

Abstract
Emerging studies indicate that hypothalamic hormonal signalling pathways and nutrient metabolism regulate glucose homeostasis in rodents. Although hypothalamic lactate-sensing mechanisms have been described to lower glucose production (GP), it is currently unknown whether the hypothalamus senses lactate in the blood circulation to regulate GP and maintain glucose homeostasis in vivo. To examine whether hypothalamic sensing of circulating lactate is required to regulate GP, we infused intravenous (i.v.) lactate in the absence or presence of inhibition of central/hypothalamic lactate-sensing mechanisms in normal rodents. Inhibition of central/hypothalamic lactate-sensing mechanisms was achieved by three independent approaches. Tracer-dilution methodology in combination with the pancreatic clamp technique was used to assess the effect of i.v. and central/hypothalamic administrations on glucose metabolism in vivo. In the presence of physiologically relevant increases in the levels of plasma lactate, inhibition of central lactate-sensing mechanisms by lactate dehydrogenase inhibitor oxamate (OXA) or ATP-sensitive potassium channels blocker glibenclamide increased GP. Furthermore, direct administration of OXA into the mediobasal hypothalamus increased GP in the presence of similar elevation of circulating lactate. Together, these data indicate that hypothalamic sensing of circulating lactate regulates GP and is required to maintain glucose homeostasis.
Keywords: brain, nutrient-sensing, glucose homeostasis
Introduction
Glucose production (GP) is regulated by gluconeogenesis and glycogenolysis. In type 2 diabetes, elevation of GP leads to fasting hyperglycaemia [1]. An elevation of plasma gluconeogenic substrate precursors such as free fatty acids (FFA) or lactate is also seen in type 2 diabetes [2–5]. These findings led to the working hypothesis that elevation of gluconeogenic substrate precursors increases gluconeogenesis and GP. This hypothesis was tested in human beings, dogs and rodents in vivo. The findings as a whole indicate that during the pancreatic clamp, when gluco-regulatory hormones and glucose levels are maintained at basal, intravenous (i.v.) infusion of FFA or lactate increases hepatic gluconeogensis but, contrary to the hypothesis, not GP because of a compensatory inhibition of glycogenolysis [6–9]. The underlying ‘nutrient-sensing’ mechanisms that inhibit glycogenolysis and counteract the direct effects of circulating FFA or lactate on hepatic gluconeogenesis remain to be explored. In this study, we began to assess the underlying nutrient-sensing mechanisms that counteract the direct effect of circulating lactate on hepatic gluconeogenesis in vivo.
The lipid-sensing mechanisms that counteract the direct effect of circulating FFA on hepatic gluconeogenesis have recently been studied [9]. It is demonstrated that the inhibition of hepatic glycogenolysis induced by circulating FFA to restrain GP is mediated by hypothalamic lipid-sensing mechanisms in rodents [9]. The hypothalamic lipid-sensing mechanisms to restrain glycogenolysis and GP are disrupted in diet-induced insulin resistance and obesity, leading to hyperglycaemia in response to systemic lipid infusions [10]. This is in line with previous reports indicating that in patients with metabolic stress conditions such as type 2 diabetes, reciprocal changes in glycogenolysis do not compensate for changes in gluconeogenesis when plasma FFA concentrations are experimentally manipulated [11].
Unlike the lipid-sensing mechanisms in the body, even in metabolic stress patients with hyperglycaemia, an elevation of circulating lactate still does not increase GP because of an inhibition of glycogenolysis [12]. Consistent with this, in contrast to hypothalamic lipid-sensing mechanisms [13], activation of central lactate metabolism via direct delivery of lactate into the hypothalamus lowers GP in normal and in early onset of diabetic and obese rodents [14, 15]. On the basis of these observations, we postulate that (i) the hypothalamic-sensing mechanism of circulating lactate that regulate GP is intact in normal, diabetic and obese individuals, and (ii) these hypothalamic ‘nutrient-sensing’ mechanisms are designed to counteract the direct stimulatory effect of circulating lactate on hepatic gluconeogenesis to maintain glucose homeostasis. Given that the ability of the hypothalamus to sense circulating lactate to regulate GP and maintain glucose homeostasis is yet to be evaluated, this will be the first study to address our working hypothesis in normal rodents.
To examine whether the hypothalamus senses circulating lactate to regulate GP and maintain glucose homeostasis in vivo, we infused i.v. L-lactate to elevate plasma lactate levels in the presence or absence of direct inhibition of brain lactate-sensing mechanisms. To directly inhibit central/hypothalamic lactate-sensing mechanisms, we independently infused (i) lactate dehydrogenase inhibitor oxamate (OXA) into the third cerebral ventricle (intracerebroventricular – i.c.v.), (ii) i.c.v. ATP-sensitive potassium (KATP) channel blocker glibenclamide (GLI) or (iii) OXA directly into the mediobasal hypothalamus (MBH) (Fig. 1A and B) that has been previously described to prevent the inhibitory effects of central lactate administration on GP [15]. Tracer-dilution methodology and the pancreatic clamp technique were used to assess the effect of i.v. and i.c.v./MBH administrations on whole body glucose kinetics independent of changes in circulating gluco-regulatory hormones. Based on this cross-discipline experimental approach, we provided the first evidence, to our knowledge, that direct inhibition of central/hypothalamic lactate-sensing mechanisms by three independent methods increased GP and disrupted glucose homeostasis in response to systemic lactate elevations in vivo.
Figure 1.
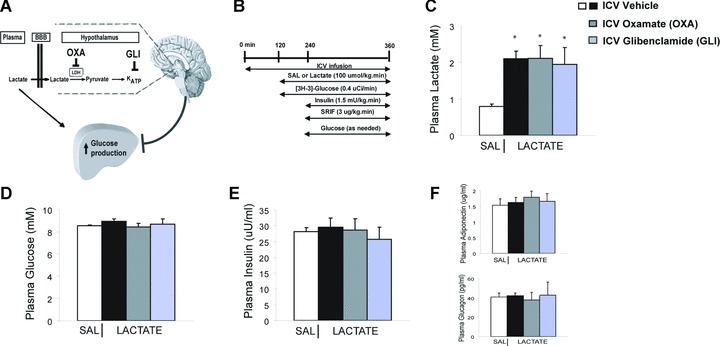
(A) Working hypothesis. The hypothalamic metabolism of lactate to pyruvate and the subsequent activation of the KATP channels are required to maintain glucose homeostasis in response to systemic lactate elevations. Lactate dehydrogenase inhibitor oxamate. KATP channel blocker glibenclamide. (B) Experimental design. Somatostatin (SRIF). During the final 30 min. of the clamps, (C) intravenous lactate infusion elevated plasma lactate levels compared to control (saline, SAL) by ∼2- to 2.5-fold. *P < 0.001 versus SAL. (D) Plasma glucose, (E) plasma insulin, (F) plasma adiponectin and glucagon levels were comparable in all groups during the clamps.
Methods
Animal preparation
Adult 8-week-old male Sprague Dawley rats (250–280 g) were obtained from Charles River Laboratories (Montreal, QC, Canada) and maintained on a standard light-dark cycle with access to rat chow and water ad libitum. Rats underwent stereotaxic surgery to insert single catheters in the third cerebral ventricle (i.c.v.) or bilateral catheters into the MBH 2 weeks before the experiments in vivo as previously described [14, 16, 17]. One week later, catheters were placed in the internal jugular vein and the carotid artery for infusion and sampling during the clamp procedures as previously described [14, 16, 18]. Recovery from surgery was monitored by measuring daily food intake and weight gain for 4–5 days after surgery. The study animal protocol was approved by the Institutional Animal Care and Use Committee of the University Health Network.
Clamp procedure in rats
All the rats were restricted to 20 g of food the night before the experiments to ensure the same nutritional status. To groups of conscious unrestrained rats, we infused i.v. sodium L-lactate (100 μmol/kg min., pH 7.0) to elevate plasma lactate concentrations by ∼2- to 2.5-fold as seen in exercise for 4 hrs (Fig. 1A–C). Central (i.c.v.) infusions consisted of the (i) lactate dehydrogenase inhibitor OXA (dissolved in artificial cerebrospinal fluid [ACSF] to 50 mM [5 μl/hr] and was first given as an i.c.v. bolus [3 μl]), (ii) KATP channel blocker GLI (dissolved in 5% DMSO to 100 μM; 5 μl/hr) or (iii) vehicle (5% DMSO or ACSF; 5 μl/hr) (Fig. 1B). MBH infusions consisted of (i) OXA (50 mM; 0.33 μl/hr and was first given as a MBH bolus [0.33 μl]) or (ii) vehicle (ACSF; 0.33 μl/hr) (Fig. 3A). It has been demonstrated that i.c.v. OXA or GLI and MBH OXA administered at these concentrations alone do not affect glucose kinetics but were sufficient to abolish the GP-lowering effect of direct brain lactate administrations [15]. The infusion studies lasted a total of 360 min. (Figs 1B and 3A). Briefly, i.c.v. vehicle (VEH) or OXA or GLI (Fig. 1B) and MBH VEH or OXA (Fig. 3A) were infused throughout the experiments. After 2 hrs of i.c.v. or MBH infusions, i.v. saline (SAL) or L-lactate infusion and a primed-continuous infusion of [3-3H]-glucose (Perkin Elmer, WoodBridge, ON, Canada; 40 μCi bolus; 0.4 μCi/min) were initiated at 120 min. and maintained throughout the study to assess the rate of GP and glucose uptake based on the tracer-dilution methodology. Samples for determination of [3-3H]-glucose specific activity were obtained at 10-min. intervals. From time 180–240 min. (steady-state basal period), the rate of GP (mg/kg min.) and plasma glucose levels (mM) were 11.9 ± 0.5 and 8.3 ± 0.1 (i.c.v. vehicle + i.v. saline), 12.7 ± 0.2 and 8.0 ± 0.2 (i.c.v. vehicle + i.v. lactate), 12.8 ± 0.2 and 8.5 ± 0.2 (i.c.v. OXA + i.v. lactate), 12.4 ± 0.6 and 8.3 ± 0.1 (i.c.v. GLI + i.v. lactate), 12.6 ± 0.2 and 8.2 ± 0.2 (MBH vehicle + i.v. lactate) and 12.2 ± 1.2 and 8.9 ± 0.6 (MBH OXA + i.v. lactate). A pancreatic clamp was performed in the final 2 hrs of the study starting at 240 min; a continuous infusion of insulin (1.5 mU/kg min.) and somatostatin (3 μg/kg min.) was administered, and a variable infusion of a 25% glucose solution was started and periodically adjusted to maintain the plasma glucose concentration at ∼8 mM. Control groups (i.e. i.c.v. OXA + i.v. saline or i.c.v. GLI + i.v. saline) were performed, and the GP during the clamps in both groups (GP: i.c.v. OXA + i.v. saline, 6.0 ± 0.8 or i.c.v. GLI + i.v. saline, 6.2 ± 0.9) was comparable to i.c.v. vehicle + i.v. saline (Fig. 2B). Plasma samples for the determination of lactate, insulin, adiponectin and glucagon concentrations were obtained at 10-min. intervals for the final 30 min. of the infusion studies. The number of infusion experiments performed in each group was n= 8 (i.c.v. vehicle + i.v. saline), n= 6 (i.c.v. vehicle + i.v. lactate), n= 5 (i.c.v. OXA + i.v. lactate), n= 5 (i.c.v. GLI + i.v. lactate), n= 5 (MBH vehicle + i.v. lactate), n= 5 (MBH OXA + i.v. lactate).
Figure 3.

Hypothalamic sensing of circulating lactate regulates glucose production (GP). (A) Experimental design. Somatostatin. In the presence of systemic lactate elevation, direct inhibition of lactate metabolism within the mediobasal hypothalamus (MBH) via MBH administration of oxamate (compared to control) (B) decreased exogenous glucose infusion rate and (C) increased GP during the final 30 min. of clamps. (D) Glucose utilization/uptake was comparable in all groups. *P < 0.001 versus lactate with MBH vehicle.
Figure 2.

Central sensing mechanisms of circulating lactate regulate glucose production (GP). In the presence of systemic elevation of lactate, direct inhibition of central lactate metabolism via i.c.v. administration of lactate dehydrogenase inhibitor oxamate (OXA) (compared to control) led to a marked (A) decrease in exogenous glucose infusion rate and (B) increase in GP during the final 30 min. of the clamps. Similarly, direct inhibition of central KATP channels via i.c.v. administration of blocker glibenclamide (A) decreased exogenous glucose infusion rate and (B) increased GP in response to systemic lactate infusions. (C) Glucose utilization/uptake was comparable in all groups. *P < 0.001 versus SAL with i.c.v. vehicle or lactate with i.c.v. vehicle.
Biochemical analyses
Plasma lactate concentrations were determined by a kit in accordance with manufacturer’s instructions (Sigma Diagnostics. St. Louis, MO, USA). Plasma glucose concentrations were measured by the glucose oxidase method (Glucose Analyzer, Analox Instruments, Lunenberg, MA, USA). Plasma insulin concentrations were measured by radioimmunoassay (RIA). Plasma adiponectin concentrations were measured by RIA (Linco Inc., Austin, TX, USA). Plasma Glucagon concentrations were measured by RIA (Linco Research, St. Charles, MO, USA).
Statistical analysis
Statistical analysis was done by ANOVA or unpaired Student’s t-test as appropriate. Statistical analysis was accepted as significant with a P-value less than 0.05. Data are presented as means + S.E.M.
Result
Here we first examined whether selectively blocking the central effects of lactate through the i.c.v. administration of lactate dehydrogenase inhibitor OXA or KATP channel blocker GLI is sufficient to alter the metabolic response of i.v. lactate infusion on GP and disrupt glucose homeostasis (Fig. 1A). Systemic infusion of lactate at 100 μmol/kg min. for 4 hrs elevated plasma lactate by ∼2- to 2.5-fold compared to i.v. saline infusion in normal rodents in vivo (Fig. 1C). In the final 2 hrs of the lactate elevation, we performed pancreatic-insulin clamps with tracer dilution methodology to assess the effects of i.v. lactate on glucose kinetics independent of changes in plasma gluco-regulatory hormones (Fig. 1B). Consistent with previous findings in human beings [8], i.v. lactate infusion did not increase GP during the pancreatic clamps when plasma glucose (Fig. 1D) and gluco-regulatory hormones (Fig. 1E and F) were maintained near basal levels. To assess whether central sensing mechanisms restrain plasma lactate from elevating GP, we administered i.c.v. OXA 2 hrs prior and maintained throughout the i.v. lactate infusions. In the same experimental conditions as i.v. saline/lactate with i.c.v. vehicle (Fig. 1C–F), central OXA administration decreased exogenous glucose infusion rate required to maintain euglycaemia (Fig. 2A) in the presence of similar degree of plasma lactate elevation (Fig. 1C). This decrease in glucose infusion rate was due entirely to an elevation of GP (Fig. 2B) rather than a change in glucose uptake (Fig. 2C). Together, these data indicate that direct inhibition of central lactate metabolism disrupted glucose homeostasis and increased GP in the presence of systemic lactate elevations.
To complement the function findings obtained with i.c.v. OXA, we administered i.c.v. KATP channels blocker GLI to alternatively negate central lactate effects and examine whether central sensing mechanisms restrain plasma lactate to increase GP. Similar to the effects of i.c.v. OXA, central GLI administration decreased the exogenous glucose infusion rate required to maintain euglycaemia (Fig. 2A) in the presence of similar degree of plasma lactate elevation as observed in the other groups (Fig. 1C). The drop in glucose infusion rate was caused by an increase in GP (Fig. 2B) and not a change in glucose uptake (Fig. 2C). These findings indicate that direct inhibition of central KATP channels disrupted glucose homeostasis in response to systemic lactate elevation.
To investigate the central neuroanatomical localization of the CNS-sensing mechanisms of circulating lactate on GP, we negated the lactate-sensing mechanisms specifically within the MBH. We combined i.v. lactate infusions (100 μmol/kg min.) with the administration of OXA (50 mM) bilaterally into the MBH (Fig. 3A). We first confirmed that similar to i.c.v. vehicle, MBH vehicle did not affect glucose kinetics in the presence of i.v. lactate infusions (Fig. 3B–D). In contrast, MBH OXA administration decreased glucose infusion rate required to maintain euglycaemia in the same degree of plasma lactate elevation (Fig. 3B). Again, the decrease in glucose infusion rate was due to increased GP (Fig. 3C), rather than a change in glucose uptake (Fig. 3D). Thus, direct inhibition of lactate metabolism within the MBH disrupted glucose homeostasis and increased GP in the presence of systemic lactate infusions. It remains to be assessed whether the hypothalamic restraining effect on GP in response to circulating lactate is mediated by an inhibition of hepatic glycogenolysis and/or gluconeogenesis.
Discussion
The hypothalamus has been recently demonstrated to detect a rise in both hormones and nutrients to regulate peripheral metabolic processes [9, 15, 19–29]. Specifically, direct activation of either (i) insulin/leptin signalling pathways [19, 21–25, 28, 29] or (ii) lipid/lactate metabolism [9, 15, 30] in the hypothalamus regulates GP. Furthermore, the demonstration of the ability of the hypothalamus to sense circulating lipids to lower hepatic glycogenolysis and restrain GP (9) has added to the existing knowledge of nutrient-sensing mechanisms in the body. These findings, at least in rodents, suggest that the hypothalamus senses circulating lipids to counteract the direct stimulatory effects of lipids on hepatic gluconeogenesis. More importantly, the hypothalamic lipid-sensing mechanisms are disrupted in the early onset of diet-induced obese rodents, leading to a rise in GP and glucose levels induced by circulating lipids [10]. These observations, if extended to human beings, could in part begin to address the inability of the body to compensate for a lipid-induced increase in gluconeogenesis in type 2 diabetes [11].
In direct contrast to lipid-sensing mechanisms, the body has the ability to inhibit hepatic glycogenolysis and restrain GP to compensate for the circulating lactate-induced increase in hepatic gluconeogenesis in both normal individuals and metabolic stress individuals with hyperglycaemia [8, 12]. In an attempt to elucidate the underlying mechanisms of lactate-sensing that are responsible for this metabolic restraining effect on GP, we assessed whether the hypothalamus senses an elevation of plasma lactate by 2- to 2.5-fold for 4 hrs to restrain GP and maintain glucose homeostasis in normal rodents. We first evaluated the metabolic effects of i.v. lactate infusion on glucose metabolism during pancreatic clamps when gluco-regulatory hormones (Fig. 1E and F) and glucose (Fig. 1D) were maintained near basal levels. Consistent with previous reports in human beings [8], an elevation of circulating gluconeogenic substrate precursor lactate did not increase GP. We postulated that the hypothalamic lactate-sensing mechanisms are responsible for this glucose homeostatic control. If this is true, direct inhibition of CNS/hypothalamic lactate-sensing mechanisms should disrupt glucose homeostasis and lead to an elevation of GP in the presence of systemic lactate infusion. To test this hypothesis, we directly inhibited CNS/hypothalamic lactate-sensing mechanisms that regulate GP by three independent approaches. During the pancreatic clamps, all three experimental interventions in the brain (comparing to control) led to a marked increase in GP in response to systemic lactate infusion. Together, these data indicated that hypothalamic sensing mechanisms restrain systemic lactate to increase GP and is required to maintain glucose homeostasis.
This is the first report that demonstrates the ability of the hypothalamus to sense circulating lactate to restrain GP and maintain glucose homeostasis in normal rodents. In light of the fact that (i) direct central lactate administration lowers GP in normal and early onset of diabetic and obese rodents [14, 15], (ii) an elevation of circulating lactate does not increase GP in normal individuals and in patients with hyperglycaemia [8, 12], we postulate that the hypothalamic nutrient sensing mechanisms of circulating lactate are intact in at least the early onset of diabetes and obesity. However, future studies are required to address this hypothesis. In summary, our data suggest that therapeutic strategies designed to activate CNS lactate-sensing mechanisms by physiological route could eventually be proven useful to maintain glucose homeostasis in vivo.
Acknowledgments
This work was supported by a research grant to T.K.T.L. from the Canadian Institute of Health Research (MOP-86554). G.W.C.C. is supported by the Banting and Best Diabetes Centre Summer Studentship at the University of Toronto. T.K.T.L. holds the John Kitson McIvor Chair in Diabetes Research at the University Health Network and University of Toronto.
References
- 1.Taylor SI. Deconstructing type 2 diabetes. Cell. 1999;97:9–12. doi: 10.1016/s0092-8674(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 2.Reaven GM, Hollenbeck C, Jeng CY, et al. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–4. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 3.Felig P, Wahren J, Hendler R. Influence of maturity-onset diabetes on splanchnic glucose balance after oral glucose ingestion. Diabetes. 1978;27:121–6. doi: 10.2337/diab.27.2.121. [DOI] [PubMed] [Google Scholar]
- 4.Hall SE, Saunders J, Sonksen PH. Glucose and free fatty acid turnover in normal subjects and in diabetic patients before and after insulin treatment. Diabetologia. 1979;16:297–306. doi: 10.1007/BF01223618. [DOI] [PubMed] [Google Scholar]
- 5.Consoli A, Nurjhan N, Capani F, et al. Predominant role of gluconeogene-sis in increased hepatic glucose production in NIDDM. Diabetes. 1989;38:550–7. doi: 10.2337/diab.38.5.550. [DOI] [PubMed] [Google Scholar]
- 6.Chen X, Iqbal N, Boden G. The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J Clin Invest. 1999;103:365–72. doi: 10.1172/JCI5479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chu CA, Sherck SM, Igawa K, et al. Effects of free fatty acids on hepatic glycogenolysis and gluconeogenesis in conscious dogs. Am J Physiol Endocrinol Metab. 2002;282:E402–11. doi: 10.1152/ajpendo.00136.2001. [DOI] [PubMed] [Google Scholar]
- 8.Jenssen T, Nurjhan N, Consoli A, et al. Failure of substrate-induced gluconeogenesis to increase overall glucose appearance in normal humans. Demonstration of hepatic autoregulation without a change in plasma glucose concentration. J Clin Invest. 1990;86:489–97. doi: 10.1172/JCI114735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam TK, Pocai A, Gutierrez-Juarez R, et al. Hypothalamic sensing of circulating fatty acids is required for glucose homeostasis. Nat Med. 2005;11:320–7. doi: 10.1038/nm1201. [DOI] [PubMed] [Google Scholar]
- 10.Pocai A, Lam TK, Obici S, et al. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J Clin Invest. 2006;116:1081–91. doi: 10.1172/JCI26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boden G, Chen X, Capulong E, et al. Effects of free fatty acids on gluconeogenesis and autoregulation of glucose production in type 2 diabetes. Diabetes. 2001;50:810–6. doi: 10.2337/diabetes.50.4.810. [DOI] [PubMed] [Google Scholar]
- 12.Tappy L, Cayeux MC, Schneiter P, et al. Effects of lactate on glucose metabolism in healthy subjects and in severely injured hyperglycemic patients. Am J Physiol. 1995;268:E630–5. doi: 10.1152/ajpendo.1995.268.4.E630. [DOI] [PubMed] [Google Scholar]
- 13.Morgan K, Obici S, Rossetti L. Hypothalamic responses to long-chain fatty acids are nutritionally regulated. J Biol Chem. 2004;279:31139–48. doi: 10.1074/jbc.M400458200. [DOI] [PubMed] [Google Scholar]
- 14.Chari M, Lam CK, Wang PY, et al. Activation of central lactate metabolism lowers glucose production in uncontrolled diabetes and diet-induced insulin resistance. Diabetes. 2008;57:836–40. doi: 10.2337/db07-1464. [DOI] [PubMed] [Google Scholar]
- 15.Lam TK, Gutierrez-Juarez R, Pocai A, et al. Regulation of blood glucose by hypothalamic pyruvate metabolism. Science. 2005;309:943–7. doi: 10.1126/science.1112085. [DOI] [PubMed] [Google Scholar]
- 16.Ross R, Wang PY, Chari M, et al. Hypothalamic PKC regulates Glucose Production. Diabetes. 2008;57:2061–5. doi: 10.2337/db08-0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam CK, Chari M, Wang PY, et al. Central lactate metabolism regulates food intake. Am J Physiol Endocrinol Metab. 2008;295:E491–6. doi: 10.1152/ajpendo.90481.2008. [DOI] [PubMed] [Google Scholar]
- 18.Wang PY, Caspi L, Lam CK, et al. Upper intestinal lipids trigger a gut-brain-liver axis to regulate glucose production. Nature. 2008;452:1012–6. doi: 10.1038/nature06852. [DOI] [PubMed] [Google Scholar]
- 19.Bence KK, Delibegovic M, Xue B, et al. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–24. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- 20.Caspi L, Wang PY, Lam TK. A balance of lipid-sensing mechanisms in the brain and liver. Cell Metab. 2007;6:99–104. doi: 10.1016/j.cmet.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 21.Coppari R, Ichinose M, Lee CE, et al. The hypothalamic arcuate nucleus: a key site for mediating leptin’s effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Gelling RW, Morton GJ, Morrison CD, et al. Insulin action in the brin contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006;3:67–73. doi: 10.1016/j.cmet.2005.11.013. [DOI] [PubMed] [Google Scholar]
- 23.Inoue H, Ogawa W, Asakawa A, et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006;3:267–75. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- 24.Kievit P, Howard JK, Badman MK, et al. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–32. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 25.Konner AC, Janoschek R, Plum L, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–49. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Lam TK, Gutierrez-Juarez R, Pocai A, et al. Brain glucose metabolism controls the hepatic secretion of triglyceride-rich lipoproteins. Nat Med. 2007;13:171–80. doi: 10.1038/nm1540. [DOI] [PubMed] [Google Scholar]
- 27.Obici S, Feng Z, Morgan K, et al. Central administration of oleic acid inhibits glucose production and food intake. Diabetes. 2002;51:271–5. doi: 10.2337/diabetes.51.2.271. [DOI] [PubMed] [Google Scholar]
- 28.Obici S, Zhang BB, Karkanias G, et al. Hypothalamic insulin signaling is required for inhibition of glucose production. Nat Med. 2002;8:1376–82. doi: 10.1038/nm1202-798. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307:375–9. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- 30.Obici S, Feng Z, Arduini A, et al. Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med. 2003;9:756–61. doi: 10.1038/nm873. [DOI] [PubMed] [Google Scholar]