

Abstract
During normal haematopoiesis, cell development and differentiation programs are accomplished by switching ‘on’ and ‘off’ specific set of genes. Specificity of gene expression is primarily achieved by combinatorial control, i.e. through physical and functional interactions among several transcription factors that form sequence-specific multiprotein complexes on regulatory regions (gene promoters and enhancers). Such combinatorial gene switches permit flexibility of regulation and allow numerous developmental decisions to be taken with a limited number of regulators. The haematopoietic-specific Ets family transcription factor PU.1 regulates many lymphoid- and myeloid-specific gene promoters and enhancers by interacting with multiple proteins during haematopoietic development. Such protein–protein interactions regulate DNA binding, subcellular localization, target gene selection and transcriptional activity of PU.1 itself in response to diverse signals including cytokines, growth factors, antigen and cellular stresses. Specific domains of PU.1 interact with many protein motifs such as bHLH, bZipper, zinc fingers and paired domain for regulating its activity. This review focuses on important protein–protein interactions of PU.1 that play a crucial role in regulation of normal as well as malignant haematopoiesis. Precise delineation of PU.1 protein-partner interacting interface may provide an improved insight of the molecular mechanisms underlying haematopoietic stem cell fate regulation. Its interactions with some proteins could be targeted to modulate the aberrant signalling pathways for reversing the malignant phenotype and to control the generation of specific haematopoietic progeny for treatment of haematopoietic disorders.
Keywords: haematopoiesis, protein–protein interaction, transcription factor, co-activator, co-repressor
Introduction
- Transcription factor PU.1: expression distribution and function
- Functional domains of PU.1 protein
- Structure of PU.1 ETS domain and its binding to DNA
- PU.1 gene regulation
- PU.1-interacting proteins
- NF-IL6β (C/EBP-δ)
- c-Jun
- CBP
- GATA-1
- Antagonism between GATA-1 and PU.1
- Synergistic interaction between PU.1 and GATA-1
- C/EBP-α
- c-Myb
- AML-1
- AML-1/ETO
- Pip/NF-EM5/IRF-4
- PU.1-protein interactions and HSC fate determination
Concluding remarks
Introduction
Haematopoiesis is a continuous and stepwise controlled process in which the pluripotent haematopoietic stem cells (HSCs) undergo differentiation to produce all mature blood cell lineages, which comprise the effector cells of both innate and acquired immunity. These cells fulfil specific roles in the host defence against invading pathogens and in the maintenance of homeostasis [1]. The regulation of haematopoiesis is essential for the replenishment of distinct blood cell types that include B and T lymphocytes; erythrocytes; megakaryocytes/platelets; basophils/mast cells; eosinophils; neutrophils/granulocytes and macrophages/mono-cytes [2]. The haematopoietic development is under stringent control from extracellular and intracellular stimuli that result in the activation of specific downstream signalling cascades. The integration of these complex but finely tuned regulatory pathways provides almost unlimited possibilities for haematopoietic regulation and results in an elaborate fail-safe mechanism for controlling gene expression and an overall cellular haematopoietic response.
Central players in this process are sequence-specific transcription factors that ‘program’ precursor cells to express growth factor receptors, which mediate proliferation and differentiation signals produced by the microenvironment [3]. The mature blood cell types acquire their characteristic properties as a result of coordinated, lineage-specific gene expression at appropriate stages of lineage commitment, proliferation, terminal differentiation and functional maturation. Therefore, haematopoietic development is largely regulated by the activities of haematopoietic transcription factors that select genes to be activated and generate a highly specific response that is determined by the precise arrangement of DNA-binding sites and the diversity of protein–protein interactions [4].
Rather than being controlled by a single master regulator, lineage-specific gene expression depends on the combination of factors that bind in overlapping functional domains. Gene transcription requires the assembly of RNA polymerase II with a multiprotein pre-initiation complex at specific DNA sequences including the TATA box in a promoter. Interactions of general transcription factors with basal promoter elements are generally essential for basal transcription but not sufficient to modulate its rate. Therefore, the activation or repression of gene transcription takes place when several transcription factors bind to their cognate sequences on the lineage-specific promoters and stimulate or inhibit transcription through protein–protein interactions with the basal transcription machinery [5]. Based on bacterial gene regulation models, it has been proposed that protein interactions between the individual enhancer-bound factors serve to promote cooperative assembly on the DNA [6]. Recent observations, however, demonstrate that major functions of typical transcription factors do not require DNA binding, suggesting that due to their protein interaction potential, transcription factors carry information beyond the stabilization of DNA contacts [7]. In addition, transcription factors interact with other trans-regulatory proteins such as co-activators, co-repressors, chromatin remodelling factors and/or bridging factors to form gene regulatory complexes. For instance, in some cases interaction between transcription factors is required to modify chromatin in such a way that a previously silent gene may become accessible to the activation machinery. Once the chromatin is favourably restructured and the gene ‘prepared’ for expression, the transcription machinery needs to be recruited.
Among various transcription factors, the ETS factors are known to play a significant role in the regulation (activation or repression) of genes associated with pathogen and tumour defence. Especially, transcription factor PU.1 appears to have the greatest impact on immunity, primarily through its control of immune cell (myeloid and B-lymphoid) development. The deregulated PU.1 function severely impairs haematopoietic development. Its complete disruption causes embryonic and/or newborn lethality in mice [8, 9] whereas its knockout leads to a profound deficit in the development of neutrophils/monocytes and B cells, followed by an impairment in T- and natural killer (NK) cells but megakaryocyte/erythroid (Meg/E) development remains intact [10, 11]. The over-expression of PU.1 in erythroid cells blocks differentiation and promotes erythroleukemic transformation, whereas its reduced expression in mice has been associated with the development of myeloid leukaemia [12, 13]. Therefore, in both erythroid and myeloid lineages, abnormally high or low levels of PU.1, respectively, contribute to the development of leukaemia, and restoration of normal PU.1 expression drives differentiation. Furthermore, heterozygous mutations in PU.1 have been reported in some patients with acute myeloid leukaemia (AML) [14].
Thus PU.1 is a multi-faceted protein that controls numerous normal and pathogenic functions within the haematopoietic system. These pleiotropic functions are attributed to its complex and dynamic expression pattern during haematopoiesis. All of its functional domains are associated with interactive protein partners. Such protein–protein interactions direct PU.1 and other transcription factors to act on promoters/enhancers in order to increase or decrease transcription of its target genes. Therefore, a detailed characterization of the molecular partnerships formed by PU.1 is the key to a profound understanding of its function in normal and malignant haematopoiesis, which will certainly facilitate the development of rationally targeted therapies for the treatment of haematological malignancies. In the following review, we have discussed the importance and mechanism of PU.1's interaction with structurally unrelated proteins that are crucial for PU.1-mediated functions such as lineage commitment and differentiation of HSCs during normal haematopoiesis. Some PU.1-protein interactions that are known to disrupt normal HSC differentiation and lead to a malignant phenotype have also been discussed here.
Transcription factor PU.1: expression distribution and function
Transcription factors of the ETS family have been studied as the key regulators of genes involved in haematopoietic cell growth and immune response. Their functional versatility emerges from their interaction with other structurally unrelated transcription factors. Indeed, this combinatorial control is the characteristic property of ETS family members [15]. PU.1 (Spi-1), a member of this family, is encoded by proto-oncogene sfpi-1 and is expressed exclusively in haematopoietic cells, with high levels detected in monocytic, granulocytic and B lymphoid lineages [16–20]. Lower levels of PU.1 are found in mature erythrocytes [21], whereas it is not expressed in mature T cells [22]. It regulates a large number of genes of the myeloid and lymphoid lineages. In myeloid lineages, the putative PU.1 target genes include G-SCF receptor [23], GM-CSF receptor [24] and M-CSF receptor [25]. Besides, PU.1 also transactivates other genes involved in the acquisition of a mature monocyte phenotype. This allows monocytes/macrophages to carry out their immunological functions following an activation signal. For instance, PU.1 controls the expression of genes encoding Fc RI [26] and Fc RIIIA [27], the high- and low-affinity receptors, respectively, for the constant region of immunoglobulin G (IgG). Once expressed on the cell surface, these molecules allow the macrophage to recognize and phagocytose IgG-opsonized bacteria. PU.1 also induces the expression of the adhesion molecules CD11b [28], CD18 [29] and CD14 [25] during myeloid differentiation. CD18 is the β chain of integrins and, when associated with CD11b, they both constitute the membrane glycoprotein Mac-1, which mediates the adhesion of monocytes to endothelium and subsequent diapedesis, and the phagocytosis of complement-opsonized particles. CD14 is a membrane glycoprotein that specifically binds to lipopolysaccharide, a structural component of the bacterial wall. This recognition is a crucial step in triggering the microbactericidal function of the macrophage. Likewise, PU.1 mediates the expression of Scavenger receptors type I and II [30], which are maximally expressed during the terminal differentiation of monocytes to macrophages and are involved in the capture and subsequent degradation of proteins that have been chemically modified at inflammation sites.
A common feature of all these genes is the absence of defined TATA boxes in their promoters; they instead have a functional PU.1 binding site located upstream of the transcriptional start site at a position corresponding to that of a TATA box, accompanied by a cluster of binding sites specific for other ubiquitous regulators such as Sp1, AML-1, CAAT/enhancer binding protein (C/EBP) and core binding factor (CBF) family members [31, 32]. Because PU.1, through its N-terminal transactivation domain (TAD), can bind to the TATA binding protein (TBP) and a set of TATA associated factors (TAFs) in vitro, it can activate almost all myeloid promoters by recruiting TBP and subsequently other components of the basal transcriptional machinery. Evidently, mutation of a PU.1 binding site located just upstream of the transcriptional start site of the FcγRIB gene abrogates myeloid expression and replacement of this mutant site with a TATA box can re-establish myeloid expression indicating the presence of a positive feedback mechanism in the regulation of the PU.1 promoter [33].
In case of the B-lymphoid genes such as Ig heavy chain (μ) [34], Ig light chains (κ and λ) [35, 36], mb- (Ig α) [37], B29 (Ig β) [38] and Ig-J chain [33], the binding of PU.1 to the enhancer regions induces the bending of DNA that facilitates the binding of other transcription factors to their respective binding sites, thus forming a synergistic multiprotein-complex to augment transcription.
Functional domains of PU.1 protein
The cDNA sequence analysis revealed that PU.1 gene has an open reading frame of 816 bases, which encodes a protein of 272 or 266 amino acids, depending upon the initiation codon for translation being used. The human homologue (264 amino acids), having five exons and four introns, exhibits a sequence identity of 85% with mouse PU.1 and is located on chromosome 11, region p11.22 [39]. The PU.1 protein comprises three distinct functional domains, through which it executes both DNA binding as well as transactivation activity by interacting with various other structurally unrelated transcriptional regulators (Fig. 1). The evolutionarily conserved DNA binding domain (also known as ETS domain) is confined to the C-terminus of PU.1 and mediates its binding to DNA as well as interaction with a number of other proteins such as c-Jun, GATA-1, GATA-2, C/EBP-α and -β and AML-1[40–42]. The N-terminus harbours a TAD that contains three acidic amino acid-rich regions and a glutamine-rich region. The TAD allows its interaction with the basal transcription factors such as TFIID and TBP (TBP) as well as other tissue-specific factors including retinoblas-toma protein, GATA-1 and GATA-2, glucocorticoid receptor and heat shock protein (HSP90) [31, 43]. In addition, PEST domain sandwiched between the above two domains is essential for many protein–protein interactions and owes its name to an abundance of proline (P), glutamic acid (E), serine (S) and threonine (T) residues. This domain interacts with interferon regulatory factors (IRFs), including Pip/NF-EM5 and interferon consensus sequence binding protein (ICSBP) [44–46].
Figure 1.
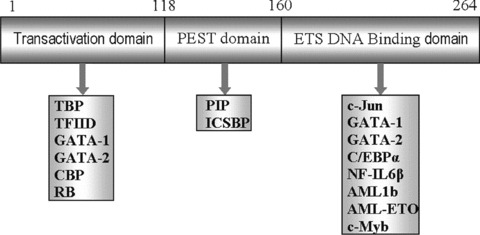
Functional domains of PU.1 protein along with their interacting proteins. The N-terminus Transactivation domain interacts with TFIID, TBP, GATA-1, GATA-2, CBP and retinoblastoma protein. The C-terminus ETS domain codes for a DNA-binding domain that recognizes the sequence 5′-GGAA-3′. The ETS domain interacts with c-Jun, c-Myb, GATA-1, GATA-2, C/EBP-α, NFIL6-β, AML-1b, AML-ETO, etc. The PEST domain interacts with PIP (PU.1 interacting partner) and ICSBP. The phosphorylation at Ser148 is essential for this interaction.
Structure of PU.1 ETS domain and its binding to DNA
The ETS domain (or DNA binding domain) of PU.1 recognizes a purine-rich core sequence (PU box) 5′-GAGGAA-3′ and binds as a monomer. In the crystal structure of PU.1-ETS domain-DNA complex, Kodandpani et al. showed that the ETS domain assumes a tight globular structure formed by three α-helices and a four-stranded anti-parallel β-sheet. It contacts DNA through a novel loop-helix-loop structure, quite similar to α+β (winged) helix-turn-helix motif. It binds to the 5′-GGAA-3′ core sequence in the major groove via the α3-recognition helix, whereas the two loops on both sides contact the phosphate backbone in the minor groove. The first loop consists of β-strands 3 and 4 (the wing), whereas the second is formed by the turn between α-helices 2 and 3 [47]. DNA in the PU.1 complex is uniformly curved by approx. 8° towards the major groove without distinct kinks [48].
In case of myeloid promoters, the binding of PU.1 close to the transcription initiation site may facilitate the recruitment of TFIID complex and promote the binding of adjoining transcription factors. Binding of TFIID to the promoter region is the first step of the assembly process that forms transcription pre-initiation complex. Even though PU.1 may make the initial contact with TFIID, downstream transcriptional factors are likely to help stabilize the TFIID complex through TAF interactions. The PU.1-initiated cooperative stabilization of TFIID may be sufficient to evoke myeloid gene-specific expression [49].
Furthermore, PU.1 serves as an architectural transcription factor for Ig light and heavy chain enhancers. In this capacity, the binding of PU.1 induces bending of DNA that promotes the binding of other transcription factors to nearby regulatory elements, thus forming a higher-order protein-enhancer complex. The PU.1-induced bending of DNA and the subsequent protein–protein interactions among other components of the multiprotein-enhancer complexes repositions the transactivation region of one or more of the other adjoining transcription factors, such as Pip, c-Jun and c-Fos, to augment transcription [50].
PU.1 gene regulation
The PU.1 gene expression is tightly regulated within the haematopoietic system, such that the relative expression level of PU.1 determines whether cells differentiate into macrophages, granulocytes or lymphocytes. As HSCs and multipotential progenitors differentiate, PU.1 is maintained or up-regulated in some lineages and down-regulated in others. For example, PU.1 is expressed in common myeloid progenitor (CMP); the level of its expression increases in granulocyte–monocyte progenitor (GMP) and their progeny (granulocytes and monocytes) but decreases in megakaryocyte–erythrocyte progenitors and their derivatives (megakaryocytes and erythroid cells) [18–20, 51, 52]. Failure to down-regulate PU.1 in erythroid cells leads to a block in erythroid development and leukaemia. Similarly, PU.1 is expressed in common lymphoid progenitor and maintained in B cells but down-regulated in T cells, and forced expression of PU.1 in early T cells can inhibit development of mature T-cell types [52–54]. Finally, ‘knockdown’ mice in which PU.1 expression in the bone marrow was decreased to 20% of wild-type levels all develop a block in macrophage and B-cell development, genomic instability and AML [13]. All of these studies indicate that control of PU.1 levels is critical for normal development and that dysregulation can lead to erythroid or myeloid leukaemia.
Previous in vitro transient transfection studies demonstrated that myeloid-specific activity is confined to a 334 bp promoter sequence extending from approximately 170 bp upstream of the major transcription start sites to 180 bp 5′ untranslated region. The major functional sites confined within this region include the Octamer (Oct-1) site at –54 bp, the Sp1 site at –39 bp, the PU.1 site at +20 bp and also a GATA-binding site at –15 bp. Among these, the PU.1 binding site plays a crucial role in the expression of the PU.1 gene itself in myeloid cells [18, 20, 55–57]. PU.1 is expressed at low levels in the undifferentiated CD34+/CD38− HSCs, prior to the up-regulation of PU.1 mRNA during myeloid development [20, 58]. The transcription factors including Oct-1, Sp1, GATA-1 and Spi-B are expressed in undifferentiated HSCs and mediate initial activation of PU.1. In addition to the activation by Oct-1 and Sp1 in myeloid and B cells, Spi-B has also been shown to bind in vitro and activate PU.1 in transfected HeLa cells [20, 59].
However, transgenic mice containing only the PU.1 promoter failed to express reporter genes in vivo, whereas a murine PU.1 P1 clone including the entire PU.1 gene locus and 35 kb each of 5′ and 3′ flanking sequences expressed exogenous PU.1 RNA in a manner similar to that of endogenous PU.1 in terms of both expression levels and cell-type specificity [60]. These data suggested that distal regulatory elements were necessary for expression of PU.1 in vivo. Subsequent DNase I hypersensitivity assays identified an element 14 kb upstream of the gene which conferred reporter gene activity more than 100-fold higher than that seen with the PU.1 promoter only in stably transfected myeloid lines [60]. These results strongly suggested that an element necessary for proper in vivo expression is located within this kb-14 region. Utilizing a combination of in vivo and in vitro assays, a PU.1 binding site was identified in the proximal (3′) conserved region of the kb-14 upstream regulatory element (URE). A mutation which abolished binding by PU.1 led to a loss of function of the URE in myeloid cells. These results also suggest that PU.1 might be positively regulated by PU.1 itself, and there could be two positive autoregulatory sites in the PU.1 gene: one in the kb-14 URE and one in the promoter region [61].
In addition, it has been reported that signalling by Notch1 transmembrane receptor up-regulates PU.1 gene expression and induces myeloid differentiation. Activated Notch1 directly increases PU.1 RNA levels, leading to a high concentration of PU.1 protein, which has been shown to direct myeloid differentiation. Thus Notch1 acts as an extrinsic regulator of myeloid commitment and PU.1 acts as a specific direct target gene of Notch1 [62].
PU.1-interacting proteins
Despite the importance of PU.1, it is not a lone determinant of immune cell development. A variety of proteins have been identified as interaction partners of PU.1. These proteins include other transcription factors, non-DNA binding cofactors, chromatin-remodelling factors and proteins involved in cell cycle regulation. The activation of gene expression mediated by transcription factor PU.1 is generally associated with synergistic interactions with other transcriptional regulators. For example, PU.1 has been shown to cooperate with NF-IL6β (C/EBP-δ) [43], c-Myb and C/EBP-α[63], c-Jun and c-Fos [40].
However, the regulation of transcription factor activity is not only restricted to synergistic combinatorial associations but negative interactions between transcription factors also play a critical role in the control of haematopoiesis. It has been presumed that transcription factors play a key role in the induction of differentiation events and the process of haematopoietic lineage commitment through antagonistic regulation of alternate lineage-specific factors [64]. For example, a negative cross-talk between PU.1 and GATA-1 plays a significant role in erythro-myeloid lineage commitment [65]. A list of PU.1-interacting proteins has been shown in Table 1.
Table 1.
PU.1 interacting proteins
| Name of the protein | PU.1’s domain of interaction | Function | References |
|---|---|---|---|
| c-Jun | β3/β4 region | Co-activator | [40] |
| NF-IL6 β (C/EBP-δ) | ETS domain | Synergistic activation | [43] |
| GATA-1 | β3/β4 region and TAD | Mutual repression | [41] |
| GATA-2 | β3/β4 region and TAD | Cooperative as well mutual antagonism | [124] |
| C/EBP-α | b3/β4 region | Antagonistic | [106] |
| c-Myb | ETS domain | Cooperative | [63] |
| CBP | TAD | Synergistic | [81] |
| AML-1B | TAD and ETS domain | Synergistic | [32] |
| AML-1/ETO | β3/β4 region | Antagonistic | [114] |
| NF-EM5/Pip/IRF-4 | PEST domain | Synergistic | [118] |
| ICSBP/IRF-8 | PEST domain | Cooperative | [125] |
Both activators and repressors may function by interacting with components of basal transcriptional apparatus, leading to modulation of transcription initiation. The function of an activator may be to recruit the basal transcriptional machinery to a promoter or to induce conformational changes in the complex either at the pre-initiation or elongation steps. However, the role of repressors is not well defined. They may act by specifically interfering, through their repressor domain, with the assembly of the transcriptional machinery (active repression), or they may mediate repression by quenching activators or co-activators (passive repression). Both activators and repressors require co-activator and co-repressors, respectively. These co-factors may function as bridging molecules between the transcription factors and the basal transcriptional machinery, facilitating or inhibiting recruitment to the promoter [66].
NF-IL6β (C/EBP-δ)
NF-IL6β is a leucine zipper transcription factor that belongs to the family of C/EBP proteins. It is a potent transactivator protein implicated in inflammatory responses [67]. Homozygous mutation of the NF-IL6β gene gives rise to mice deficient in macrophage bactericidal and tumoricidal activities. PU.1 and NF-IL6β synergistically cooperate to activate transcription [43]. It has been found that potential PU.1 and NF-IL6β sites are present in some naturally occurring promoters, including the IL-6 and RANTES gene promoters. These genes are responsive to a number of signalling pathways or to inflammatory agents in a variety of tissues, including those that express PU.1. Maximal interaction between them requires PU.1 carboxyl-terminal ETS domain and an intact NF-IL6β leucine zipper domain (Fig. 2). Although both can simultaneously bind to DNA to yield a complex containing both proteins, there is no considerable alteration in the kinetics or affinity of protein–DNA interaction. Instead the transcriptional synergy results from each protein independently influencing components of basal transcriptional complex. Furthermore, because maximal interaction between PU.1 and NF-IL6β requires sequences near their respective DNA binding domains, it is likely that their physical association can inhibit DNA binding by either protein. The deletion of carboxyl-terminal 28 amino acid residues of PU.1 caused a dramatic loss of its interaction with NF-IL6β[43].
Figure 2.

PU.1 interacts through its c-terminal ETS domain with the leucine zipper domain of NF-IL6β. Both the proteins simultaneously bind to DNA during complex formation and the transcriptional synergy results from each protein independently influencing components of basal transcriptional complex.
A related transcription factor, C/EBP-β (NF-IL6) has also been shown to interact with PU.1 during the transcriptional regulation of IL-1β gene and other monocyte-specific genes. The two factors strongly cooperate on IL-1β core promoter (−59/+12) in the absence of direct C/EBP-β binding to DNA. Transient transfection assays, using mutated IL-1β core promoters showed that PU.1, but not NF-IL6 binding site is absolutely required for functional cooperativity. Furthermore, the NF-IL6 transactivation domain is functionally indispensable and more critical than that of PU.1 [67, 68].
C-Jun
c-Jun belongs to the bZIP (basic leucine zipper) group of DNA binding proteins and is a component of AP-1 transcription factor complexes [69]. It forms homodimers or can heterodimerize with other Jun family members or with other bZIP proteins. It plays a significant role in monocytic differentiation [70]. c-Jun mRNA is up-regulated upon monocytic differentiation of bipotential myeloid cell lines [71, 72]. c-Jun enhances the ability of PU.1 to transactivate the human monocyte-specific M-CSF receptor promoter even when it does not bind to the M-CSF receptor promoter (as it does not contain any consensus AP-1 binding sites) [25]. In contrast, the macrosialin promoter and macrophage scavenger receptor promoter contain PU.1 and AP-1 binding sites, which are critical for their monocyte-specific expression [30, 73].
Because c-Jun physically binds to PU.1 at the β3/β4 region of its ETS domain and functionally activates it independently of JNK phosphorylation, it was concluded that c-Jun acts as a JNK-independent co-activator of PU.1 (Fig. 3). The transactivation domain of PU.1 (amino acids 1–118) is necessary for the basal transactivation of the M-CSF receptor promoter by PU.1. However, the mutants that lack the transactivation domain cannot interact with c-Jun [40]. It has been shown that during 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced monocytic differentiation of U937 cells, the mRNA expression of c-Jun and M-CSF receptor increases [74]. It has been demonstrated that the growth factors or other signals activate the Ras pathway, which in turn increases the c-Jun expression in monocytic progenitors. Increased c-Jun expression then activates PU.1, resulting in increased M-CSF receptor expression and hence increased survival, proliferation and differentiation of the monocytic lineage. In c-Jun-deficient F9 cells there is no effect of Ras on PU.1, whereas Ras enhances the ability of PU.1 to transactivate the M-CSF receptor promoter in c-Jun-containing CV-1 cells [75].
Figure 3.

c-Jun acts as an important co-activator of transcription factor PU.1 during the gene regulation of various myeloid gene promoters such as M-CSF receptor promoter and macrosialin promoter. c-Jun physically interacts with the β3/β4 region in the ETS domain of PU.1 and functionally activates it. Generally, c-Fos heterodimerizes with c-Jun but it blocks the coactivation of PU.1 by c-Jun.
Usually, c-Jun forms heterodimers with c-Fos in AP-1 transcription factor complexes [69, 70]. However, c-Fos does not cooperate with c-Jun in its co-activator function. In contrast, c-Fos completely blocks the co-activation of PU.1 by c-Jun (Fig. 3). Because c-Fos does not physically bind to PU.1, it might compete with PU.1 for the binding partner c-Jun.
CBP
CREB (cAMP response element binding protein) binding protein (CBP) serves as a co-activator for a variety of transcription factors involved in growth and differentiation. CBP and the related p300 function by bridging between sequence-specific transcriptional activators and general transcription factors of the basal transcription machinery [76] (Fig. 4). They are believed to enhance transcription by targeted acetylation of specific chromatin domains (amino acid lysine in histones) with their intrinsic histone acetyltransferase activities [77]. The acetylation and deacetylation of histones are thought to be the key machinery of transcriptional activation and repression. Histone acetylation weakens the interaction of histones with DNA and induces alterations in nucleosome structure. This enhances the accessibility of targeted promoters to components of the transcription machinery, thereby increasing transcription.
Figure 4.

CBP/p300 functions by bridging between sequence-specific transcriptional activators and general transcription factors of the basal transcription machinery. CBP/p300 enhances transcription by targeted acetylation of specific chromatin domains with their intrinsic histone acetyltransferase activities. The region of CBP spanning residues 1283–1915 interacts with a portion of the TAD of PU.1 (aa residues 74–122) directly and acts as its co-activator. CBP also binds and stimulates the activity of erythroid-specific transcription factor GATA-1 by acetylating its two highly conserved lysine-rich motifs near each of the two zinc fingers.
GST binding assays revealed that a portion of the transcriptional activation domain of PU.1 (amino acids 74–122) directly interacts with the region spanning residues 1283–1915 of CBP (Fig. 4). CBP enhances PU.1-mediated transcription of multimerized PU-box luciferase reporter constructs, suggesting that CBP acts as a co-activator for the transcription factor PU.1. Because the amount of CBP is limited in cells, CBP may mediate positive and negative cross-talk between PU.1 and other transcription factors in the process of haematopoietic cell differentiation [78].
CBP binds and stimulates the activity of erythroid-specific transcription factor GATA-1 by acetylating its two highly conserved lysine-rich motifs near each of the two zinc fingers [79]. Mutations in the acetylation sites impair the ability of GATA-1 to trigger differentiation of erythroid cells, suggesting that acetylation is important for GATA-1 function in vivo[80]. Hong et al. have shown that PU.1 inhibits CBP-mediated acetylation of GATA-1 in vitro and in vivo, which is one of the mechanisms by which PU.1 blocks erythroid lineage commitment. During differentiation of murine erythroleukaemia (MEL) cells there is an inverse correlation between PU.1 levels and CBP-acetyl transferase activity [81]. Structure-function analysis of PU.1 in vitro and in vivo suggests that both the activation domain and the ETS domain are required for full inhibitory activity towards CBP and p300.
It was shown through gel shift experiments using extracts from MEL and MEL-PU.1 cells that there is no detectable difference in GATA-1 DNA-binding activity before and after differentiation despite significant changes in PU.1 protein levels. Thus inhibition of GATA-1 activity results from inhibition of its co-activator (i.e. CBP) rather than from blocking its DNA contacts. Moreover, it has been reported previously that inhibition of GATA-1 DNA binding requires very high concentrations of PU.1 [82, 83]. Furthermore, CBP acetyltransferase activity has been reported to increase during MEL cell differentiation as PU.1 levels decline and is inhibited by sustained PU.1 expression [81], suggesting the importance of a regulated balance between CBP and PU.1 during erythroid cell differentiation.
GATA-1
GATA-1 is a key erythroid transcription factor required for the development of normal erythroid and megakaryocytic lineages in which it regulates the expression of many specific genes, such as, α- and β-globin genes [84, 85], erythropoietin receptor [86], erythroid Kruppel-like factor [87], α-spectrin [88], platelet factor 4 [89], glycoprotein IIb [90] and so on. GATA-1 also plays an essential role in eosinophil development. GATA-1 null mouse embryos die from severe anaemia between embryonic day E10.5 and E11.5 [91].
Antagonism between GATA-1 and PU.1
Several lines of evidence suggest that GATA-1 and PU.1 functionally antagonize each other through direct physical interaction via the DNA-binding domains of both proteins [41, 42, 65]. But the mechanisms by which these transcription factors antagonize each other are quite distinct. Zhang et al. demonstrated that the N-terminal TAD of PU.1 physically interacts with the conserved C-terminal zinc finger of GATA-1 and blocks its DNA-binding ability in vitro and in vivo, thereby repressing GATA-1-mediated transcription of its target genes [83] (Fig. 5). Both the DNA binding and transactivation domains of PU.1 are required for inhibition of GATA-1-dependent transcription. In Xenopus embryos, ectopic expression of PU.1 blocks erythropoiesis during normal development. Introduction of exogenous GATA-1 can trigger Xenopus embryos to resume differentiation and undergo terminal cell division in order to lose their tumorigenicity [41]. Thus, the stoichiometry of these two mutually antagonistic transcription factors is important not only during normal erythroid development but also during leukemogenesis [92]. On the contrary, GATA-1 represses the transcriptional activity of PU.1 by blocking the binding of its co-activator c-Jun to the β3/β4 region in its DNA-binding domain [40, 65] (Fig. 5). The carboxy-terminal zinc finger of GATA-1 interacts with the β3/β4 region of PU.1, and disrupts the PU.1/c-Jun interaction in a competitive manner, thereby blocking c-Jun from co-activating PU.1 [41].
Figure 5.

Cross-antagonism between PU.1 and GATA-1. GATA-1 represses PU.1 function by interacting through its c-terminal zinc finger with the β3/β4 region of PU.1 and displacing its co activator c-Jun. On the other hand, PU.1 represses GATA-1 function by interacting through its transactivation domain with the c-terminal zinc finger of GATA-1 and thereby inhibiting its binding to the cognate DNA sequence.
In many acute leukaemias and some lymphomas, aberrant differentiation is a major feature of the malignant phenotype that often results from a single genetic alteration and hence provides a site-specific target for therapy. For example, deregulation of PU.1 in erythroid precursors can cause erythroleukaemias in mice. Differentiation induction of haematological malignant cells as well as normal HSC differentiation is mediated by a stochiometric ratio of transcription factors GATA-1 and PU.1 [41], which broadly determine lineage and cooperate with more specific transcription factors such as RAR receptor and CEBP family members [59, 93, 94]. Erythropoietin-induced erythroid differentiation of leukemic cells is associated with GATA-1 induction and down-regulation of PU.1 and other transcription factors that direct myeloid lineage during normal differentiation. Furthermore, PU.1 and GATA-1 both potentially exhibit autoregulation, whereas at the same time interactions between these two mediate inhibition of each other’s function. Therefore, the reciprocal inhibition and autoregulation of these two regulators provides a mechanism for lineage choice during early blood stem cell and progenitor cell differentiation into either myeloid or erythroid cells [65, 83].
Synergistic interaction between PU.1 and GATA-1
In contrast to the functional antagonism reported for PU.1 and GATA-1 for the activation of the M-CSFR promoter and various erythroid genes, these factors have also been shown to cooperate synergistically in the activation of the eosinophil MBP-P2 (major basic protein) promoter. Key combinatorial interactions of GATA-1, PU.1 and C/EBP-δ isoforms mediate either synergy or antagonism of eosinophil gene transcription during myeloid development and terminal differentiation [95].
The synergistic effect of PU.1 on GATA-1-mediated transactivation is mediated by a conformational change in GATA-1 when it binds through both of its C- and N-terminal zinc fingers to the two canonical non-overlapping tandem GATA-binding sites present in the MBP-P2 promoter. Involvement of the N-terminal zinc finger by binding to the other GATA site in the MBP-P2 promoter might trigger a conformational change leading to decreased DNA binding activity of the C-terminal zinc finger and a decrease in transactivating activity for the β-globin silencer [96]. A low amount of PU.1 may interact differentially or preferentially with the N-terminal zinc finger of GATA-1 and prevent this decrease in transactivating potential; thus accounting for PU.1/GATA-1 synergy in transactivation of the MBP-P2 promoter.
On the contrary, expression of a high amount of PU.1 could lead to interactions with both zinc fingers, thus interfering with the DNA binding activity of GATA-1 and abrogating the synergy. Of note, PU.1 has been shown to interact with both the C- and N-terminal zinc fingers of GATA-1 [41]. Only the C-terminal zinc finger of GATA-1 is capable of independently binding to a GATA consensus site and stimulating transcription, whereas the N-terminal finger shows no independent DNA binding activity but can modify binding specificity (stabilize or disrupt binding) at some naturally occurring dual GATA sites that have been shown to be critical for gene expression [97–99].
Interestingly, C/EBP family members C/EBP-ɛ and C/EBP-β, were found to antagonize both GATA-1 transactivation and GATA-1/PU.1 synergistic activation of the eosinophil MBP-P2 promoter, as well as PU.1-mediated transactivation of PU.1 target genes/promoters such as the M-CSFR, suggesting a potent repressor function for this isoform in myeloid development [95].
C/EBP-α
C/EBP-α (CAAT/EBP), a bZip transcription factor, regulates not only a variety of hepatocyte and adipocyte genes, but several myeloid-specific genes as well [100]. For example, it regulates M-CSF receptor, G-CSF receptor and GM-CSF receptor α promoters. C/EBP-α expression is prominent in immature myeloid cells [24, 101]. C/EBP-α-null mice lack the entire granulocyte lineage but develop normal monocytes [102].
Combinations of inhibition and autoregulation involving PU.1 and C/EBP-α have been hypothesized to mediate the decision of myeloid progenitors to differentiate into either granulocytes or monocytes [103, 104]. The haematopoietic progenitors require PU.1 to initiate monocyte differentiation and C/EBP-α to initiate granulopoiesis. Laslo et al. have shown that nonlinear positive feedback regulates differentiation of CMPs into macrophages or neutrophils [105]. Earlier it was suggested that the relative difference in expression of the transcription factors PU.1 and C/EBP-α regulates the differentiation of CMPs [103]. They found that macrophage differentiation is favoured when the level of PU.1 is higher than that of C/EBP-α, whereas neutrophil differentiation is favoured when the level of C/EBP-α is higher than that of PU.1. Thus a mutual corepression was thought to exist between PU.1 and C/EBP-α that drives CMPs towards one fate or the other. Each factor has been shown to synergize on various promoters, including M-CSF receptor promoter and neutrophil elastase (NE) promoter [24, 63]. Each is expressed in a bipotential myeloid cell; C/EBP-α is capable of functionally blocking the PU.1 protein, and this interference is mediated through interaction between the β3/β4 region of the PU.1 DNA-binding domain and the leucine zipper in the DNA-binding domain of C/EBP-α, which in turn displaces PU.1’s co-activator c-Jun [106, 107].
Additionally, a mutual corepression was found between Egr-2/Nab-2, a complex of genes activated by PU.1, and Gfi-1, a gene activated by C/EBP-α. Because both Egr-2 and Gfi-1 are known to promote the expression of genes specific to macrophages and neutrophils, respectively, their corepression may indeed be the basis of a positive feedback loop that promotes and stabilizes a particular cell fate during CMP differentiation. For instance, when PU.1 and C/EBP-α are expressed at low levels as is the case in undifferentiated CMPs, positive feedback between Egr-2/Nab-2 and Gfi-1 is sufficiently weak such that neither is amplified and the mixed-lineage stage persists. In contrast, when PU.1 is expressed at a much higher level than C/EBP-α, the mixed-lineage state of gene expression is resolved and the system is monostable, promoting differentiation of the CMP into a macrophage (or, if the ratio is reversed, into a neutrophil) [105]. C/EBP-α also functionally interacts with the activation domain of PU.1, which might disrupt possible protein–protein interactions important for the PU.1-induced differentiation program. One such candidate is CBP/p300, a co-activator of PU.1 [78], which binds to the transactivation domain of PU.1. This would further block PU.1 activity to induce dendritic cell formation and enhance the capacity of C/EBP-α to induce granulocytes. Interestingly, C/EBP-α is also known to cooperate with PU.1 in controlling the expression of the GM-CSF receptor [108]. The GM-CSF receptor α promoter contains functional PU.1-specific binding site between positions –53 and –41. A C/EBP-α site is located upstream of the PU.1 site between positions –70 and –54. Point mutations of either the PU.1 site or of the C/EBP-α site that abolish the binding of the respective factors result in a significant decrease of GM-CSF receptor α promoter activity in myelomonocytic cells.
c-Myb
c-Myb is predominantly expressed in immature cells of haematopoietic lineages and mice lacking the c-Myb gene have defective foetal haematopoiesis [109]. C-Myb has been implicated in the activation of mammalian myeloid genes, both alone and cooperatively with C/EBP-α and/or PU.1. There are potential C/EBP-, c-Myb- and PU.1-binding sites in the azurocidin and myeloblastin genes. The presence of functional sites matching the c-Myb, C/EBP and PU.1 consensus sequences in the murine myeloperoxidase gene [110, 111] indicates that cooperation between these factors is relevant to the activation of a substantial number of early myeloid genes. In most experiments, the activation seen with C/EBP-α, c-Myb and PU.1 was equivalent to the multiplication of the individual activations obtained with these factors. This interaction has been termed as ‘cooperative’ instead of synergistic because synergism exists when a factor combination produces much more than a multiplicative effect [112].
A 91-bp NE promoter region contains three evolutionarily conserved cis elements, which are essential for activation of the promoter in differentiating 32Dcl3 myeloid cells. These elements bound c-Myb (at 249), C/EBP-α (at 257) and PU.1 (at 282) (Fig. 6). In NIH 3T3 cells, the NE promoter was activated by c-Myb, C/EBP-α and PU.1, via their respective binding sites. Cooperative activation was seen by any combination of c-Myb, C/EBP-α and PU.1, including all three together, again via their DNA-binding sites. In CV-1 cells, but not in NIH 3T3 cells, cooperation between Myb and C/EBP-α depended on the integrity of the PU.1-binding site [78].
Figure 6.

The activation of neutrophil elastase gene is regulated by the cooperative interaction between c-Myb, C/EBP-α and PU.1 through their respective DNA binding domains.
AML-1
AML-1 is a member of the CBF or polyoma EBP (PEBP2) family [113]. During monocytic commitment and differentiation, AML-1B regulates the myeloid-specific expression of M-CSF receptor in synergistic association with C/EBP-α and PU.1 by forming a ternary complex on DNA [32] (Fig. 7).
Figure 7.

The M-CSF receptor gene is regulated by transcription factor PU.1 in combination with C/EBP-α and AML-1B. PU.1, C/EBP-α and AML-1B interact through their respective DNA binding domains and transactivation domains. AML-1 exhibits cooperative DNA binding with C/EBP-α but not with PU.1. The co-activator CBF-β serves to amplify the activation by increasing transcription efficiency.
AML-1 and PU.1 interact physically and this interaction occurs primarily through their DNA binding domains. AML-1 synergizes with PU.1 to activate the M-CSF receptor promoter and this activity requires DNA-binding sites for both factors and regions contained within the transactivation domains of AML-1 and PU.1. The deletion of the activation domain of either PU.1 or C/EBP-α abrogates synergy with AML-1B. Although AML-1 and PU.1 exhibit a relatively weak synergistic activation of the promoter, but because the effect seen in the presence of both PU.1 and AML-1 is more than additive, the interaction is defined as synergistic [32].
Detailed analysis of the physical and functional interaction of AML-1 with PU.1 and C/EBP-α has revealed that the proteins contact one another through their DNA-binding domains and that AML-1 exhibits cooperative DNA binding with C/EBP-α but not with PU.1. This difference in DNA-binding abilities may explain, in part, the differences observed in synergistic activation. Furthermore, the activation domains of all three factors are required for synergistic activation, and the region of AML-1 required for synergy with PU.1 is distinct from that required for synergy with C/EBP-α. These observations present the possibility that synergistic activation is mediated by secondary proteins contacted through the activation domains of AML-1, C/EBP-α and PU.1.
PU.1, C/EBP-α and AML-1 form a transcriptional unit or a primary complex, on the DNA and this primary complex makes multiple and specific contacts with a second, perhaps ubiquitous, complex composed of co-activators. The role of the DNA-binding proteins is to confer tissue-specific, temporal regulation, whereas the co-activators (such as CBF-β) serve to amplify the activation by increasing transcription efficiency [32].
AML-1/ETO
PU.1 plays a major role in leukemogenesis as suggested by the heterozygous PU.1 mutations reported in some patients with AML [14]. However, PU.1 was not found to be mutated in AML patients with translocation t(8; 21), which indicates that distinct pathways of inactivation of PU.1 might be occurring in t(8; 21). The fusion protein AML-1/ETO physically interacts with PU.1 and thereby causes inactivation of the transcriptional activity of PU.1 by displacing its co-activator c-Jun [114] (Fig. 8).
Figure 8.

The fusion protein AML-1/ETO inhibits the function of PU.1 by displacing its co-activator c-Jun from its β3/β4 region.
AML-1/ETO down-regulates the transcriptional activity of PU.1 in myeloid cells and physically interacts at the β3/β4 region in the DNA-binding domain of PU.1. The competitive protein–protein interaction experiments with in vitro-translated proteins indicated that AML-1/ETO disrupts PU.1/c-Jun interaction in a competitive manner, thus blocking c-Jun from co-activating PU.1, a mechanism similar to GATA-1 and C/EBP-α. The physical interaction between AML-1/ETO and PU.1 did not abolish the DNA-binding capacity of PU.1, although AML-1/ETO interacts with the PU.1 DNA-binding domain. It has been shown in the crystal structure of PU.1 that its β3/β4 region does not interact with DNA, but is exposed to the solvent [47]. This structural ability allows PU.1 to retain its DNA binding though being functionally repressed. Interaction of PU.1 with AML-1/ETO and subsequent suppression of PU.1 target genes might contribute to the phenotypic changes seen in t(8; 21).
Because the expression of AML-1 and PU.1 genes was observed even in a leukemic condition or in presence of AML-1/ETO, like in Kasumi-1 cells, it was suggested that the presence of AML-1/ETO does not completely repress the expression levels of these genes, but may block their functions by protein–protein interactions. Overexpression of PU.1 in t(8; 21)-Kasumi-1 cells directs their differentiation towards the monocytic lineage by overcoming the functional block imposed by AML-1/ETO. PU.1 and C/EBP-α are important factors for myeloid differentiation and AML-1/ETO down-regulating these two factors could be an important step towards leukaemia. This also suggests the possibility of using these two factors independently or in combination for therapy of t(8; 21) myeloid leukaemias. Expression of AML-1/ETO fusion protein in the myeloid progenitor cell line 32Dcl3 prevents G-CSF-induced granulocytic differentiation [115].
Pip/NF-EM5/IRF-4
Pip/NF-EM5/IRF-4, a member of IRF family, is a lymphoid-restricted transcription factor that is recruited to a composite element (IRF/ETS-binding site) within Ig light chain enhancers through a specific interaction with PU.1 protein [116, 117]. The formation of the ternary complex requires both protein–protein and protein–DNA interactions. A model was proposed for PU.1/Pip ternary complex. In this two-step recruitment mechanism, PU.1/Pip interaction is DNA template-directed and involves two distinct protein–protein interaction surfaces: (i) the ETS (PU.1) and IRF (Pip) DNA binding domains and (ii) the phosphorylated PEST domain of PU.1 and a putative α helix in Pip [118, 119] (Fig. 9). Specifically, the first step involves an interaction between the DNA binding domains of PU.1 and Pip. In the second step, phosphorylation of PU.1 at Ser 148 results in a conformational change in Pip (either before or after interaction with Pip) allowing cooperative DNA binding [117]. This leads to the alteration or unmasking of the Pip auto-inhibitory c-terminal DNA binding domain, enabling Pip recruitment to DNA. Protein–protein contacts that occur between the two DNA binding domains may then contribute to additional specificity and stability to the ternary complex. The recent molecular modelling study of the PU.1-Pip DNA binding domain complex reveals that the DNA recognition helix α3 of PU.1 Ets domain might interact with Pip wHTH (winged helix-turn-helix) domain to allow cooperative DNA binding. Another highly homologous IRF family member, IRF-8/ICSBP, competes with Pip/IRF-4 for binding to PU.1 on the enhancer element and thus represses the transactivation activity of the complex [46]. Although IRF-4/Pip is known to respond to any extrinsic stimuli, IRF-8/ICSBP is strongly induced by interferon [120]. Thus, interferon largely controls the presence of IRF-8/ICSBP in the ternary complex on the enhancer.
Figure 9.
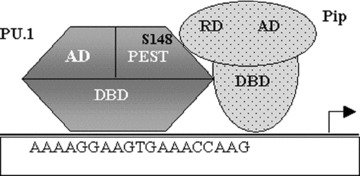
PU.1 recruits PIP (PU.1 interacting partner) by binding with its regulatory domain through its PEST domain. For PU.1’s interaction with Pip, phosphorylation of Ser148 residue is essential. RD, regulatory domain; AD, activation domain; DBD, DNA binding domain.
PU.1-protein interactions and HSC fate determination
The development of different lineages of blood cells is triggered not only by a combination of particular sets of transcription factors but also by their levels relative to one another. The successive steps in the maturation of haematopoietic progenitor cells are thought to involve the up-regulation of factors promoting a particular lineage and the repression of factors promoting alternate lineages. For example, both PU.1 and GATA-1 direct lineage commitment not only by transactivating their individual genetic targets but also by disrupting the functions of opposing transcription factors. When the concentration of GATA-1 is relatively high, it represses PU.1’s function by inhibiting PU.1’s interaction with its co-activator c-Jun, which leads to a reduction in myelopoiesis and a concomitant increase in erythropoiesis because of increased GATA-1 activity. Likewise an antagonism exists between PU.1 and C/EBP-α whereby the relative concentration of each transcription factor is important for differentiating GMPs into either monocytic or granulocytic cells. A higher concentration of C/EBP-α also down-regulates PU.1 by inhibiting its interaction with c-Jun and as C/EBP-α is up-regulated, GMPs are directed towards granulopoiesis. Furthermore, a fusion protein AML-1/ETO is also known to inhibit PU.1’s activity again by inhibiting its interaction with co-activator c-Jun. This in turn leads to a maturation arrest of myeloblasts ultimately causing myeloid leukaemia. Taken together, the above three proteins, namely GATA-1, C/EBP-α and AML-1/ETO, down-regulate PU.1 by displacing its co-activator c-Jun thereby modulating HSC fate in altogether different directions, i.e. erythrocytes, granulocytes and myeloid leukaemia, respectively (Fig. 10). This suggests that PU.1-protein interactions certainly play a significant role in HSC lineage commitment. A slow fluctuation or cycling of gene activity tends to maintain cells in a stable state, while also priming them to differentiate when conditions are right. Progenitor cells commit to one of their potential paths and do not switch to different lineages or revert to less differentiated progenitor cell types. Such stability is certainly important for immune cells, which receive a transient lineage-specific differentiation stimulus in the bone marrow and otherwise stay undifferentiated in the absence of this stimulus in the blood stream or other tissues [121].
Figure 10.

PU.1-protein interactions play a vital role in HSC fate determination. Co-activator c-Jun interacts with the β3/β4 region of PU.1 and enhances its transcriptional ability that in turn leads to increased formation of monocyte/macrophages. When the relative concentration of GATA-1 is higher, it inhibits the PU.1-c-Jun interaction by displacing c-Jun from its binding site and thus blocking the formation of monocytes. The up-regulated activity of GATA-1 in turn leads to enhanced erythropoiesis. Similarly, transcription factor C/EBP-α also inhibits PU.1-c-Jun interaction and leads to the formation of granulocytes. Additionally, a fusion protein AML-1/ETO also represses PU.1 by inhibiting its interaction with co-activator c-Jun, leading to maturation arrest of myeloblasts and thus causing myeloid leukaemia.
Concluding remarks
Although PU.1 is one of the most intensively studied of all mammalian transcription factors, there is still much that we do not understand about the mechanisms through which it regulates gene expression. Understanding how PU.1 controls immune cell development could have important medical applications. Once we understand the regulatory circuitry that controls immune cell development we could exploit that knowledge to develop treatments that direct progenitor cells to differentiate into desired cell types for therapeutic purposes. Also many cancers of the immune system involve interruption of blood cell differentiation. Therapeutic targeting of protein–protein interactions that have been identified as mediators of transcriptional repression that block haematopoietic differentiation should be further explored. For example, in myeloid leukaemia cells that express an AML-1/ETO fusion protein, PU.1 activity is reduced, which is characterized by maturation arrest of myeloblasts [114]. Likewise, the expression of the PML-RAR-α fusion protein in acute promyelocytic leukaemia (APL) reduces PU.1 expression [122]. Disruption of PU.1’s interaction with repressor proteins AML-1/ETO and PML-RAR-α can restore PU.1 expression in AML and APL cells thereby inducing monocytic and neutrophil differentiation respectively. Similarly, GATA-1-mediated repression of PU.1 blocks myeloid differentiation. Therefore, abolishing protein–protein interaction between PU.1 and GATA-1 in myeloid cells would lead to an increase in PU.1’s activity and hence immune cell differentiation. Therefore, it is a challenge for the future to determine the extent to which dysfunction of these transcription factor antagonisms contributes to human leukaemias and how this knowledge can be exploited to benefit cancer therapy.
Further studies are required to delineate the precise interactions of transcription factor PU.1 with basal transcriptional machinery, co-repressors or co-activators and other bridging proteins that collectively are critical for lympho-myeloid transcriptional responses. Efforts are needed to determine precise characterization of its association with different protein partners and to determine how these interactions influence target gene selection and transcription activation or repression. Overlap between specific protein–protein interactions may provide a mechanism to control the diverse functions of this factor. As it is important to correlate specific physical interactions with physiological processes, more sensitive methods need to be developed for characterization of transient interactions and for direct assessment of the biological consequences of specific interactions. Crystallographic structure determination of multiple protein complexes will tremendously help in understanding the combinatorial control of PU.1-mediated transcription. Ultimately, the protein–protein interface may provide a unique target for intervention, thus, providing a novel approach for blocking aberrant signalling pathways or reversing the malignant phenotype associated with oncogenic transcription factors [123].
Acknowledgments
We are grateful to the Director, INMAS for his encouragement and support. P.G. in particular thanks Indian Council of Medical Research (ICMR) for the award of Research Fellowship.
References
- 1.Barreda DR, Belosevic M. Transcriptional regulation of hematopoiesis. Dev Comp Immunol. 2001;25:763–89. doi: 10.1016/s0145-305x(01)00035-0. [DOI] [PubMed] [Google Scholar]
- 2.Szilvassy SJ. The biology of hematopoietic stem cells. Arch Med Res. 2003;34:446–60. doi: 10.1016/j.arcmed.2003.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Simon MC. PU.1 and hematopoiesis: lessons learned from gene targeting experiments. Semin Immunol. 1998;10:111–8. doi: 10.1006/smim.1998.0112. [DOI] [PubMed] [Google Scholar]
- 4.Tjian R, Maniatis T. Transcriptional activation: a complex puzzle with few easy pieces. Cell. 1994;77:5–8. doi: 10.1016/0092-8674(94)90227-5. [DOI] [PubMed] [Google Scholar]
- 5.Goodrich JA, Cutler G, Tjian R. Contacts in context: promoter specificity and macromolecular interactions in transcription. Cell. 1996;84:825–30. doi: 10.1016/s0092-8674(00)81061-2. [DOI] [PubMed] [Google Scholar]
- 6.Carey M. The enhanceosome and transcriptional synergy. Cell. 1998;92:5–8. doi: 10.1016/s0092-8674(00)80893-4. [DOI] [PubMed] [Google Scholar]
- 7.Reichardt HM, Kaestner KH, Tuckermann J, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell. 1998;93:531–41. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- 8.Scott EW, Simon MC, Anastasi J, et al. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–7. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 9.McKercher SR, Torbett BE, Anderson KL, et al. Targeted disruption of the PU.1 gene results in multiple hematopoietic abnormalities. EMBO J. 1996;15:5647–58. [PMC free article] [PubMed] [Google Scholar]
- 10.Spain LM, Guerriero A, Kunjibettu S, et al. T cell development in PU.1-deficient mice. J Immunol. 1999;163:2681–7. [PubMed] [Google Scholar]
- 11.Colucci F, Samson SI, DeKoter RP, et al. Differential requirement for the transcription factor PU.1 in the generation of natural killer cells versus B and T cells. Blood. 2001;97:2625–32. doi: 10.1182/blood.v97.9.2625. [DOI] [PubMed] [Google Scholar]
- 12.Cook WD, McCaw BJ, Herring C, et al. PU.1 is a suppressor of myeloid leukemia, inactivated in mice by gene deletion and mutation of its DNA binding domain. Blood. 2004;104:3437–44. doi: 10.1182/blood-2004-06-2234. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbauer F, Wagner K, Kutok JL, et al. Acute myeloid leukemia induced by graded reduction of a lineage-specific transcription factor, PU.1. Nat Genet. 2004;36:624–30. doi: 10.1038/ng1361. [DOI] [PubMed] [Google Scholar]
- 14.Mueller BU, Pabst T, Osato M, et al. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood. 2002;100:998–1007. doi: 10.1182/blood.v100.3.998. [DOI] [PubMed] [Google Scholar]
- 15.Verger A, Duterque-Coquillaud M. When Ets transcription factors meet their partners. Bioessays. 2002;24:362–70. doi: 10.1002/bies.10068. [DOI] [PubMed] [Google Scholar]
- 16.Moreau-Gachelin F, Tavitian A, Tambourin P. Spi-1 is a putative oncogene in virally induced murine erythroleukaemias. Nature. 1988;331:277–80. doi: 10.1038/331277a0. [DOI] [PubMed] [Google Scholar]
- 17.Goebl MK. The PU.1 transcription factor is the product of the putative oncogene Spi-1. Cell. 1990;61:1165–6. doi: 10.1016/0092-8674(90)90676-6. [DOI] [PubMed] [Google Scholar]
- 18.Klemsz M, McKercher SR, Celada A, et al. The macrophage and B cell-specific transcription factor PU.1 is related to the ets oncogene. Cell. 1990;61:113–24. doi: 10.1016/0092-8674(90)90219-5. [DOI] [PubMed] [Google Scholar]
- 19.Hromas R, Orazi A, Neiman RS, et al. Hematopoietic lineage-restricted and stage-restricted expression of the ETS oncogene family member PU.1. Blood. 1993;82:2998–3004. [PubMed] [Google Scholar]
- 20.Chen H, Ray-Gallet D, Zhang P, et al. PU.1 (Spi-1) autoregulates its expression in myeloid cells. Oncogene. 1995;11:1549–60. [PubMed] [Google Scholar]
- 21.Galson DL, Hensold JO, Bishop TR, et al. Mouse beta-globin DNA binding protein B1 is identical to a proto-oncogene, the transcription Spi-1/PU.1, and is restricted in expression to hematopoietic cells and the testis. Mol Cell Biol. 1993;13:2929–41. doi: 10.1128/mcb.13.5.2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ray D, Bosselut R, Ghysdael J, et al. Characterization of Spi-B, a transcription factor related to the putative oncoprotein Spi-1/PU.1. Mol Cell Biol. 1992;12:4297–304. doi: 10.1128/mcb.12.10.4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith LA, Gonzalez DA, Hohaus S, et al. The myeloid specific granulocyte colony-stimulating factor G-CSF receptor promoter contains a functional site for the myeloid transcription factor PU.1 (Spi-1) Blood. 1994;84:372a. [Google Scholar]
- 24.Hohaus S, Petrovick MS, Voso MT, et al. PU.1 Spi-1 and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol. 1995;15:5830–45. doi: 10.1128/mcb.15.10.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang DE, Hetherington CJ, Chen HM, et al. The macrophage transcription factor PU.1 directs tissue-specific expression of the macrophage colony-stimulating factor receptor. Mol Cell Biol. 1994;14:373–81. doi: 10.1128/mcb.14.1.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perez C, Coeffier E, Moreau-Gachelin F, et al. Involvement of the transcription factor PU.1/Spi-1 in myeloid cell-restricted expression of an interferon-inducible gene encoding the human high-affinity Fc gamma receptor. Mol Cell Biol. 1994;14:5023–31. doi: 10.1128/mcb.14.8.5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Feinman R, Qiu WQ, Pearse RN, et al. PU.1 and an HLH family member contribute to the myeloid-specific expression of the Fc gamma RIIIA promoter. EMBO J. 1994;13:3852–60. doi: 10.1002/j.1460-2075.1994.tb06696.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pahl HL, Rosmarin AG, Tenen DG. Characterization of the myeloid-specific CD11b promoter. Blood. 1992;79:865–70. [PubMed] [Google Scholar]
- 29.Rosmarin AG, Levy R, Tenen DG. Cloning and analysis of the CD18 promoter. Blood. 1992;79:2598–604. [PubMed] [Google Scholar]
- 30.Moulton KS, Semple K, Wu H, et al. Cell-specific expression of the macrophage scavenger receptor gene is dependent on PU.1 and a composite AP-1/ets motif. Mol Cell Biol. 1994;14:4408–18. doi: 10.1128/mcb.14.7.4408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hagemeier C, Bannister AJ, Cook A, et al. The activation domain of transcription factor PU.1 binds the retinoblastoma (RB) protein and the transcription factor TFIID in vitro: RB shows sequence similarity to TFIID and TFIIB. Proc Natl Acad Sci USA. 1993;90:1580–4. doi: 10.1073/pnas.90.4.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Petrovick MS, Hiebert SW, Friedman AD, et al. Multiple functional domains of AML1: PU.1 and C/EBPα synergize with different regions of AML1. Mol Cell Biol. 1998;18:3915–25. doi: 10.1128/mcb.18.7.3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shin MK, Koshland ME. Ets-related protein PU.1 regulates expression of the immunoglobulin J-chain gene through a novel Ets-binding element. Genes Dev. 1993;7:2006–15. doi: 10.1101/gad.7.10.2006. [DOI] [PubMed] [Google Scholar]
- 34.Nelsen B, Tian G, Erman B, et al. Regulation of lymphoid-specific immunoglobulin mu heavy chain gene enhancer by ETS-domain proteins. Science. 1993;261:82–6. doi: 10.1126/science.8316859. [DOI] [PubMed] [Google Scholar]
- 35.Pongubala JM, Nagulapalli S, Klemsz MJ, et al. PU.1 recruits a second nuclear to a site important for immunoglobulin kappa 3′ enhancer activity. Mol Cell Biol. 1992;12:368–78. doi: 10.1128/mcb.12.1.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenbeis CF, Singh H, Storb U. PU.1 is a component of a multiprotein complex, which binds an essential site in the murine immunoglobulin lambda 2–4 enhancer. Mol Cell Biol. 1993;13:6452–61. doi: 10.1128/mcb.13.10.6452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feldhaus AL, Mbangkollo D, Arvin KL, et al. BLyF, a novel cell-type and stage-specific regulator of the B-lymphocyte gene mb-1. Mol Cell Biol. 1992;12:1126–33. doi: 10.1128/mcb.12.3.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Omori SA, Wall R. Multiple motifs regulate the B-cell-specific promoter of the B29 gene. Proc Natl Acad Sci USA. 1993;90:11723–7. doi: 10.1073/pnas.90.24.11723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen VC, Ray D, Gross MS, et al. Localization of the human oncogene Spi1 on chromosome 11, p11.22. Hum Genet. 1990;84:542–6. doi: 10.1007/BF00210807. [DOI] [PubMed] [Google Scholar]
- 40.Behre G, Whitmarsh AJ, Coghlan MP, et al. c-Jun is a JNK-independent coactivator of the PU.1 transcription factor. J Biol Chem. 1999;274:4939–46. doi: 10.1074/jbc.274.8.4939. [DOI] [PubMed] [Google Scholar]
- 41.Rekhtman N, Radparvar F, Evans T, et al. Direct interaction of hematopoietic transcription factors PU.1 and GATA-1: functional antagonism in erythroid cells. Genes Dev. 1999;13:1398–411. doi: 10.1101/gad.13.11.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nerlov C, Querfurth E, Kulessa H, et al. GATA-1 interacts with the myeloid PU.1 transcription factor and represses PU.1-dependent transcription. Blood. 2000;95:2543–51. [PubMed] [Google Scholar]
- 43.Nagulapalli S, Pongubala JM, Atchison ML. Multiple proteins physically interact with PU.1. Transcriptional synergy with NF-IL6 beta. J Immunol. 1995;155:4330–8. [PubMed] [Google Scholar]
- 44.Pongubala JM, Beveren CV, Nagulapalli S, et al. Effect of PU.1 phosphorylation on interaction with NF-EM5 and transcriptional activation. Science. 1993;259:1622–5. doi: 10.1126/science.8456286. [DOI] [PubMed] [Google Scholar]
- 45.Pongubala JM, Atchison ML. PU.1 can participate in an active enhancer complex without its transcriptional activation domain. Proc Natl Acad Sci USA. 1997;94:127–32. doi: 10.1073/pnas.94.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eisenbeis CF, Singh H, Storb U. Pip, a novel IRF family member, is a lymphoid-specific, PU.1-dependent transcriptional activator. Genes Dev. 1995;9:1377–87. doi: 10.1101/gad.9.11.1377. [DOI] [PubMed] [Google Scholar]
- 47.Kodandpani R, Pio F, Ni CZ, et al. A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature. 1996;380:456–60. doi: 10.1038/380456a0. [DOI] [PubMed] [Google Scholar]
- 48.Pio F, Kodandpani R, Ni CZ, et al. New insights on DNA recognition by ets proteins from the crystal structure of the PU.1 ETS domain-DNA complex. J Biol Chem. 1996;271:23329–37. doi: 10.1074/jbc.271.38.23329. [DOI] [PubMed] [Google Scholar]
- 49.Pugh BF. Mechanism of transcriptional complex assembly. Curr Opin Cell Biol. 1996;8:303–11. doi: 10.1016/s0955-0674(96)80002-0. [DOI] [PubMed] [Google Scholar]
- 50.Fisher RC, Scott EW. Role of PU.1 in hematopoiesis. Stem Cells. 1998;16:25–37. doi: 10.1002/stem.160025. [DOI] [PubMed] [Google Scholar]
- 51.Akashi K, Traver D, Miyamoto T, et al. A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature. 2000;404:193–7. doi: 10.1038/35004599. [DOI] [PubMed] [Google Scholar]
- 52.Miyamoto T, Iwasaki H, Reizis B, et al. Myeloid or lymphoid promiscuity as a critical step in hematopoietic lineage commitment. Dev Cell. 2002;3:137–47. doi: 10.1016/s1534-5807(02)00201-0. [DOI] [PubMed] [Google Scholar]
- 53.Anderson MK, Hernandez-Hoyos G, Diamond RA, et al. Precise developmental regulation of Ets family transcription factors during specification and commitment to the T cell lineage. Development. 1999;126:3131–48. doi: 10.1242/dev.126.14.3131. [DOI] [PubMed] [Google Scholar]
- 54.Anderson MK, Weiss AH, Hernandez-Hoyos G, et al. Constitutive expression of PU.1 in fetal hematopoietic progenitors blocks T cell development at the pro-T cell stage. Immunity. 2002;16:285–96. doi: 10.1016/s1074-7613(02)00277-7. [DOI] [PubMed] [Google Scholar]
- 55.Moreau-Gachelin F, Ray D, Mattei MG, et al. The putative oncogene Spi-1: murine chromosomal localization and transcriptional activation in murine acute erythroleukemias. Oncogene. 1989;4:1449–56. [PubMed] [Google Scholar]
- 56.Chen HM, Zhang P, Radomska HS, et al. Octamer binding factors and their coactivators can activate the murine PU.1 (Spi-1) promoter. J Biol Chem. 1996;271:15743–52. doi: 10.1074/jbc.271.26.15743. [DOI] [PubMed] [Google Scholar]
- 57.Eklund EA, Kakar R. Recruitment of CREB-binding protein by PU.1, IFN-regulatory factor-1, and the IFN consensus sequence-binding protein is necessary for IFN-g-induced p67phox and gp91phox expression. J Immunol. 1999;163:6095–105. [PubMed] [Google Scholar]
- 58.Cross MA, Heyworth CM, Murrell AM, et al. Expression of lineage restricted transcription factors precedes lineage specific differentiation in a multipotent hematopoietic progenitor cell line. Oncogene. 1994;9:3013–6. [PubMed] [Google Scholar]
- 59.Tenen DG, Hromas R, Licht JD, et al. Transcription factors, normal myeloid development, and leukemia. Blood. 1997;90:489–519. [PubMed] [Google Scholar]
- 60.Li Y, Okuno Y, Zhang P, et al. Regulation of the PU.1 gene by distal elements. Blood. 2001;98:2958–65. doi: 10.1182/blood.v98.10.2958. [DOI] [PubMed] [Google Scholar]
- 61.Okuno Y, Huang G, Rosenbauer F, et al. Potential autoregulation of transcription factor PU.1 by an upstream regulatory element. Mol Cell Biol. 2005;25:2832–45. doi: 10.1128/MCB.25.7.2832-2845.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schroeder T, Kohlhof H, Rieber N, et al. Notch signaling induces multilineage myeloid differentiation and up-regulates PU.1 expression. J Immunol. 2003;170:5538–48. doi: 10.4049/jimmunol.170.11.5538. [DOI] [PubMed] [Google Scholar]
- 63.Oelgeschlager M, Nuchprayoon I, Luscher B, et al. C/EBP, c-Myb and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol. 1996;16:4717–25. doi: 10.1128/mcb.16.9.4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kehrl JH. Hematopoietic lineage commitment: role of transcription factors. Stem Cells. 1995;13:223–41. doi: 10.1002/stem.5530130304. [DOI] [PubMed] [Google Scholar]
- 65.Zhang P, Behre G, Pan J, et al. Negative cross-talk between hematopoietic regulators: GATA-1 proteins repress PU.1. Proc Natl Acad Sci USA. 1999;96:8705–10. doi: 10.1073/pnas.96.15.8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Moldonado E, Hampsey M, Reinberg D. Repression: targeting the heart of the matter. Cell. 1999;99:455–8. doi: 10.1016/s0092-8674(00)81533-0. [DOI] [PubMed] [Google Scholar]
- 67.Konishita S, Akira S, Kishimoto T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc Natl Acad Sci USA. 1992;89:1473–6. doi: 10.1073/pnas.89.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang Z, Wara-aswapati N, Chen C, et al. NF-IL6 (C/EBPbeta) vigorously activates IL1b gene expression via a Spi-1 (PU.1) protein-protein tether. J Biol Chem. 2000;275:21272–7. doi: 10.1074/jbc.M000145200. [DOI] [PubMed] [Google Scholar]
- 69.Bohmann D, Bos TJ, Admon A, et al. Human proto-oncogene c-jun encodes a DNA binding protein with structural and functional properties of transcription factor AP-1. Science. 1987;238:1386–92. doi: 10.1126/science.2825349. [DOI] [PubMed] [Google Scholar]
- 70.Whitmarsh AJ, Davis RJ. Transcription factor AP-1 regulation by mitogen-activated protein kinase signal transduction pathways. J Mol Med. 1996;74:589–607. doi: 10.1007/s001090050063. [DOI] [PubMed] [Google Scholar]
- 71.Lord KA, Abdollahi A, Hoffman-Liebermann B, et al. Proto-oncogenes of the fos/jun family of transcription factors are positive regulators of myeloid differentiation. Mol Cell Biol. 1993;13:841–51. doi: 10.1128/mcb.13.2.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mollinedo F, Gajate C, Tugores A, et al. Differences in expression of transcription factor AP-1 in human promyelocytic HL-60 cells during differentiation towards macrophages versus granulocytes. Biochem J. 1993;294:137–44. doi: 10.1042/bj2940137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li AC, Guidez FRB, Collier JG, et al. The macrosialin promoter directs high levels of transcriptional activity in macrophages dependent on combinatorial interactions between PU.1 and c-Jun. J Biol Chem. 1998;273:5389–99. doi: 10.1074/jbc.273.9.5389. [DOI] [PubMed] [Google Scholar]
- 74.Hass R, Prudovsky I, Kruhoffer M. Differential effects of phorbol ester on signaling and gene expression in human leukemia cells. Leuk Res. 1997;21:589–94. doi: 10.1016/s0145-2126(97)00010-6. [DOI] [PubMed] [Google Scholar]
- 75.Sistonen L, Holtta E, Makela TP, et al. The cellular response to induction of the p21 c-Ha-ras oncoprotein includes stimulation of jun gene expression. EMBO J. 1989;8:815–22. doi: 10.1002/j.1460-2075.1989.tb03442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Janknecht R, Hunter T. Transcription: a growing coactivator network. Nature. 1996;383:22–3. doi: 10.1038/383022a0. [DOI] [PubMed] [Google Scholar]
- 77.Ogryzko VV, Schiltz LR, Russanova V, et al. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell. 1996;87:953–9. doi: 10.1016/s0092-8674(00)82001-2. [DOI] [PubMed] [Google Scholar]
- 78.Yamamoto H, Kihara-Negishi F, Yamada T, et al. Physical and functional interactions between the transcription factor PU.1 and the coactivator CBP. Oncogene. 1999;18:1495–501. doi: 10.1038/sj.onc.1202427. [DOI] [PubMed] [Google Scholar]
- 79.Blobel GA, Nakajima T, Eckner R, et al. CREB-binding protein (CBP) cooperates with transcription factor GATA-1 and is required for erythroid differentiation. Proc Natl Acad Sci USA. 1998;95:2061–6. doi: 10.1073/pnas.95.5.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hung HL, Lau J, Kim AY, et al. CREB-binding protein (CBP) acetylates hematopoietic transcription factor GATA-1 at functionally important sites. Mol Cell Biol. 1999;19:3496–505. doi: 10.1128/mcb.19.5.3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hong W, Kim AY, Ky S, et al. Inhibition of CBP-mediated protein acetylation by the Ets family oncoprotein PU.1. Mol Cell Biol. 2002;22:3729–43. doi: 10.1128/MCB.22.11.3729-3743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yamada T, Kihara-Negishi F, Yamamoto H, et al. Reduction of DNA binding activity of the GATA-1 transcription factor in the apoptotic process induced by overexpression of PU.1 in murine erythroleukemia cells. Exp Cell Res. 1998;245:186–94. doi: 10.1006/excr.1998.4251. [DOI] [PubMed] [Google Scholar]
- 83.Zhang P, Zhang X, Iwama A, et al. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000;96:2641–8. [PubMed] [Google Scholar]
- 84.Wall L, DeBoer E, Grosveld F. The human beta-globin gene 3′enhancer contains multiple binding sites for an erythroid-specific protein. Genes Dev. 1988;2:1089–100. doi: 10.1101/gad.2.9.1089. [DOI] [PubMed] [Google Scholar]
- 85.Anguita E, Hughes J, Heyworth C, et al. Globin gene activation during haemopoiesis is driven by protein complexes nucleated by GATA-1 and GATA-2. EMBO J. 2004;23:2841–52. doi: 10.1038/sj.emboj.7600274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zon LI, Youssoufian H, Mather C, et al. Activation of the erythropoietin receptor promoter by transcription factor GATA-1. Proc Natl Acad Sci USA. 1991;88:10638–41. doi: 10.1073/pnas.88.23.10638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Anderson KP, Crable SC, Lingrel JB. Multiple proteins binding to a GATA-E box-GATA motif regulate the erythroid Kruppel-like factor (EKLF) gene. J Biol Chem. 1998;273:14347–54. doi: 10.1074/jbc.273.23.14347. [DOI] [PubMed] [Google Scholar]
- 88.Boulanger L, Sabatino DE, Wong EY, et al. Erythroid expression of the human a-spectrin gene promoter is mediated by GATA-1- and NF-E2-binding proteins. J Biol Chem. 2002;277:41563–70. doi: 10.1074/jbc.M208184200. [DOI] [PubMed] [Google Scholar]
- 89.Minami T, Tachibana K, Imanishi T, et al. Both Ets-1 and GATA-1 are essential for positive regulation of platelet factor 4 gene expression. Eur J Biochem. 1998;258:879–89. doi: 10.1046/j.1432-1327.1998.2580879.x. [DOI] [PubMed] [Google Scholar]
- 90.Martin F, Prandini MH, Thevenon D, et al. The transcription factor GATA-1 regulates the promoter activity of the platelet glycoprotein IIb gene. J Biol Chem. 1993;268:21606–12. [PubMed] [Google Scholar]
- 91.Fujiwara Y, Browne CP, Cunniff K, et al. Arrested development of embryonic red cell precursors in mouse embryos lacking transcription factor GATA-1. Proc Natl Acad Sci USA. 1996;93:12355–8. doi: 10.1073/pnas.93.22.12355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr Opin Genet Dev. 2001;11:513–9. doi: 10.1016/s0959-437x(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 93.Matushansky I, Radparvar F, Skoultchi AI. Reprogramming leukemic cells to terminal differentiation by inhibiting specific cyclin-dependent kinases in G1. Proc Natl Acad Sci USA. 2000;97:14317–22. doi: 10.1073/pnas.250488697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park DJ, Chumakov AM, Vuong PT, et al. CCAAT/enhancer binding protein epsilon is a potential retinoid target gene in acute promyelocytic leukemia treatment. J Clin Invest. 1999;103:1399–408. doi: 10.1172/JCI2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Du J, Stankiewicz MJ, Liu Y, et al. Novel combinatorial interactions of GATA-1, PU.1 and C/EBP isoforms regulate transcription of the gene encoding eosinophil granule major basic protein. J Biol Chem. 2002;277:43481–94. doi: 10.1074/jbc.M204777200. [DOI] [PubMed] [Google Scholar]
- 96.Trainor CD, Ghirlando R, Simpson MA. GATA zinc finger interactions modulate DNA binding and transactivation. J Biol Chem. 2000;275:28157–66. doi: 10.1074/jbc.M000020200. [DOI] [PubMed] [Google Scholar]
- 97.Martin DI, Orkin SH. Transcriptional activation and DNA binding by the erythroid factor GF-1/NF-E1/Eryf 1. Genes Dev. 1990;4:1886–98. doi: 10.1101/gad.4.11.1886. [DOI] [PubMed] [Google Scholar]
- 98.Tsai SF, Strauss E, Orkin SH. Functional analysis and in vivo footprinting implicate the erythroid transcription factor GATA-1 as a positive regulator of its own promoter. Genes Dev. 1991;5:919–31. doi: 10.1101/gad.5.6.919. [DOI] [PubMed] [Google Scholar]
- 99.Evans T, Felsenfeld G. Trans-activation of a globin promoter in nonerythroid cells. Mol Cell Biol. 1991;11:843–53. doi: 10.1128/mcb.11.2.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Landschulz WH, Johnson PF, McKnight SL. The leucine zipper: a hypothetical structure common to a new class of DNA binding proteins. Science. 1988;240:1759–64. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 101.Radomska HS, Huettner CS, Zhang P, et al. CCAAT/enhancer binding protein α is a regulatory switch sufficient for induction of granulocytic development from bipotential myeloid progenitors. Mol Cell Biol. 1998;18:4301–14. doi: 10.1128/mcb.18.7.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhang DE, Zhang P, Wang ND, et al. Absence of granulocyte colony-stimulating factor signaling and neutrophil development in CCAAT enhancer binding protein alpha-deficient mice. Proc Natl Acad Sci USA. 1997;94:569–74. doi: 10.1073/pnas.94.2.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dahl R, Walsh JC, Lancki D, et al. Regulation of macrophage and neutrophil cell fates by the PU.1: C/EBPalpha ratio and granulocyte colony-stimulating factor. Nat Immunol. 2003;4:1029–36. doi: 10.1038/ni973. [DOI] [PubMed] [Google Scholar]
- 104.McIvor Z, Hein S, Fiegler H, et al. Transient expression of PU.1 commits multipotent progenitors to a myeloid fate whereas continued expression favors macrophage over granulocyte differentiation. Exp Hematol. 2003;31:39–47. doi: 10.1016/s0301-472x(02)01017-2. [DOI] [PubMed] [Google Scholar]
- 105.Laslo P, Spooner CJ, Warmflash A, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–66. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 106.Reddy VA, Iwama A, Iotzova G, et al. The granulocytic inducer C/EBPα inactivates the myeloid master gene PU.1: possible role in lineage commitment decisions. Blood. 2002;100:483–90. doi: 10.1182/blood.v100.2.483. [DOI] [PubMed] [Google Scholar]
- 107.Rangatia J, Vangala RK, Treiber N, et al. Downregulation of c-Jun expression by transcription factor C/EBPα is critical for granulocytic lineage commitment. Mol Cell Biol. 2002;22:8681–94. doi: 10.1128/MCB.22.24.8681-8694.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nerlov C, Graf T. PU.1 induces myeloid lineage commitment in multipotent hematopoietic progenitors. Genes Dev. 1998;12:2403–12. doi: 10.1101/gad.12.15.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene. 1999;18:3017–33. doi: 10.1038/sj.onc.1202839. [DOI] [PubMed] [Google Scholar]
- 110.Suzow J, Friedman AD. The murine myeloperoxidase promoter contains several functional elements, one, which binds a cell type-restricted transcription factor, myeloid nuclear factor 1 (MyNF1) Mol Cell Biol. 1993;13:2141–51. doi: 10.1128/mcb.13.4.2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zhu J, Bennett CA, MacGregor AD, et al. A myeloid-lineage-specific enhancer upstream of the mouse myeloperoxidase (MPO) gene. Leukemia. 1994;8:717–23. [PubMed] [Google Scholar]
- 112.Herschlag D, Johnson FB. Synergism in transcriptional activation: a kinetic view. Genes Dev. 1993;7:173–9. doi: 10.1101/gad.7.2.173. [DOI] [PubMed] [Google Scholar]
- 113.Nuchprayoon I, Meyers S, Scott LM, et al. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2b/CBFb oncoproteins, regulates the murine MPO and neutrophil elastase genes in immature myeloid cells. Mol Cell Biol. 1994;14:5558–68. doi: 10.1128/mcb.14.8.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Vengala RK, Heiss-Neumann MS, Rangatia JS, et al. The myeloid master regulator transcription factor PU.1 is inactivated by AML-ETO in t(8; 21) myeloid leukemia. Blood. 2003;101:270–7. doi: 10.1182/blood-2002-04-1288. [DOI] [PubMed] [Google Scholar]
- 115.Westendorf JJ, Yamamoto CM, Lenny N, et al. The t(8; 21) fusion product, AML-1-ETO, associates with C/EBPα, inhibits C/EBPα-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol. 1998;18:322–33. doi: 10.1128/mcb.18.1.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Marecki S, Fenton MJ. PU.1/Interferon regulatory factor interactions: mechanisms of transcriptional regulation. Cell Biochem Biophys. 2000;33:127–48. doi: 10.1385/CBB:33:2:127. [DOI] [PubMed] [Google Scholar]
- 117.Brass AL, Kehrli E, Eisenbeis CF, et al. Pip, a lymphoid-restricted IRF, contains a regulatory domain that is important for autoinhibition and ternary complex formation with the Ets factor PU.1. Genes Dev. 1996;10:2335–47. doi: 10.1101/gad.10.18.2335. [DOI] [PubMed] [Google Scholar]
- 118.Perkel JM, Atchison ML. A two-step mechanism for recruitment of Pip by PU.1. J Immnuol. 1998;160:241–52. [PubMed] [Google Scholar]
- 119.Brass AL, Zhu AQ, Singh H. Assembly requirements of PU.1-Pip (IRF-4) activator complexes: inhibiting function in vivo using fused dimmers. EMBO J. 1999;18:977–91. doi: 10.1093/emboj/18.4.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kanno Y, Kozak CA, Schindler C, et al. The genomic structure of the murine ICSBP gene reveals the presence of the gamma interferon-responsive element, to which an ISGF3 alpha subunit (or similar) molecule binds. Mol Cell Biol. 1993;13:3951–63. doi: 10.1128/mcb.13.7.3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Muzzey D, Qudenaarden AV. When it comes to decisions, myeloid progenitors crave positive feedback. Cell. 2006;126:650–2. doi: 10.1016/j.cell.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 122.Mueller BU, Pabst T, Fos J, et al. ATRA resolves the differentiation block in t(15; 17) acute myeloid leukemia by restoring PU.1 expression. Blood. 2006;107:3330–8. doi: 10.1182/blood-2005-07-3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Li R, Pei H, Watson DK. Regulation of Ets function by protein-protein interactions. Oncogene. 2000;19:6514–23. doi: 10.1038/sj.onc.1204035. [DOI] [PubMed] [Google Scholar]
- 124.Walsh JC, DeKoter RP, Lee HJ, et al. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17:665–76. doi: 10.1016/s1074-7613(02)00452-1. [DOI] [PubMed] [Google Scholar]
- 125.Rehli M, Poltorak A, Schwarzfischer L, et al. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 Gene. J Biol Chem. 2000;275:9773–81. doi: 10.1074/jbc.275.13.9773. [DOI] [PubMed] [Google Scholar]