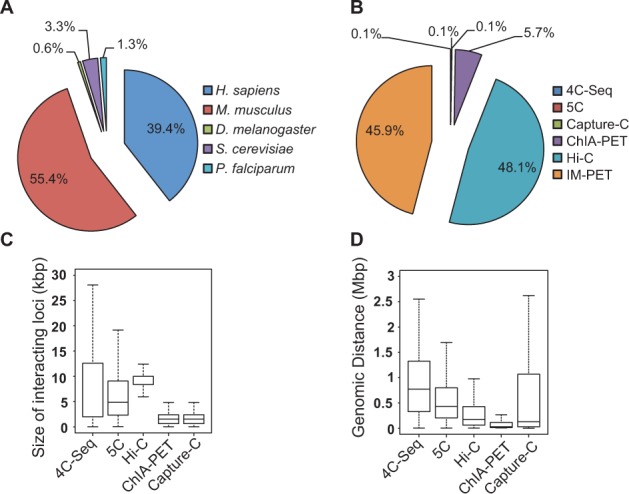
Content of 4DGenome and general characteristics of database records. (A) Percentage of interactions by organism. (B) Percentage of interactions by detection method. Interactions detected by 3C are not included in the pie chart because its percentage is too small to show. (C) Resolution of interactions. Shown are the sizes of interacting genomic regions by various methods. (D) Distribution of linear genomic distance between interacting regions. Distance is computed as the center-to-center distance between two interactors
3.1 The database web interface
4DGenome can be accessed via the web site http://4dgenome.int-med.uiowa.edu. The user can query database records by a number of annotation dimensions, download database records and view a summary statistics for each dataset (Supplementary Fig. S3).
3.2 Database query
We provide two modes to query the database: query by genomic regions and query by genes. Up to 10 genomic regions or genes can be inputted in the query box. For query by genomic regions, the genomic coordinates of the region can be provided in the input text box, using the same format as that used in the UCSC genome browser, e.g. chr1:1000–20 000. For query by genes, RefSeq gene symbols or Ensembl gene IDs can be used. Wild card is accepted in this query mode. Using the dropdown boxes, the user can retrieve interactions that match specific combination of organism, cell/tissue type and detection method.
Besides using the input box, users can also conduct batch queries by uploading a tab-delimited text file. Up to 100 genomic regions or genes can be included in each text file. Each row of the file contains a query region or gene. For query by genomic regions, the first three columns of the file (chromosome number, left genomic coordinate, right genomic coordinate) are required. For query by genes, the first column of the file (gene symbol or ID) is required. Additional columns are allowed in the files but are not used during the search.
In the query by regions mode, to retrieve database records that overlap with the query, an overlapping criterion is needed. Currently, we use two overlapping rules: ‘Any overlap’ and ‘Center overlap’ (Supplementary Fig. S4). ‘Any overlap’ is a loose measure which only requires the query region to overlap with either of two interacting regions by at least 1 bp. ‘Center overlap’ is more stringent and requires the center-to-center distance between the query and record regions to be less than half of the length of the smaller region. In other words, this means the smaller genomic region (either query or record) is encompassed inside the larger region.
3.3 Understanding the query result
Retrieved records can be downloaded as a tab-delimited text file (Fig. 2A top). The output file can be directly imported into a number of programming languages for further analyses, such as R. To facilitate comparison of database content over time, the file name contains the date when the query is performed. Query results are also shown on the ‘Search Results’ page. Interaction records are grouped by detection methods and are shown as tables (Fig. 2A bottom). Under each detection method, the user can use the ‘Details’ button to toggle between summary and detailed views of each record. By default, each viewing window shows 20 rows of the table. Users can navigate through the pages by clicking on the page numbers at the bottom of each viewing window.
Fig. 2.

An example query to 4DGenome. (A) Results page using the example query gene SLC2A13. (B) UCSC genome browser view of the returned interactions involving the query gene SLC2A13. Interactions are color-coded by their detection methods, 3C, red; 4C-Seq, magenta; 5C, green; Hi-C, brown; ChIA-PET, black; Capture-C, bright blue; IM-PET, navy blue. Shades of color encode confidence scores with darker shades indicating higher confidence scores
3.4 Display of interactions along with additional genomics datasets
To facilitate data interpretation and integration, interactions can be displayed (via the magnifying glass icon next to each record) in the UCSC genome browser, providing additional functional views of the interactions (Fig. 2B). Interactions are color-coded by their detection methods. Confidence scores of the interactions are encoded by the shade of the color. The custom interaction track can be displayed along with a vast array of annotation tracks available in the genome browser. By default, we only display gene annotation track, chromatin modification and accessibility, transcription factor ChIP-Seq tracks generated by the encyclopedia of DNA elements (ENCODE) project, and genomic sequence conservation track.
3.5 Export database records
We have organized the interactions into downloadable files by organisms and detection methods to suit different needs of the users. These files will be constantly updated at each release of the database. Data can be downloaded either from the database webpage or by anonymous FTP.
3.6 An example user case
Accumulating evidence suggests that chromatin interactions are highly dynamic across cell types and developmental stages. To investigate this issue systematically, we used the chromatin interaction data across multiple human and mouse cell types stored in 4DGenome. For each gene, we counted its total number of interactors across all cell types and its unique number of interactors (i.e. occur only in one cell type). We then computed the ratio of the number of unique interactions versus the number of the total interactions. Larger ratio means the gene tends to interact with different sets of loci across cell types, suggesting that its interaction with other genomic loci is more dynamic. As shown in Figure 3A, the median ratios are approximately 0.8 for both human and mouse cell types. Thus the systematic analysis further confirmed the notion that chromatin interactions are highly dynamic across cell types. Figure 3B shows an example of dynamic chromatin interactions involving the gene MLLT4 across three human cell types, MCF-7, K562 and IMR90. Interactions in MCF-7 and K562 were detected by ChIA-PET; whereas interactions in IMR90 were detected by Hi-C. The thickness of the arcs is proportional to the confidence score of the interactions. As can be seen, there are both shared and unique interactions among the three cell types. Some of unique interactions may be due to difference in the detection methods. However, even for interactions in MCF-7 and K562 cells, which were generated using the same method and by the same group (Li et al., 2012), there are still interactions unique to each cell type, suggesting that these interactions truly reflect the dynamics of chromatin interactions.
Fig. 3.
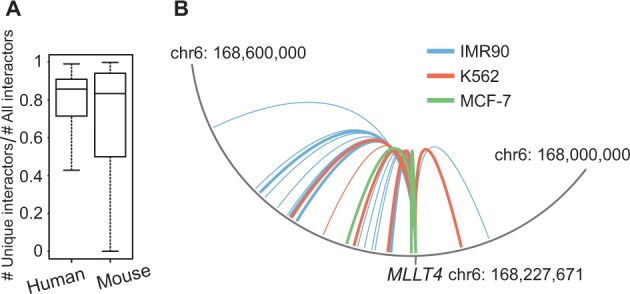
Use 4DGenome to investigate dynamics of chromatin interactions. (A) Dynamics of chromatin interactions across cell types in human and mouse. (B) An example of dynamic chromatin interaction involving the gene MLLT4 across three cell types in human. MCF-7 and K562, ChIA-PET data; IMR90, Hi-C data. Thickness of the arcs is proportional to the confidence score of the interactions
4 Discussion
Two alternative tools have been developed before. However, they either only curate a particular data type, Hi-C (Li et al., 2014) or is specialized for visualizing interactions (Zhou et al., 2013). To the best of our knowledge, 4DGenome is the first database that comprehensively document and curate chromatin interactions generated by both experimental and computational approaches. Another unique feature of 4Dgenome is that it provides contact frequency and confidence measure for each interaction, whenever such a measure is possible. As a result, interactions can be prioritized in various analyses. Interactions documented in 4DGenome were detected using different experimental technologies. Although these technologies rely on similar molecular biology principle, caveats need to be taken when comparing interactions involving the same loci but generated by different technologies. On the other hand, if an interaction involving the same loci is detected by multiple technologies, it increases our confidence in the interaction. In the future, integraing interactions detected by different technologies in a statistical sound way should be an important research area.
We anticipate that 4DGenome will be a valuable resource for investigating the spatial structure-and-function relationship of genomes as research in this burgeoning area intensifies. We envision that a wide range of investigations will benefit from a carefully curated database such as 4DGenome. A few examples include standardization and benchmarking of interaction data qualities by different detection technologies, relationship between genome 3D structure and epigenetic state, dynamics of chromatin interaction across cell/tissue types and organisms, and genome 3D structure and pathogenic genetic variations.
Besides continued curation effort to add new interactions, our future research will focus on two areas in order to improve the quality and utility of database records. First, we plan to improve the integration of 4DGenome with other molecular interaction databases, such as BioGrid and STRING. Second, we plan to conduct systematic comparisons to identify and apply the most accurate means to assign confidence measures to interaction data generated by different technologies.
Supplementary Material
Acknowledgements
We thank members of the Tan lab for helpful discussion. We thank Lucas Van Tol and the University of Iowa Institute for Clinical and Translational Science for providing computing support.
Funding
This study was supported by the National Institutes of Health grants [HG006130], [GM108716] and [GM104369] to K.T.
Conflict of Interest: none declared.
References
- Ay F., et al. (2014) Statistical confidence estimation for Hi-C data reveals regulatory chromatin contacts. Genome Res., 24, 999–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., et al. (2013) The BioGRID interaction database: 2013 update. Nucleic Acids Res., 41, D816–D823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O., et al. (2014) Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res., 24, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J., et al. (2002) Capturing chromosome conformation. Science, 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- Dostie J., et al. (2006) Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res., 16, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J., Kellis M. (2010) Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat. Biotechnol., 28, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullwood M.J., et al. (2009) An oestrogen-receptor-alpha-bound human chromatin interactome. Nature, 462, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B., et al. (2014) Global view of enhancer-promoter interactome in human cells. Proc. Natl Acad. Sci. U.S.A., 111, E2191–E2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes J.R., et al. (2014) Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet., 46, 205–212. [DOI] [PubMed] [Google Scholar]
- Li C., et al. (2014) The 3DGD: a database of genome 3D structure. Bioinformatics, 30, 1640–1642. [DOI] [PubMed] [Google Scholar]
- Li G., et al. (2012) Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell, 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R., et al. (2012) A travel guide to Cytoscape plugins. Nat. Methods, 9, 1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splinter E., et al. (2011) The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev., 25, 1371–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadhouders R., et al. (2013) Multiplexed chromosome conformation capture sequencing for rapid genome-scale high-resolution detection of long-range chromatin interactions. Nat. Protoc., 8, 509–524. [DOI] [PubMed] [Google Scholar]
- Thurman R.E., et al. (2012) The accessible chromatin landscape of the human genome. Nature, 489, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., et al. (2013) Exploring long-range genome interactions using the WashU Epigenome Browser. Nat. Methods, 10, 375–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.