

Abstract
Motivation: Post-sequencing DNA analysis typically consists of read mapping followed by variant calling. Especially for whole genome sequencing, this computational step is very time-consuming, even when using multithreading on a multi-core machine.
Results: We present Halvade, a framework that enables sequencing pipelines to be executed in parallel on a multi-node and/or multi-core compute infrastructure in a highly efficient manner. As an example, a DNA sequencing analysis pipeline for variant calling has been implemented according to the GATK Best Practices recommendations, supporting both whole genome and whole exome sequencing. Using a 15-node computer cluster with 360 CPU cores in total, Halvade processes the NA12878 dataset (human, 100 bp paired-end reads, 50× coverage) in <3 h with very high parallel efficiency. Even on a single, multi-core machine, Halvade attains a significant speedup compared with running the individual tools with multithreading.
Availability and implementation: Halvade is written in Java and uses the Hadoop MapReduce 2.0 API. It supports a wide range of distributions of Hadoop, including Cloudera and Amazon EMR. Its source is available at http://bioinformatics.intec.ugent.be/halvade under GPL license.
Contact: jan.fostier@intec.ugent.be
Supplementary information: Supplementary data are available at Bioinformatics online.
1 Introduction
The speed of DNA sequencing has increased considerably with the introduction of next-generation sequencing platforms. For example, modern Illumina systems can generate several hundreds of gigabases per run (Zhang et al., 2011) with a high accuracy. This, in turn, gives rise to several hundreds of gigabytes of raw sequence data to be processed.
Post-sequencing DNA analysis typically consists of two major phases: (i) alignment of reads to a reference genome and (ii) variant calling, i.e. the identification of differences between the reference genome and the genome from which the reads were sequenced. For both tasks, numerous tools have been described in literature, see e.g. Fonseca et al. (2012) and Nielsen et al. (2011) for an overview. Especially for whole genome sequencing, applying such tools is a computational bottleneck. To illustrate this, we consider the recently proposed Best Practices pipeline for DNA sequencing analysis (Van der Auwera et al., 2013) that consists of the Burrow-Wheeler Aligner (BWA) (Li and Durbin, 2009) for the alignment step, Picard (http://picard.sourceforge.net) for data preparation and the Genome Analysis Toolkit (GATK) (Depristo et al., 2011; McKenna et al., 2010) for variant calling. On a single node, the execution of this pipeline consumes more time than the sequencing step itself: a dataset consisting of 1.5 billion paired-end reads (Illumina Platinum genomes, NA12878, 100 bp, 50-fold coverage, human genome) requires over 12 days using a single CPU core of a 24-core machine (dual socket Intel Xeon E5-2695 v2 @ 2.40 GHz): 172 h for the alignment phase, 35 h for data preparation (Picard steps) and 80 h for GATK, including local read realignment, base quality score recalibration and variant calling. When allowing the involved tools to run multithreaded on the same machine, the runtime decreases only by a factor of roughly 2.5 to ∼5 days, indicative of a poor scaling behavior in some of the steps of the pipeline.
To overcome this bottleneck, we developed Halvade, a modular framework that enables sequencing pipelines to be executed in parallel on a multi-node and/or multi-core compute infrastructure. It is based on the simple observation that read mapping is parallel by read, i.e. the alignment of a certain read is independent of the alignment of another read. Similarly, variant calling is conceptually parallel by chromosomal region, e.g. variant calling in a certain chromosomal region is independent of variant calling in a different region. Therefore, multiple instances of a tool can be run in parallel on a subset of the data. Halvade relies on the MapReduce programming model (Dean and Ghemawat, 2008) to execute tasks concurrently, both within and across compute nodes. The map phase corresponds to the read mapping step while variant calling is performed during the reduce phase. In between both phases, aligned reads are sorted in parallel according to genomic position. By making use of the aggregated compute power of multiple machines, Halvade is able to strongly reduce the runtime for post-sequencing analysis. A key feature of Halvade is that it achieves very high parallel efficiency which means that computational resources are efficiently used to reduce runtime. Even on a single, multi-core machine, the runtime can be reduced significantly as it is often more efficient to run multiple instances of a tool, each instance with a limited number of threads, compared with running only a single instance of that tool with many threads. As an example, both whole genome and whole exome variant calling pipelines were implemented in Halvade according to the GATK Best Practices recommendations (i.e. using BWA, Picard and GATK).
The MapReduce programming model has been used before in CloudBurst (Schatz, 2009) and DistMap (Pandey and Schlötterer, 2013) to accelerate the read mapping process and in Crossbow (Langmead et al., 2009a) to accelerate a variant calling pipeline based on modified versions of Bowtie (Langmead et al., 2009b) and SOAPsnp (Li et al., 2008). The Halvade framework extends these ideas, enabling the implementation of complex pipelines while supporting different tools and versions. The software is designed to achieve a good load balance, maximize data locality and minimize disk I/O by avoiding file format conversions. As a result, Halvade achieves much higher parallel efficiencies compared with similar tools.
More recently, MapReduce-like scripts were used in MegaSeq (Puckelwartz et al., 2014), a workflow for concurrent multiple genome analysis on Beagle, a Cray XE6 supercomputer at Argonne National Laboratories. Like Halvade, MegaSeq implements a whole genome analysis pipeline based on the GATK Best Practices recommendations. However, whereas MegaSeq focuses on a high throughput of many genomes using a specific, extreme-scale compute platform, Halvade aims to maximally reduce the analysis runtime for the processing of a single genome, while supporting a wide variety of computer clusters. This approach is particularly of use in a clinical setting, where the analysis step will typically be performed on a local cluster within a hospital environment, and where the time between obtaining a DNA sample from a patient and diagnosing should be kept as small as possible. The source code of Halvade is publicly available.
2 Methods
2.1 Halvade framework
Halvade relies on the MapReduce programming model (Dean and Ghemawat, 2008) to enable parallel, distributed-memory computations. This model consists of two major phases: the map and reduce phase. During the map phase, different map tasks are executed in parallel, each task independently processing a chunk of the input data and producing as output a number of intermediate <key, value> pairs. Next, the intermediate <key, value> pairs emitted by all map tasks are sorted, in parallel, according to key by the MapReduce framework. During the reduce phase, different reduce tasks are executed in parallel, each reduce task independently processing a single key and its corresponding values.
Conceptually, a read alignment and variant calling pipeline can be cast into the MapReduce framework: read alignment is then performed in the map phase where the different map tasks are processing part of the input FASTQ files in parallel, while the variant calling and, if required, additional data preparation steps, are handled in the reduce phase where the different reduce tasks are processing chromosomal regions in parallel. Using the MapReduce framework, the reads are sorted according to their aligned position and grouped by chromosomal region in between the two phases. The Halvade framework provides access to data streams for individual tools that run in parallel during read mapping and variant calling. An overview of the Halvade framework is depicted in Figure 1. The different computational steps are described in more detail below.
Fig. 1.

Overview of the Halvade framework. The entries of pairs of input FASTQ files (containing paired-end reads) are interleaved and stored as smaller chunks. Map tasks are executed in parallel, each task taking a single chunk as input and aligning the reads to a reference genome using an existing tool. The map tasks emit <key, value> pairs where the key contains positional information of an aligned read and the value corresponds to a SAM record. The aligned reads are grouped and sorted per chromosomal region. Chromosomal regions are processed in parallel in the reduce phase, this includes data preparation and variant detection again using tools of choice. Each reduce task outputs the variants of the region it processed. These variants can optionally be merged into a single VCF file. Note that the names of the tools shown correspond to those of the GATK Best Practices DNA-seq implementation in Halvade
2.1.1 Input data preparation
The input data typically consist of paired-end reads stored in two distinct, compressed FASTQ files. We provide a separate tool called ‘Halvade Uploader’ which interleaves the paired-end reads, storing paired reads next to each other, and splits the data in chunks of ∼60 MB of compressed data. These chunks are transferred on-the-fly to the file system from which they can be accessed by the worker nodes. In case a generic Hadoop system is used, this is the Hadoop Distributed File System (HDFS); in case Amazon EMR is used, chunks are uploaded to the Amazon Simple Storage Service (Amazon S3) using the Amazon S3 API. The number of input chunks corresponds to the number of map tasks that will be executed during the map phase. The rationale behind the Halvade Uploader is that data have to be copied or uploaded onto the compute infrastructure anyhow, and that decompressing, interleaving, splitting and again compressing can easily overlap with this transfer, thus reducing file I/O to its minimum. The interleaving of paired-end reads ensures that both pairs are accessible in a single task, which is required for the read alignment. The Halvade Uploader is multithreaded and operates on data streams, which means that its execution can overlap with the data generation (i.e. sequencing) step itself.
Prior to the actual execution of the MapReduce job, additional preparatory steps are required. First, the reference genome is partitioned into a pre-determined number of non-overlapping chromosomal regions of roughly equal size. The number of chromosomal regions corresponds to the total number of reduce tasks that will be executed during the reduce phase and can be configured by the user based on the size of the reference genome in question. Next, Halvade ensures that all required binaries and configuration files are available on each worker node. It does so by adding all required files, in a compressed archive, to the distributed cache which is then copied to each worker node and again decompressed. Note that when these files are persistently stored onto the worker nodes, this preparatory step can be omitted.
2.1.2 Map phase read alignment
For the map phase, one map task is created per input FASTQ chunk. These tasks are in turn executed in parallel on the worker nodes by a number of mappers. Typically, the number of map tasks ≫ the number of mappers which means that each mapper will process many tasks. Key to the MapReduce model is that a map task will preferably be executed by a worker node that contains the input chunk locally on disk (as a part of the HDFS) in order to minimize remote file access and thus network communication. Each mapper first checks if the indexed reference genome is locally available and retrieves it from HDFS or Amazon S3 when this is not the case. At this point, the reference genome itself is not partitioned, i.e. the complete index is used by each individual mapper. The input FASTQ chunks are read from HDFS or S3 and parsed into input <key, value> pairs using the Hadoop-BAM (Niemenmaa et al., 2012) API. The values of these pairs contain the FASTQ entries and are streamed to an instance of the alignment tool.
Halvade requires that the alignments are accessible in SAM format and provides a SAM stream parser that will create the required intermediate <key, value> pairs. This intermediate key represents a composite object that contains the chromosome number and alignment position of a read, along with the identifier of the chromosomal region to which it aligns. The value contains the corresponding SAM record, i.e. the read itself and all metadata. Reads that cannot be aligned are optionally discarded. For individual or paired-end reads that span the boundary of adjacent chromosomal regions, two intermediate <key, value> pairs are created, one for each chromosomal region. This redundancy ensures that all required data for the data preparation and variant calling is available for each reduce task.
After all map tasks are completed, the MapReduce framework sorts, in parallel, the intermediate pairs according to chromosomal region (as part of the key). This way, all reads that align to the same chromosomal region are grouped together thus forming the input of a single reduce task. Halvade uses secondary sorting to further sort the SAM records for each chromosomal region by genomic position. Both grouping and sorting effectively replace the sorting of SAM records typically performed by tools such as Picard or SAMtools (Li et al. 2009) and are performed in a highly efficient manner by the MapReduce framework.
2.1.3 Reduce phase—variant calling
When all data have been grouped and sorted, the different reduce tasks are executed in parallel by different reducers. Again, the number of reduce tasks ≫ the number of reducers. Before the reads are processed, Halvade can copy to each worker node additional files or databases that are required for variant calling. A specific task takes as input all (sorted) intermediate <key, value> pairs for a single chromosomal region and converts it to an input stream in SAM format. Halvade iterates over the SAM records and creates a BED file (Quinlan and Hall, 2010) containing position intervals that cover all SAM records in the chromosomal region. This file can optionally be used to specify relevant intervals on which tools need to operate. Finally, instances of these tools are created in order to perform the actual variant calling.
Typically, at the end of each reduce task, a Variant Call Format (VCF) file has been produced which contains all variants identified in the corresponding chromosomal region. Halvade provides the option to merge these VCF files using an additional MapReduce job. In this second job, the map phase uses the individual VCF files as input and the variants are parsed as <key, value> pairs using Hadoop-BAM. The key contains the chromosome identifier and the position of the variant, while the value contains all other meta-information. These values are collected in a single reduce task, which writes the aggregated output to either HDFS or Amazon S3. At the interface of adjacent chromosomal regions, it is possible that overlapping variants are called twice by GATK (once in each chromosomal region). This is due to SAM records that span this interface and that were thus sent to both regions. During the VCF merging step, Halvade assigns a unique name to each of the overlapping variants, thus keeping all of them, or, optionally, retains only the variant with the highest Phred-scaled quality score. Such overlapping variants are rarely observed. Note that this second MapReduce job is very light-weight.
2.2 Best practices DNA-seq implementation
In Halvade, a DNA-seq variant calling pipeline has been implemented according to the Best Practices recommendations by Van der Auwera et al. (2013). Table 1 lists the different steps involved.
Table 1.
Overview of the steps and tools involved in the DNA-sequencing pipeline according to the GATK Best Practices recommendations described by Van der Auwera et al. (2013)
| step | program | input | output |
|---|---|---|---|
| align reads | BWA | FASTQ | SAM |
| convert SAM to BAM | Picard | SAM | BAM |
| sort reads | Picard | BAM | BAM |
| mark duplicates | Picard | BAM | BAM |
| identify realignment intervals | GATK | BAM | Intervals |
| realign intervals | GATK | BAM and intervals | BAM |
| build BQSR table | GATK | BAM | table |
| recalibrate base quality scores | GATK | BAM and table | BAM |
| call variants | GATK | BAM | VCF |
During the map phase, read alignment is performed by BWA; both BWA-mem and BWA-aln with BWA-sampe are supported in our implementation. In case BWA-aln is used, paired-end reads are again separated and aligned individually by two instances of BWA-aln after which BWA-sampe is used to join these partial results. The standard output stream of either BWA-sampe or BWA-mem is captured, and its SAM records are parsed into intermediate <key, value> pairs.
In the reduce phase, the SAM stream is first prepared according to GATK’s requirements, i.e. the readgroup information is added, read duplicates (i.e. reads that are sequenced from the same DNA molecule) are marked and the data is converted to the binary, compressed BAM format. Note that in the Best Practices recommendations, readgroup information is added during read alignment. In Halvade, this is postponed to the reduce phase in order to avoid sending this extra meta-information (as part of the SAM record) over the network during sorting. For data preprocessing, Halvade can use either Picard or elPrep (http://github.com/exascience/elprep) with SAMtools (Li et al., 2009). ElPrep is a tool that combines all data preparation steps and outputs a SAM file that conforms to the GATK requirements. When using elPrep, the input SAM records are streamed directly to elPrep for marking duplicate reads and adding of readgroup information. Its resulting SAM file is then converted to BAM format using SAMtools. When using Picard, Halvade first writes the input SAM stream to local disk in a compressed BAM file and then invokes the Picard MarkDuplicates and AddReadGroups modules. Note that both options (elPrep/SAMtools or Picard) produce identical output. However, the combination of elPrep and SAMtools is considerably faster than Picard.
Next, the actual GATK modules are executed. To correct potentially misaligned bases in reads due to the presence of insertions or deletions, the RealignerTargetCreator module is used to identify intervals that require realignment followed by the IndelRealigner module to perform the actual realignment. Next, using the dbSNP database of known variants (Sherry et al., 2001), the BaseRecalibrator module is used to generate co-variation data tables that are then used by the PrintReads module to recalibrate the base quality scores of the aligned reads. Finally, the actual variant calling is done using either the HaplotypeCaller or UnifiedGenotyper module.
The DNA-seq analysis pipeline implementation in Halvade supports both whole genome and exome sequencing analysis. One important difference to note is that an additional BED file is required, containing the coordinates of the exome regions to be processed by GATK. Additionally, the dbSNP database file used for the base quality score recalibration must be compatible with the exome being targeted.
2.3 Optimizations
In order to get the best performance out of the available resources, two crucial factors come into play. First, one needs to determine the optimal number of mappers and reducers per node. This determines the number of map and reduces tasks that will be executed concurrently on a worker node. Second, the user needs to select appropriate map and reduce task sizes. This determines the total number of map and reduce tasks that will be executed. Both factors are described below.
To exploit parallelism in a workstation with one or more multi-core CPUs, one can either run a single instance of a tool with multithreading on all available CPU cores, or run multiple instances of that tool, each instance using only a fraction of the CPU cores. To illustrate the difference in performance, the parallel speedup for different GATK modules as a function of number of threads was benchmarked on a 16-core machine (dual socket Intel Xeon CPU E5-2670 @ 2.60 GHz) with 94 GB of RAM (see Fig. 2). The benchmarks show that the maximum speedup gained by any of the GATK modules, using 16 threads, is <10, with one module exhibiting no speedup at all. Most modules show good scaling behavior up to 4 or 8 threads, but show only moderate reduction in runtime when the number of threads is increased further. It is thus more beneficial to start multiple instances of GATK, each instance using only a limited number of threads. Note that this is also true for Picard (which is single-threaded) and to a lesser extent, also for BWA. This concept is used in Halvade, which leads to better use of available resources and a higher overall parallel efficiency (see Supplementary Data S1.1). On the other hand, the maximum number of parallel instances of a tool than can be run on a machine might be limited due to memory constraints.
Fig. 2.
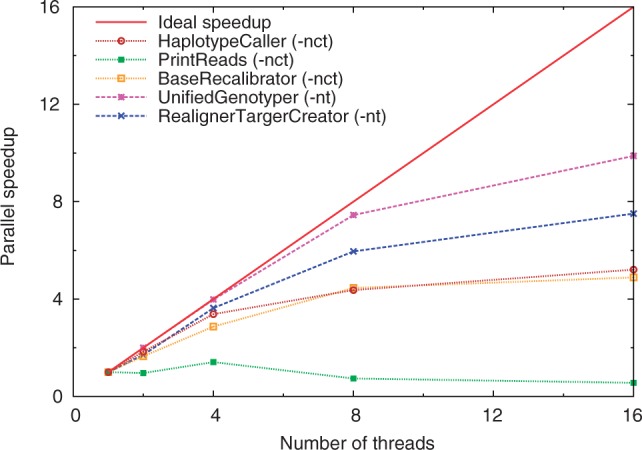
The parallel speedup (multithreading) of five GATK modules used in the Best Practices pipeline on a 16-core node with 94 GB of RAM. The limited speedup prevents the efficient use of this node with more than a handful of CPU cores. Option -nt denotes data threads while option -nct denotes CPU threads (cfr. GATK manual)
A second important factor is the optimal task size, which in turn determines the total number of tasks. For the map phase, the task size is determined by the size of a FASTQ chunk. Very few, large chunks will lead to a high per-task runtime but an unevenly balanced workload, whereas many little files will result in a large task scheduling and tool initialization overhead. After extensive testing, we determined that a file size of ∼60 MB leads to the lowest runtime (see Supplementary Data S1.2). Such chunk size is sufficiently big to define a meaningful task size, and small enough for a chunk to fit into a single HDFS block (default = 64 MB) which is entirely stored on a single worker node. If that worker node processes the corresponding map task, network traffic is avoided. Similarly, for the reduce phase, the task size is determined by the size of the chromosomal regions.
For data preparation, Picard can be replaced by elPrep. This tool combines all data preparation steps needed in the Best Practices pipeline. Whereas Picard requires file I/O for every preparation step, elPrep avoids file I/O by running entirely in memory and merges the computation of all steps in a single pass over the data. Using elPrep for data preparation again gives a significant speedup for this phase in Halvade.
3 Results
Halvade was benchmarked on a whole genome human dataset (NA12878 from Illumina Platinum Genomes). The dataset consists of 1.5 billion 100 bp paired-end reads (50-fold coverage) stored in two 43 GB compressed (gzip) FASTQ files. The software was benchmarked on two distinct computer clusters, an Intel-provided big-data cluster located in Swindon, UK and a commercial Amazon EMR cluster. Table 2 provides an overview of the runtime on these clusters. For these benchmarks, GATK version 3.1.1, BWA (-aln and -sampe) version 0.7.5a, BEDTools version 2.17.0, SAMtools version 0.1.19 and Picard version 1.112 were used. The dbSNP database and human genome reference found in the GATK hg19 resource bundle (ftp://ftp.broadinstitute.org/bundle/2.8/hg19/) were used. The reference genome was stripped of all alternate allele information and contains chromosomes 1 through 22, M, X and Y.
Table 2.
Runtime as a function of the number of parallel tasks (mappers/reducers) on the Intel Big Data cluster and Amazon EMR
| Cluster | No. worker nodes | No. parallel tasks | No. CPU cores | Runtime |
|---|---|---|---|---|
| Intel Big Data cluster | 1 | 3 | 18 | 47 h 59 min |
| 4 | 15 | 90 | 9 h 54 min | |
| 8 | 31 | 186 | 4 h 50 min | |
| 15 | 59 | 354 | 2 h 39 min | |
| Amazon EMR | 1 | 4 | 32 | 38 h 38 min |
| 2 | 8 | 64 | 20 h 19 min | |
| 4 | 16 | 128 | 10 h 20 min | |
| 8 | 32 | 256 | 5 h 13 min | |
| 16 | 64 | 512 | 2 h 44 min |
The time for uploading data to S3 over the internet is not included in the runtimes for Amazon EMR.
3.1 Intel big data cluster benchmark
This cluster consists of 15 worker nodes, each containing 24 CPU cores (dual-socket Intel Xeon CPU E5-2695 v2 @ 2.40 GHz) and 62 GB of RAM. The nodes each dispose of four hard drives with a total capacity of 4 TB, intended as HDFS storage and a single 800 GB solid-state drive (SSD) that is intended for local storage during MapReduce jobs. The nodes are interconnected by a 10 Gbit/s Ethernet network. Cloudera 5.0.1 b which supports MapReduce 2.3 was used. Initially, the input FASTQ files were present on a local disk of a single node. Using the Halvade Uploader, both files were decompressed, interleaved, compressed into separate files of ∼60 MB each (1552 chunks in total) and copied onto the local HDFS storage. This pre-processing step required ∼1.5 h using eight threads and can, in principle, overlap with the generation of the sequence data itself. For the chromosomal regions, a size of 2.5 Mbp was used, corresponding to 1261 reduce tasks in total.
On this cluster, Halvade runs four mappers/reducers per node in parallel to achieve optimal performance (see Supplementary Data S1.3), each mapper/reducer having 6 CPU cores and ∼15.5 GB of memory at its disposal. The scalability of Halvade was assessed by running the analysis pipeline with an increasing number of 1–15 nodes. As Cloudera reserves one slot for scheduling and job management, this corresponds to running 3 parallel tasks (1 node) to 59 parallel tasks (15 nodes) in total. Figure 3 depicts the parallel speedup as a function of the number of parallel tasks and provides an accurate view of the scalability and efficiency. When using 59 tasks (15 nodes), we observe a speedup of a factor 18.11 compared with using 3 tasks (1 node). This corresponds to a parallel efficiency of 92.1%. In absolute terms, the runtime reduces from ∼48 h (single node) to 2 h 39 min.
Fig. 3.
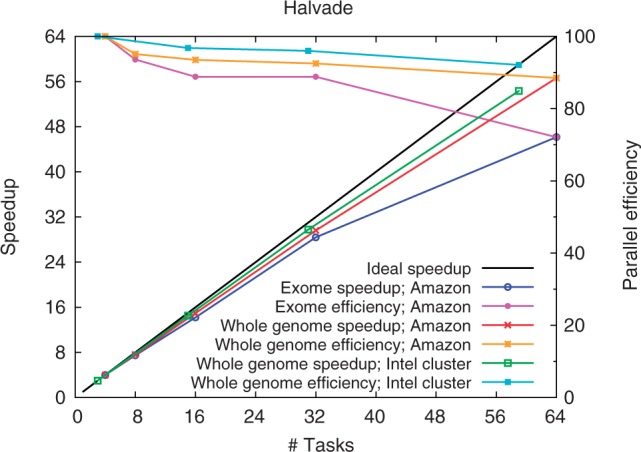
The speedup (primary y-axis) and parallel efficiency (secondary y-axis) of Halvade as a function of number of parallel tasks (cluster size) on both an Intel Big Data cluster and Amazon EMR
It is important to note that Halvade already attains a significant speedup when applied to a single node (3 tasks and 18 CPU cores), compared with the scenario of running the multithreaded versions of the individual tools using all 24 CPU cores. Indeed, whereas Halvade requires ∼48 h on a single node, ∼120 h are required when Halvade is not applied (speedup of a factor 2.5). This is due to the limited multithreaded scaling behavior of certain tools or modules (see ‘Methods’ section). It is hence far more efficient to run multiple instances of e.g. GATK with a limited number of threads per instance than letting GATK make use of all available cores. Ultimately, Halvade achieves a ∼45-fold speedup when applied to 15 nodes (2 h 39 min) compared with running the pipeline on a single node using only multithreading (120 h).
3.2 Amazon EMR benchmark
Amazon Elastic Compute Cloud (Amazon EC2) provides, as a web service, a resizeable compute cluster in the cloud. MapReduce can be used on this compute cluster with a service called Amazon EMR. This provides access to a Hadoop MapReduce cluster which can be chosen according to the requirements of the task at hand, e.g. the number of nodes and the node type. The EMR cluster was initialized with the Amazon Machine Images v3.1.0, providing access to Hadoop MapReduce v2.4, which takes ∼5 min. One node is reserved for job scheduling and management while the remainder is used as worker nodes.
Using the Halvade Uploader, the input data were preprocessed and uploaded to S3, the Amazon cloud storage system, again as 1552 chunks. The uploading speed is highly dependent on internet network conditions, which varies greatly. Again, this step can overlap with the generation of the sequence data itself. As data stored on S3 is directly accessible by worker nodes, one can chose whether or not to copy these data to HDFS prior to starting the MapReduce job. According to Deyhim (2013), data can be copied between S3 and HDFS at a rate of 50 GB per 19 min. However, for these benchmarks, data was read directly from S3, as copying would increase the overall runtime considerably.
For this benchmark, worker nodes of the type c3.8×large were used. Each node provides 32 CPU cores (Intel Xeon E5-2680 v2 @ 2.80 GHz), 60 GB of RAM and two 320 GB SSDs which are available for both HDFS and intermediate data. To obtain optimal performance, Halvade again assigned four parallel tasks per node, with each task disposing of 8 CPU cores and 15 GB of memory. The scalability was assessed by running Halvade with an increasing number of 1–16 worker nodes. When using 64 tasks (16 nodes), a speedup of a factor 14.16 is achieved compared with using 4 tasks (1 node) (see Fig. 3). This corresponds to a parallel efficiency of 88.5%. In absolute terms, the total runtime is reduced from 38 h 38 min (4 tasks) to 2 h 44 min (64 tasks) (see Table 2).
The runtime on Amazon EMR is slightly higher than that obtained using the Intel Big Data cluster, even though a higher number of CPU cores was used. This is because the Intel Big Data cluster is configured with a persistent HDFS, whereas for the Amazon cluster, the HDFS is created on-the-fly when the MapReduce job starts. On the Intel Big Data cluster, we can therefore instruct the Halvade Uploader to copy data directly to the HDFS after which only limited network communication is required for map tasks to access the data. On Amazon, Halvade accesses the data straight from S3, which requires network communication and explains the increased runtime. With a total runtime of <3 h, the financial cost of a whole genome analysis on Amazon EMR with 16 worker nodes amounts to 111.28 US dollar (based on the pricing of May 2014), which is in a similar price range as running the pipeline on a single, smaller Amazon instance without the use of Halvade (based on an expected runtime of ∼5 days on a c3.4×large instance).
3.3 Exome sequencing analysis benchmark
To assess the performance of Halvade on an exome sequencing dataset (Illumina HiSeq NA12878), the same Amazon EMR cluster was used. The dataset consists of 168 million 100 bp paired-end reads stored in eight ∼1.6 GB compressed (gzip) FASTQ files.
Using an Amazon EMR cluster with eight worker nodes (32 parallel tasks), Halvade can call the variants in under 1 h for a total cost of 19.64 US dollar (based on the pricing of May 2014). As the input for exome analysis is considerably smaller, the load balancing is more challenging as there are only 225 map tasks and 469 reduce tasks in total. A high parallel efficiency of ∼90% is obtained when using 8 worker nodes; the efficiency drops to ∼70% when using 16 worker nodes (see Fig. 3).
4 Discussion and conclusion
Especially for whole genome sequencing, the post-sequencing analysis (runtime of ∼12 days, single-threaded) is more time-consuming than the actual sequencing (several hours to a few days). Individual tools for mapping and variant calling are maturing and pipeline guidelines such as the GATK Best Practices recommendations are provided by their authors. However, existing tools currently have no support for multi-node execution. Even though some of them support multithreading, the parallel speedup that can be attained might be limited, especially when the number of CPU cores is high. As whole genome analysis is increasingly gaining attention, the need for a solution is apparent.
Halvade provides a parallel, multi-node framework for read alignment and variant calling that relies on the MapReduce programming model. Read alignment is then performed during the map phase, while variant calling is handled in the reduce phase. A variant calling pipeline based on the GATK Best Practices recommendations (BWA, Picard and GATK) has been implemented in Halvade and shown to significantly reduce the runtime. On both a Cloudera cluster (15 worker nodes, 360 CPU cores) and a commercial Amazon EMR cluster (16 worker nodes, 512 CPU cores), Halvade is able to process a 50-fold coverage whole genome dataset in under 3 h, with a parallel efficiency of 92.1 and 88.5%, respectively. To the best of our knowledge, these are the highest efficiencies reported to date (see Supplementary Data S1.4 for a comparison with Crossbow). Like Halvade, MegaSeq supports a pipeline which is based on the GATK Best Practices recommendations. As the source code of MegaSeq is not publicly available, a direct comparison to Halvade is difficult. Puckelwartz et al. (2014) estimate, based on benchmarks on 61 whole genomes, that 240 whole genomes could be processed in 50.3 h, using a supercomputer with 17 424 CPU cores (AMD Magny-Cours @ 2.1 GHz). When rescaling this to 360 CPU cores @ 2.4 GHz, an average runtime of 8.9 h is obtained for a single genome. Halvade processes such dataset in 2 h and 39 min and thus provides for a more cost-effective way to minimize runtime. It should be noted that (i) MegaSeq used the GATK HaplotypeCaller whereas Halvade relied on the (much) faster UnifiedGenotyper during benchmarking, (ii) additional variant annotation was performed in MegaSeq, (iii) a comparison based on the number of CPU cores and clock frequency alone has its limitations as also disk speed, available RAM, network speed and other hardware aspects may play a role.
To enable multi-node parallelization of sequencing pipelines, Halvade provides and manages parallel data streams to multiple instances of existing tools that run on the different nodes. No modifications to these tools are required; they can thus easily be replaced by newer versions. The same holds for replacing the current tools by alternatives: if input/output formats are not affected, the interchange of tools is straightforward. For the map phase (read alignment), the use of tools other than the natively supported BWA(-aln and -mem) should be possible with minimal effort, provided that they output SAM records either to disk or standard output. For the reduce phase (variant calling), it is more invasive to make modifications as the steps involved and the (intermediate) input and output formats are not standardized across tools. Making major changes to this analysis step will require modification to the source code (Java) of Halvade. However, Halvade provides all functionality to partition the reference genome in chunks and provides functionality to copy external dependencies (files or databases) to the worker nodes. This should greatly facilitate the implementation of variant calling pipelines using other tools.
To achieve optimal performance on computer clusters with multi-core nodes, Halvade can be configured to run multiple parallel instances of a tool per node, each instance using a limited number of threads. This approach significantly increases the per-node performance as the multithreaded scaling behavior of certain tools is limited. Indeed, on a single 24-core node with three parallel tasks, Halvade already attains a speedup of 2.5 compared with a multithreaded execution of the same tools in a single task. A second key factor in attaining a high efficiency is the choice of appropriate task sizes. Few, large tasks might result in an unevenly balanced load whereas lots of small tasks result in scheduling and tool initialization overhead.
In Halvade, it is assumed that read alignment is parallel by read and that variant calling is parallel by chromosomal region. Certain tools, however, produce slightly different results when they operate on only part of the data. We have analyzed these sources of variation in detail (see Supplementary Data S1.5). As for the accuracy of whole genome analysis, the variants found by Halvade match >99% with variants in the validation set created by a sequential run of the GATK Best Practices pipeline. Additionally, almost all the 1% different variants have a very low variant score.
In the implementation of the GATK Best Practices pipeline, the latest versions of BWA, Picard and GATK are supported. Both whole genome and exome analysis are supported. An RNA-seq variant calling pipeline is currently in development. Halvade is built with the Hadoop MapReduce 2.0 API and thus supports all distributions of Hadoop, including Amazon EMR and Cloudera, the latter which can be installed on a local cluster. The Halvade source code is available at http://bioinformatics.intec.ugent.be/halvade under GPL license.
Funding
This work is funded by Intel, Janssen Pharmaceutica and by the Institute of the Promotion of Innovation through Science and Technology in Flanders (IWT). Some benchmarks were run at the Intel Big Data Lab, Swindon, UK. We acknowledge the support of Ghent University (Multidisciplinary Research Partnership ‘Bioinformatics: From Nucleotides to Networks').
Conflict of Interest: none declared.
Supplementary Material
References
- Dean J., Ghemawat S. (2008) MapReduce: simplified data processing on large clusters. Commun. ACM, 51, 107–113. [Google Scholar]
- Depristo M.A., et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491––498.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyhim P. (2013). Best Practices for Amazon EMR. Technical report, Amazon Web Services Inc. [Google Scholar]
- Fonseca N.A., et al. (2012) Tools for mapping high-throughput sequencing data. Bioinformatics, 28, 3169–3177. [DOI] [PubMed] [Google Scholar]
- Langmead B., et al. (2009a) Searching for SNPs with cloud computing. Genome Biol., 10, R134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., et al. (2009b) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., et al. ; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., et al. (2008) SOAP: short oligonucleotide alignment program. Bioinformatics, 24, 713–714. [DOI] [PubMed] [Google Scholar]
- McKenna A., et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R., et al. (2011) Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet., 12, 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemenmaa M., et al. (2012) Hadoop-BAM: Directly manipulating next generation sequencing data in the cloud. Bioinformatics, 28, 876–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey R.V., Schlötterer C. (2013) DistMap: a toolkit for distributed short read mapping on a hadoop cluster. PLoS One, 8, doi:10.1371/journal.pone.0072614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckelwartz M., et al. (2014) Supercomputing for the parallelization of whole genome analysis. Bioinformatics, 30, 1508–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz M.C. (2009) CloudBurst: highly sensitive read mapping with MapReduce. Bioinformatics, 25, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry S.T., et al. (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res., 29, 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera G.A., et al. (2013) From FastQ data to high-confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinformatics, 43, 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., et al. (2011) The impact of next-generation sequencing on genomics. J. Genet. Genom., 38, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.