

Abstract
B-type natriuretic peptide (BNP) is a cardiac hormone, which plays a major role in body fluid and cardiovascular homeostasis. Produced by cardiac ventricles, its expression is highly regulated by various mediators. Canine cardiac fibroblasts have been identified as a source of BNP. Cardiac fibroblasts are key regulators of myocardial structure and function. We treated cultured human adult cardiac fibroblasts (HACF) with 2000 U/ml tumour necrosis factor-α (TNF-α), 200 U/ml interleukin-1α (IL-1α) or 50 ng/ml transforming growth factor-β (TGF-β) in the presence or absence of 500 nM fluvastatin. N-terminal pro-BNP (Nt-proBNP) concentration was determined by a competitive enzyme immunoassay. RealTime polymerase chain reaction (real-time PCR) was performed to investigate changes in BNP mRNA expression. Nt-proBNP peptide was present in the conditioned media of HACF and incubation with fluvastatin significantly reduced Nt-proBNP peptide levels. Treatment of HACF with TNF-α, IL-1α or TGF-β significantly increased Nt-proBNP levels compared with untreated cells. This effect was completely abolished in the presence of fluvastatin. Real-time PCR analysis confirmed these changes at the level of mRNA expression. Our data suggest that cardiac fibroblasts are a potential source of BNP in the human heart. Pro-inflammatory cytokines, associated with ventricular dysfunction and cardiac fibrosis, seem to be major inducers of BNP production in cardiac fibroblasts. This effect can be reverted by a statin. Based on our data, we speculate that elevated plasma BNP levels might not only reflect increased myocardial stretch but also inflammatory and remodelling processes. A possible benefit of statin-induced reduction in BNP production requires further studies.
Keywords: B-type natriuretic peptides, human adult cardiac fibroblasts, statins, pro-inflammatory cytokines
Introduction
B-type natriuretic peptide (BNP) is a cardiac hormone with potent diuretic, natriuretic and vasodilatory effects that plays a major role in body fluid and cardiovascular homeostasis [1]. It is predominantly produced in cardiac ventricles and is secreted after myocardial stretch mainly from the left ventricular wall [1]. Various biomolecules such as cardiotrophin-1, endothelin-I, angiotensin-II, lipopolysaccharide, interleukin-1 (IL-1), triiodothyronine and α-adrenergic agonists have been shown to increase BNP expression in cardiac myocytes [1, 2]. Recently, canine cardiac fibroblasts have been identified as a source for BNP [3]. Cardiac fibroblasts are activated in pathological conditions and play an important role in the inflammatory response and in the regulation of matrix remodelling in response to myocardial injury [4].
In this paper, we show that human cardiac fibroblasts express BNP and that its expression is up-regulated by cytokines such as tumour necrosis factor-α (TNF-α), IL-1α and transforming growth factor (TGF)-β that have been implicated in the pathogenesis of ventricular remodelling and progression of cardiac dysfunction and whose plasma levels are elevated in patients with heart failure or myocardial infarction and correlate with disease severity and progression [5–8]. Furthermore, we provide evidence that this up-regulation of BNP by inflammatory mediators is counteracted by fluvastatin, a 3-hydroxy-3-methylglutaryl-coenzyme-A-reductase inhibitor.
Materials and methods
Materials
Fluvastatin (kindly provided by Novartis, Basel, Switzerland) was handled as described previously [9]. Recombinant human (rh) TNF-α, rh IL-1α, rh TGF-β were purchased from Roche (Basel, Switzerland) and rh IL-6, rh IL-11, rh oncostatin M (OSM) and rh leukaemia inhibitory factor (LIF) were purchased from R&D Systems (Minneapolis, MN).
Methods
Human adult cardiac fibroblasts (HACF) and human adult cardiac myocytes (HACMs) were isolated, characterized and cultured as previously published [10]. More than 95% of HACF stained positive for fibroblast-specific antigen. HACF neither stained for the cardiac myocyte markers troponin I, tropomyosin, cardiotin and myocardial muscle actin, nor for the endothelial marker vWF or for smooth muscle actin, ruling out contamination with myocytes, smooth muscle cells and endothelial cells, respectively. More than 95% of HACM stained positive for the cardiac myocyte markers troponin I, tropomyosin, cardiotin and myocardial muscle actin. HACM neither stained for fibroblast-specific antigen nor for the endothelial marker vWF or for smooth muscle actin, ruling out contamination with fibrobasts, smooth muscle cells and endothelial cells, respectively. All human material was obtained and processed according to the recommendations of the hospital’s Ethics Committee, including informed consent.
Twenty-four hours prior to the experiments, cells were starved with M199 containing 0.1% BSA (both Sigma, St. Louis, MO). For stimulation experiments with cytokines, IL-1α, TNF-α or TGF-β with and without fluvastatin were added to the cells at the concentrations indicated. To characterize a possible effect of fluvastatin in additional experiments, HACF were simultaneously treated with the respective cytokine and 500 nM fluvastatin alone or together with 100 μM mevalonate (Sigma) or 10 μM geranyl-geranyl pyrophosphate (GGPP; Sigma) or pre-treated with 5 μM Y26372 [(+)-(R)-trans-4-(1-aminoethyl)-N-(4-pyridyl)cyclohexanecarboxamide dihydrochloride] (Sigma), a specific inhibitor of Rho-associated kinases (ROCKs) for 1 hr before addition of the respective cytokine. Nt-proBNP levels in the conditioned media of HACF were measured by a competitive enzyme immunoassay using a sheep antibody specific for proBNP 8-29 according to the manufacturer’s instruction (Biomedica, Vienna, Austria). The detection limit of this assay is 5 fmol/ml.
For specific proBNP mRNA quantification cells were treated with cytokines as described earlier and mRNA was isolated using QuickPrep™ Micro mRNA Purification Kit (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer’s instructions. Real-time PCR was performed using LightCycler-RNA Master SYBR Green I (Roche) according to the manufacturer’s instructions. Primers are shown in Table 1 and were designed using the LightCycler Probe Design Software Version 1.0 (Roche) and the Primer3 Software (http://frodo.wi.mit.edu/). The amplification conditions consisted of an initial incubation at 61°C for 20 min., followed by incubation at 95°C for 30 sec., 50 cycles of 95°C for 1 sec., 65°C for 10 sec. and 72°C for 10 sec., a melting step from 45°C to 95°C increasing 0.1°C per second and a final cooling to 40°C. Specific mRNA for the receptors gp130, IL-6 receptor (IL-6R), IL-11R, OSMR and LIFR was determined by Real-time PCR using conditions as described recently [11]. Real-time PCR for IL-1 receptor-1 (IL-1R1), IL-1R2, TGF-βR1, TGF-β2, TNFR superfamily 1A (TNFRSF1A) and TNFRSF2A was performed using LightCycler® TaqMan® Master (Roche) according to the manufacturer’s instructions. Primers, shown in Table 1, were designed using the Roche Universal ProbeLibrary Assay Design Center (http://www.universalprobelibrary.com/). The amplification conditions consisted of an initial incubation at 95°C for 10 min., followed by 45 cycles of 95°C for 10 sec., 63°C for 20 sec. and 72°C for 6 sec. and a final cooling to 40°C. Data were analysed using LightCycler Software Version 3.5 (Roche).
Table 1.
Primers used for real-time PCR
| Primer | fwd-Primer (corresponding position) | Rev-primer (corresponding position) | UPL probe (amplicon size [bp]) |
|---|---|---|---|
| proBNP | 5′-CAG CCT CGG ACT TGG AAA-3′ (200–217) | 5′-ATC TTG GGG CTT CGT GGT-3′ (416–398) | (199) |
| GAPDH | 5′-GAGGTGTGAGTGGGATGGTGG-3′ (681–702) | 5′-GCCTGCTTCACCACCTTCTTG-3′ (890–869) | (189) |
| IL1R1 | 5′-GTT CAT TTA TGG AAG GGA TGA-3′ (1350–1371) | 5′-TCT GCT TTT CTT TAC GTT TTC ATT-3′ (1403–1427) | #60 (78) |
| IL1R2 | 5′-TAC GCA CCA CAG TCA AGG AA-3′ (1239–1258) | 5′-AAG AAG GCC AGT GAA AGT GG-3′ (1295–1314) | #2 (76) |
| TGFBR1 | 5′-AAA TTG CTC GAC GAT GTT CC-3′ (1305–1324) | 5′-CAT AAT AAG GCA GTT GGT AAT CTT CA-3′ (1339–1364) | #31 (60) |
| TGFBR2 | 5′-GGG AAA TGA CAT CTC GCT GTA-3′ (1821–1841) | 5′-CAC CTT GGA ACC AAA TGG AG-3′ (1872–1891) | #7 (71) |
| TNFRSF1A | 5′-GAG AGG CCA TAG CTG TCT GG-3′ (261–280) | 5′-GAG GGG TAT ATT CCC ACC AAC-3′ (335–355) | #59 (95) |
| TNFRSF1B | 5′-GCA GTG CGT TGG ACA GAA G-3′ (1093–1111) | 5′-CCA CCA GGG GAA GAA TCT G-3′ (1192–1210) | #5 (118) |
To determine possible cytotoxic effects of fluvastatin, lactate dehydrogenase (LDH) leakage was measured in cultures treated with the statin using a commercially available assay for photometric determination of LDH activity (Sigma). In addition, cells were counted at the end of the respective experiments using a haemocytometer.
We used t-test for independent variables to compare Nt-proBNP levels in the conditioned media of treated cells with those of control cells as well as to detect significant effects of fluvastatin on BNP secretion. Statistical significance was present if P < 0.05.
Results
Nt-proBNP concentration increased dose dependently in condition media of HACF treated with IL-1α, TNF-α or TGF-β, whereas IL-6, IL-11, OSM or LIF had no effect (Fig. 1A and B). In further analyses, we used the most effective concentrations of IL-1α (200 U/ml), TNF-α (2000 U/ml) or TGF-β (50 ng/ml). In addition, specific mRNA IL-1R1, IL-1R2, TGF-βR1, TGF-β2, TNFRSF1A, TNFRSF2A, gp130, IL-6R, IL-11R, OSMR and LIFR was detected in these cells by real-time PCR (data not shown). As can be also seen from Fig. 1A, HACMs produced approximately 1/10 of the amount of Nt-proBNP secreted by HACF under the same culture conditions. Furthermore, Nt-proBNP production was not affected by IL-1α or TNF-α in HACM.
Figure 1.

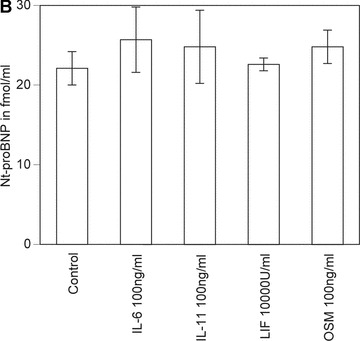
Effect of IL-1α, TNF-α, TGF-β, IL-6, IL-11, OSM and LIF on proBNP production in HACF and HACM. Confluent monolayers of HACF (open bars) were incubated for 24 hrs with or without 2, 20 and 200 U/ml IL-1α; 20, 200 and 2000 U/ml TNF-α; or 0.5, 5 and 50 ng/ml TGF-β, respectively (panel A). Confluent monolayers of HACM (hatched bars) were incubated for 24 hrs with or without 200 U/ml IL-1α; or 2000 U/ml TNF-α (panel A). Confluent monolayers of HACF were incubated for 24 hrs with or without 100 ng/ml IL-6; 100 ng/ml IL-11; 10,000 U/ml LIF; or 100 ng/ml OSM, respectively (panel B). Nt-proBNP was determined in conditioned media using a specific ELISA and values are given in fmol/ml and represent mean values ± S.D. of three independent determinations. Experiments were performed three times with cells isolated from three different donors. A representative experiment is shown. ***P < 0.001, **P < 0.005, *P < 0.05 as compared with untreated control cells.
Fluvastatin dose dependently reduced baseline Nt-proBNP secretion of untreated HACF. A significant reduction of Nt-proBNP was seen with concentrations of fluvastatin between 10 nM and 500 nM (Fig. 2A). Furthermore, the increase in Nt-proBNP secretion by HACF induced by IL-1α, TNF-α and TGF-β, respectively, was reduced to Nt-proBNP levels seen in untreated control cells by the presence of 500 nM fluvastatin. A significant reduction of Nt-proBNP was also seen with 100 nM fluvastatin (Fig. 2B–D). To determine whether this effect of fluvastatin on BNP secretion depends on its capacity to inhibit mevalonate synthesis by HMG-CoA reductase, HACF were simultaneously treated with the respective cytokine and 500 nM fluvastatin alone or together with 100 μM mevalonate or 10 μM GGPP. In these experiments, the inhibiting effect of fluvastatin on IL-1α-, TNF-α- and TGF-β-induced Nt-proBNP secretion was completely reversed by mevalonate or GGPP. Mevalonate or GGPP alone had no effect on Nt-proBNP production by HACF (data not shown). When cells were incubated with fluvastatin for 48 hrs at a concentration of 500 nM, cell viability was not significantly affected by this treatment as LDH leakage did not exceed 105% of control. In addition, no changes in cell numbers were observed. A cytotoxic effect was only observed at concentrations greater than 5 μM of the statin (data not shown). As can be seen from Fig. 2E, the ROCKs inhibitor Y26372 mimicked the effect of fluvastatin as it inhibited IL-1α-, TNF-α- and TGF-β-stimulated Nt-proBNP secretion. Y26372 at the concentration of 5 μM did neither affect cell viability as judged by LDH leakage nor change cell numbers.
Figure 2.



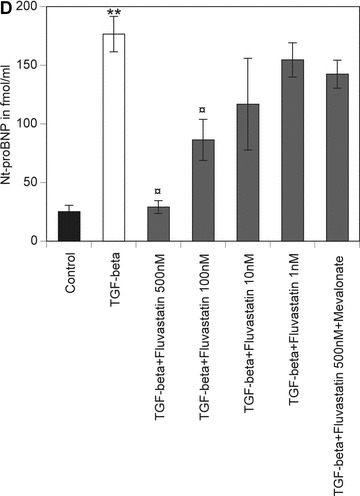

Effect of TNF-α, IL-1α, TGF-β and fluvastatin on proBNP production in HACF. Confluent monolayers of HACF were incubated for 24 hrs without (panel A) or with 200 U/ml IL-1α (panel B), 2000 U/ml TNF-α (panel C) or 50 ng/ml TGF-β (panel D), respectively, in the absence (open bars) or presence of 500 nM, 100 nM, 10 nM or 1 nM fluvastatin without or with 100 μM mevalonate or 10 μM GGPP (hatched bars). In panel E, confluent monolayers of HACF were pre-treated with medium (open bars) or 5 μM Y27632 (hatched bars) for 1 hr and then incubated for 24 hrs without or with 200 U/ml IL-1α, 2000 U/ml TNF-α or 50 ng/ml TGF-β, respectively. Nt-proBNP was determined in conditioned media using a specific ELISA and values are given in fmol/ml and represent mean values ± S.D. of three independent determinations. Experiments were performed 3 times with cells isolated from three different donors. A representative experiment is shown. **P < 0.001, *P < 0.05 as compared with untreated control cells; P¤ < 0.001 as compared with cells incubated without fluvastatin or Y27632.
Incubation of HACF for 24 hrs with IL-1α (200 U/ml), TNF-α (2000 U/ml) or TGF-β (50 ng/ml), respectively, significantly increased BNP mRNA levels, whereas fluvastatin (500 nM) blunted the effects of these cytokines (Table 2). It should be noted that fluvastatin at a concentration of 500 nM did not systematically decrease mRNA levels in cardiac fibroblasts as it had no down-regulating effect on mRNA levels specific for IL-6, IL-8, plasminogen activator inhibitor type-1 (PAI-1) and vascular endothelial growth factor (VEGF) in these cells. When HACF were incubated for 24 hrs with 500 nM fluvastatin the respective mRNA levels increased slightly for IL-6, IL-8 and PAI-1 (1.3-fold, 1.5-fold and 1.3-fold, respectively) and 2.3-fold for VEGF. Furthermore, mRNA specific for GAPDH did not change significantly (1.1-fold over control) when the cells were treated with 500 nM fluvastatin for 24 hrs.
Table 2.
Effect of TNF-α, IL-1α, TGF-β and fluvastatin on proBNP mRNA expression in HACF
| Without fluvastatin | With fluvastatin 500 nM | |
|---|---|---|
| Control | 1.0 ± 0.1 | 0.6 ± 0.1 |
| IL-1α 2000 U/ml | 2.9 ± 0.5* | 0.8 ± 0.1† |
| TNF-α 200 U/ml | 2.9 ± 0.7* | 0.9 ± 0.2† |
| TGF-β 50 ng/ml | 2.6 ± 0.5* | 0.9 ± 0.2† |
Confluent monolayers of HACF were incubated for 24 hrs without or with 200 U/ml IL-1·, 2000 U/ml TNF-α or 50 ng/ml TGF-β, respectively, in the absence or presence of 500 nM fluvastatin. Real-time PCR with primers specific for BNP and GAPDH was performed. BNP mRNA levels were normalized according to the respective GAPDH levels. Values are given as fold of control and represent mean values ± S.D. of three independent determinations. Experiments were performed 2 times with HACF isolated from two different donors. A representative experiment is shown.
P < 0.001 as compared with untreated control cells,
P < 0.001 as compared with cells incubated without fluvastatin.
Discussion
Initial results on BNP production originate from in vitro data of cultivated neonatal rat cardiac myocytes. Limited data are, however, available on human myocardial cells [12]. In this study, we provide evidence for the first time that similar to canine cardiac fibroblast [3], cultured human adult cardiac fibroblasts also express BNP-mRNA and secrete BNP and that inflammatory mediators such as TNF-α, IL-1-α and TGF-β are potent inducers of BNP mRNA expression and protein secretion by these cells. HACF also express the respective receptors for TNF-α, IL-1-α and TGF-β. Other inflammatory cytokines such as IL-6, IL-11, OSM or LIF did not affect BNP production in HACF. The respective receptors, however, were expressed by these cells. In contrast, human adult cardiac myocytes produced only 1/10 of the amount of BNP secreted by cardiac fibroblasts and BNP production in human cardiac myocytes was not affected by TNF-α or IL-1-α. It should be emphasized that cultured rat cardiac myocytes produced only minute amounts of BNP as compared with mixed cultures of cardiac myocytes and non-myocytes. In addition, only these mixed cultures responded to IL-1 with a significant increase in BNP production [13].
TNF-α, IL-1-α and TGF-β, shown to up-regulate BNP in cardiac fibroblasts in our study, are known to be associated with cardiac dysfunction and remodelling of the myocardium and are elevated in the plasma of patients with heart failure or myocardial infarction [5–8]. Thus, one could speculate that BNP might not only reflect reduced ventricular function but also inflammatory and/or remodelling processes within the myocardium.
Statin therapy in patients with heart failure improves ventricular function and symptoms of heart failure and reduces ventricular dimensions as well as plasma concentrations of BNP, TNF-α and IL-6 independent of plasma cholesterol levels [14, 15]. In the present study, fluvastatin between 10 nM and 500 nM significantly and dose dependently decreased baseline levels of BNP. Fluvastatin at concentrations of 100 nM and 500 nM also reduced the cytokine-induced expression of BNP whereby fluvastatin at the latter concentration reduced the cytokine-induced expression of BNP to control levels. In this respect, it should be noted that in patients undergoing 6 weeks of statin therapy with 40 mg of fluvastatin daily, plasma concentrations of approximately 250 nM were measured [16]. In our study, the inhibiting effect of fluvastatin on the IL-1α-, TNF-α- and TGF-β-induced up-regulation of Nt-proBNP production by cardiac fibroblasts was completely reversed by mevalonate and GGPP, suggesting that this effect was brought about by inhibition of the mevalonic acid pathway and protein prenylation. In particular, GGPP is essential for the isoprenylation of the GTPases Rap, Rab and Rho. Blocking isoprenylation of these GTPases causes their inactivation and thus prevents ROCK signalling [17]. Indeed, statins have been shown to inhibit the prenylation of ROCK [18–20]. Of note, Rho is crucial for the activation of rac, which is involved in IL-1-dependent regulation of the human BNP promoter [21]. We show here that the ROCK inhibitor Y27632 inhibits the IL-1α-, TNF-α- and TGF-β-induced up-regulation of Nt-proBNP similarly to fluvastatin. These results suggest that fluvastatin exerts its effect on IL-1α-, TNF-α- and TGF-β-induced up-regulation of Nt-proBNP by inhibiting the ROCK signalling pathway. The stimulating effect of IL-1α, TNF-α and TGF-β on BNP expression and its inhibition by fluvastatin was also seen at the level of specific BNP-mRNA expression in human cardiac fibroblasts. It should be emphasized, however, that we could show here that fluvastatin does not systematically decrease mRNA levels in cardiac fibroblasts as in these cells it slightly increased mRNA levels specific for the IL-6, IL-8 and PAI-1, whereas mRNA levels specific for VEGF doubled after incubation with fluvastatin in cardiac fibroblasts. Furthermore, mRNA specific for the housekeeping gene GAPDH was not affected significantly by fluvastatin. However, at this point further studies are needed to determine if the down-regulatory effect of fluvastatin on basal and cytokine-induced BNP expression in human cardiac fibroblasts is brought about by modulating directly gene transcription, stability of mRNA or post-translational modification. In that respect, it is of interest that it was shown recently that atorvastatin decreases the stability of mRNA for monocyte chemoattractant protein-1 (MCP-1) [22].
Our finding that human cardiac fibroblasts secrete BNP in considerable amount reveals a previously unknown function of these cells. Cardioprotective effects of BNP, like natriuresis, diuresis and inhibition of the renin–angiotensin–aldosterone system have been shown in healthy individuals and in patients with heart failure suggesting that elevated BNP expression is an adaptive mechanism of the heart in order to maintain cardiac performance during ventricular overload [2]. On the other hand, BNP has been shown to enhance hypoxia-induced apoptosis of myocardial cells [23], to have negative inotropic effects within the myocardium and to adversely affect cardiac contractility through inhibition of the expression of sarcoplasmic reticulum Ca2+ ATPase [24, 25]. BNP also inhibits expression of several pro-inflammatory cytokines, which are essential to protect the heart against reperfusion injury and ischaemic myocardial damage [26, 27], increases the expression of matrix metalloproteinases involved in remodelling [3], promotes the accumulation of inflammatory cells in the infarcted myocardium [28], suppresses the mitochondrial respiration in the myocardium, reduces the effects of catecholamines, induces systemic vasodilation and potentiates the generation of nitric oxide, which in turn is a known inhibitor of myocardial contractility [25, 29–32]. Thus, inhibiting the excessive expression of BNP, as shown by our study, by administration of statins might at least in theory result in prevention of ventricular dilation, and could improve myocardial performance and outcome of patients. Certainly, this possibility requires further intensive investigations, in particular in the light of a recent study published by the GISSI-group, which did not find any benefit for patients suffering from chronic heart failure who received rosuvastatin [33].
Sources of funding
The Fund for the Promotion of Scientific Research (Grant No. S9409-B11) and the Association for the Promotion of Research in Arteriosclerosis, Thrombosis and Vascular Biology supported this work.
References
- 1.Jarai R, Wojta J, Huber K. Circulating B-type natriuretic peptides in patients with acute coronary syndromes. Pathophysiological, prognostical and therapeutical considerations. Thromb Haemost. 2005;94:926–32. doi: 10.1160/TH05-06-0395. [DOI] [PubMed] [Google Scholar]
- 2.Hall C. Essential biochemistry and physiology of (NT-pro)BNP. Eur J Heart Fail. 2004;6:257–60. doi: 10.1016/j.ejheart.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 3.Tsuruda T, Boerrigter G, Huntley BK, et al. Brain natriuretic peptide is produced in cardiac fibroblasts and induces matrix metalloproteinases. Circ Res. 2002;91:1127–34. doi: 10.1161/01.res.0000046234.73401.70. [DOI] [PubMed] [Google Scholar]
- 4.Manabe I, Shindo T, Nagai R. Gene expression in fibroblasts and fibrosis: involvement in cardiac hypertrophy. Circ Res. 2002;91:1103–13. doi: 10.1161/01.res.0000046452.67724.b8. [DOI] [PubMed] [Google Scholar]
- 5.Bradham WS, Moe G, Wendt KA, et al. TNF-alpha and myocardial matrix metalloproteinases in heart failure: relationship to LV remodeling. Am J Physiol Heart Circ Physiol. 2002;282:H1288–95. doi: 10.1152/ajpheart.00526.2001. [DOI] [PubMed] [Google Scholar]
- 6.Oral H, Sivasubramanian N, Dyke DB, et al. Myocardial proinflammatory cytokine expression and left ventricular remodeling in patients with chronic mitral regurgitation. Circulation. 2003;107:831–7. doi: 10.1161/01.cir.0000049745.38594.6d. [DOI] [PubMed] [Google Scholar]
- 7.Lim H, Zhu YZ. Role of transforming growth factor-beta in the progression of heart failure. Cell Mol Life Sci. 2006;63:2584–96. doi: 10.1007/s00018-006-6085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen D, Assad-Kottner C, Orrego C, et al. Cytokines and acute heart failure. Crit Care Med. 2008;36:S9–16. doi: 10.1097/01.CCM.0000297160.48694.90. [DOI] [PubMed] [Google Scholar]
- 9.Wiesbauer F, Kaun C, Zorn G, et al. HMG CoA reductase inhibitors affect the fibrinolytic system of human vascular cells in vitro: a comparative study using different statins. Br J Pharmacol. 2002;135:284–92. doi: 10.1038/sj.bjp.0704454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weiss TW, Kvakan H, Kaun C, et al. The gp130 ligand oncostatin M regulates tissue inhibitor of metalloproteinases-1 through ERK1/2 and p38 in human adult cardiac myocytes and in human adult cardiac fibroblasts: a possible role for the gp130/gp130 ligand system in the modulation of extracellular matrix degradation in the human heart. J Mol Cell Cardiol. 2005;39:545–51. doi: 10.1016/j.yjmcc.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 11.Weiss TW, Speidl WS, Kaun C, et al. Glycoprotein 130 ligand oncostatin-M induces expression of vascular endothelial growth factor in human adult cardiac myocytes. Cardiovasc Res. 2003;59:628–38. doi: 10.1016/s0008-6363(03)00463-2. [DOI] [PubMed] [Google Scholar]
- 12.Kapoun AM, Liang F, O’Young G, et al. B-type natriuretic peptide exerts broad functional opposition to transforming growth factor-beta in primary human cardiac fibroblasts: fibrosis, myofibroblast conversion, proliferation, and inflammation. Circ Res. 2004;94:453–61. doi: 10.1161/01.RES.0000117070.86556.9F. [DOI] [PubMed] [Google Scholar]
- 13.Harada E, Nakagawa O, Yoshimura M, et al. Effect of interleukin-1 beta on cardiac hypertrophy and production of natriuretic peptides in rat cardiocyte culture. J Mol Cell Cardiol. 1999;31:1997–2006. doi: 10.1006/jmcc.1999.1030. [DOI] [PubMed] [Google Scholar]
- 14.Node K, Fujita M, Kitakaze M, et al. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation. 2003;108:839–43. doi: 10.1161/01.CIR.0000084539.58092.DE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khush KK, Waters DD, Bittner V, et al. Effect of high-dose atorvastatin on hospitalizations for heart failure: subgroup analysis of the Treating to New Targets (TNT) Study. Circulation. 2007;115:576–83. doi: 10.1161/CIRCULATIONAHA.106.625574. [DOI] [PubMed] [Google Scholar]
- 16.Smit JW, Wijnne HJ, Schobben F, et al. Effects of alcohol and fluvastatin on lipid metabolism and hepatic function. Ann Intern Med. 1995;122:678–80. doi: 10.7326/0003-4819-122-9-199505010-00006. [DOI] [PubMed] [Google Scholar]
- 17.Rikitake Y, Liao JK. Rho GTPases, statins, and nitric oxide. Circ Res. 2005;97:1232–5. doi: 10.1161/01.RES.0000196564.18314.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shiga N, Hirano K, Hirano M, et al. Long-term inhibition of RhoA attenuates vascular contractility by enhancing endothelial NO production in an intact rabbit mesenteric artery. Circ Res. 2005;96:1014–21. doi: 10.1161/01.RES.0000165483.34603.91. [DOI] [PubMed] [Google Scholar]
- 19.Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- 20.Degraeve F, Bolla M, Blaie S, et al. Modulation of COX-2 expression by statins in human aortic smooth muscle cells. Involvement of geranylgeranylated proteins. J Biol Chem. 2001;276:46849–55. doi: 10.1074/jbc.M104197200. [DOI] [PubMed] [Google Scholar]
- 21.He Q, LaPointe MC. Interleukin-1beta regulation of the human brain natriuretic peptide promoter involves Ras-, Rac-, and p38 kinase-dependent pathways in cardiac myocytes. Hypertension. 1999;33:283–9. doi: 10.1161/01.hyp.33.1.283. [DOI] [PubMed] [Google Scholar]
- 22.Tanimoto A, Murata Y, Wang KY, et al. Monocyte chemoattractant protein-1 expression is enhanced by granulocyte-macrophage colony-stimulating factor via Jak2-Stat5 signaling and inhibited by atorvastatin in human monocytic U937 cells. J Biol Chem. 2008;283:4643–51. doi: 10.1074/jbc.M708853200. [DOI] [PubMed] [Google Scholar]
- 23.Wang TN, Ge YK, Li JY, et al. B-type natriuretic peptide enhances mild hypoxia-induced apoptotic cell death in cardiomyocytes. Biol Pharm Bull. 2007;30:1084–90. doi: 10.1248/bpb.30.1084. [DOI] [PubMed] [Google Scholar]
- 24.Kogler H, Schott P, Toischer K, et al. Relevance of brain natriuretic peptide in preload-dependent regulation of cardiac sarcoplasmic reticulum Ca2+ ATPase expression. Circulation. 2006;113:2724–32. doi: 10.1161/CIRCULATIONAHA.105.608828. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Q, Moalem J, Tse J, et al. Effects of natriuretic peptides on ventricular myocyte contraction and role of cyclic GMP signaling. Eur J Pharmacol. 2005;510:209–15. doi: 10.1016/j.ejphar.2005.01.031. [DOI] [PubMed] [Google Scholar]
- 26.Goukassian DA, Qin G, Dolan C, et al. Tumor necrosis factor-alpha receptor p75 is required in ischemia-induced neovascularization. Circulation. 2007;115:752–62. doi: 10.1161/CIRCULATIONAHA.106.647255. [DOI] [PubMed] [Google Scholar]
- 27.Vellaichamy E, Kaur K, Pandey KN. Enhanced activation of pro-inflammatory cytokines in mice lacking natriuretic peptide receptor-A. Peptides. 2007;28:893–9. doi: 10.1016/j.peptides.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kawakami R, Saito Y, Kishimoto I, et al. Overexpression of brain natriuretic peptide facilitates neutrophil infiltration and cardiac matrix metalloproteinase-9 expression after acute myocardial infarction. Circulation. 2004;110:3306–12. doi: 10.1161/01.CIR.0000147829.78357.C5. [DOI] [PubMed] [Google Scholar]
- 29.Dejam A, Hunter CJ, Tremonti C, et al. Nitrite infusion in humans and nonhuman primates. Endocrine effects, pharmacokinetics, and tolerance formation. Circulation. 2007;116:1821–31. doi: 10.1161/CIRCULATIONAHA.107.712133. [DOI] [PubMed] [Google Scholar]
- 30.Erusalimsky JD, Moncada S. Nitric oxide and mitochondrial signaling. From physiology to pathophysiology. Arterioscler Thromb Vasc Biol. 2007;27:2524–31. doi: 10.1161/ATVBAHA.107.151167. [DOI] [PubMed] [Google Scholar]
- 31.Cosby K, Partovi KS, Crawford JH, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 32.Van Der Zander K, Houben AJ, Kroon AA, et al. Nitric oxide and potassium channels are involved in brain natriuretic peptide induced vasodilatation in man. J Hypertens. 2002;20:493–9. doi: 10.1097/00004872-200203000-00025. [DOI] [PubMed] [Google Scholar]
- 33.Gissi-HFI . Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet. 2008;372:1231–9. doi: 10.1016/S0140-6736(08)61240-4. [DOI] [PubMed] [Google Scholar]